

Abstract
In a restricted group of opportunistic fungal pathogens the universal leucine CUG codon is translated both as serine (97%) and leucine (3%), challenging the concept that translational ambiguity has a negative impact in living organisms. To elucidate the molecular mechanisms underlying the in vivo tolerance to a nonconserved genetic code alteration, we have undertaken an extensive structural analysis of proteins containing CUG-encoded residues and solved the crystal structures of the two natural isoforms of Candida albicans seryl-tRNA synthetase. We show that codon reassignment resulted in a nonrandom genome-wide CUG redistribution tailored to minimize protein misfolding events induced by the large-scale leucine-to-serine replacement within the CTG clade. Leucine or serine incorporation at the CUG position in C. albicans seryl-tRNA synthetase induces only local structural changes and, although both isoforms display tRNA serylation activity, the leucine-containing isoform is more active. Similarly, codon ambiguity is predicted to shape the function of C. albicans proteins containing CUG-encoded residues in functionally relevant positions, some of which have a key role in signaling cascades associated with morphological changes and pathogenesis. This study provides a first detailed analysis on natural reassignment of codon identity, unveiling a highly dynamic evolutionary pattern of thousands of fungal CUG codons to confer an optimized balance between protein structural robustness and functional plasticity.
Keywords: aminoacyl-tRNA synthetase, morphogenesis, mitogen-activated protein kinase pathway, Ras1, X-ray crystallography
Genetic code alterations and ambiguity are widespread in nature even though it is not yet clear how their negative impact is overcome (1). Expansion of the genetic code to selenocysteine (Sec) and pyrrolysine (Pyl) provides, however, a glimpse of advantages that may explain the evolution of codon reassignments under negative selective pressure (2). Sec is inserted into the genetic code of bacteria and eukaryotes by a specific selenocysteyl-tRNASec, which places selenocysteine in the catalytic site of selenoproteins increasing their chemical reaction rate relative to cysteine-containing homologues (3). Similarly, Pyl is cotranslationally introduced into the active center of methyltransferases of Methanosarcineace spp. and of Desulfitobacterium hafniense, a symbiont of the gutless worm Olavius algarvensis, where it plays a fundamental role in methane biosynthesis (2, 4). Another interesting case involves the mammalian methionyl-tRNA synthetase (MetRS), which is modified under environmental stress and misacylates noncognate tRNAs with reactive oxygen species (ROS)-scavenging methionine (5), therefore protecting proteins from oxidative damage. A similar adaptive mechanism apparently drove mitochondrial reassignment of Ile AUA codons to methionine (6).
In Candida albicans and in most other CTG clade species a mutant serine tRNA (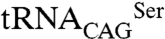 ) has the peculiarity of decoding leucine CUG codons both as serine and leucine (7, 8). SerRS is the main charging enzyme of
) has the peculiarity of decoding leucine CUG codons both as serine and leucine (7, 8). SerRS is the main charging enzyme of 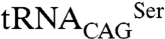 and the leucyl-tRNA synthetase (LeuRS) is a poor competitor (7), resulting in the incorporation of both leucine (3%) and serine (97%) at CUG sites during ribosome decoding (9). This genetic code alteration occurred over 100 ± 15 million years, during which the original CUG codons were erased from the genome of the CTG clade ancestor and approximately 26,000 unique CUG codons arose from serine or other polar amino acid codons in extant CTG clade species (10, 11). Leucine incorporation can be increased up to 28% in C. albicans without visible effects on growth rate but with an impressive impact in cell morphology (7), but the molecular mechanisms of ambiguity tolerance, which could explain the CUG reassignment pathway remain unclear.
and the leucyl-tRNA synthetase (LeuRS) is a poor competitor (7), resulting in the incorporation of both leucine (3%) and serine (97%) at CUG sites during ribosome decoding (9). This genetic code alteration occurred over 100 ± 15 million years, during which the original CUG codons were erased from the genome of the CTG clade ancestor and approximately 26,000 unique CUG codons arose from serine or other polar amino acid codons in extant CTG clade species (10, 11). Leucine incorporation can be increased up to 28% in C. albicans without visible effects on growth rate but with an impressive impact in cell morphology (7), but the molecular mechanisms of ambiguity tolerance, which could explain the CUG reassignment pathway remain unclear.
Our data show that reintroduction of CUG codons in CTG clade genomes was not random, avoiding structurally sensitive sites where Ser/Leu ambiguity could induce protein misfolding. The crystal structures of C. albicans SerRS isoforms, containing serine or leucine at its single CUG position unveil the unique architectural features of a cytoplasmic eukaryotic SerRS and show that incorporation of leucine or serine induces only localized structural changes. Coexpression of the 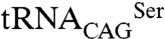 –SerRS orthogonal pairs and in vitro serine activation assays demonstrate that both SerRS isoforms are functional, although with differences in serine activation/serylation activity. In addition, identification of CUG-encoded residues in conserved functional sites within proteins involved in signal transduction pathways further highlights a correlation between codon ambiguity and functional plasticity in multiple molecular targets associated with morphological changes and pathogenesis.
–SerRS orthogonal pairs and in vitro serine activation assays demonstrate that both SerRS isoforms are functional, although with differences in serine activation/serylation activity. In addition, identification of CUG-encoded residues in conserved functional sites within proteins involved in signal transduction pathways further highlights a correlation between codon ambiguity and functional plasticity in multiple molecular targets associated with morphological changes and pathogenesis.
Results
Unique Distribution of CUG-Encoded Residues in C. albicans Proteins.
Ambiguous translation of CUG codons affects 67% (4168) of C. albicans protein-coding genes (7). To elucidate the structural and functional implications of such ambiguity, the genome of C. albicans was screened for proteins containing at least one CUG-encoded residue and for which orthologs were identified in Saccharomyces cerevisiae. The resulting 680 protein sequences (2,071 CUG-encoded residues in C. albicans; 5,053 CUG-encoded leucines in S. cerevisiae) were aligned with the orthologs from six related fungi, revealing two distinct CUG-codon distribution profiles (Fig. 1A and Dataset S1) (10, 11). In C. albicans CUG codons are mainly located in nonconserved sequence positions (90%) (Fig. S1) where both aliphatic leucine and polar serine can be accommodated without major disruption of protein structure and function. In contrast, CUG-encoded leucines in S. cerevisiae proteins display a considerably more uniform distribution (22% conserved; 27% semiconserved) (Fig. 1A, Fig. S1, and Dataset S1). The CUG-encoded residue distribution pattern observed for C. albicans is strikingly preserved across the CTG clade species despite the lack of CUG codon positional conservation amongst orthologous genes (Fig. 1B and Fig. S1).
Fig. 1.

Misfolding-minimizing distribution of CUG-encoded residues in C. albicans and CTG clade ORFs. (A) In S. cerevisiae CUG-encoded residues are uniformly distributed in the protein sequence, whereas in C. albicans they are mainly located at nonconserved positions (only 10% in conserved or strictly conserved sites). (B) The peculiar distribution of CUG-encoded residues in nonconserved regions of the proteins is preserved within the CTG clade. (C) In C. albicans and other CTG clade ORFs most CUG-encoded residues are solvent accessible, whereas over 60% are buried in S. cerevisiae proteins. (D) CTG clade CUG-encoded residues in nonconserved regions of the protein sequence are predominantly exposed, whereas those in conserved sites are evenly distributed between the protein surface and its core.
Analysis of the position and relative surface accessibility of the CUG-encoded residues in the three-dimensional models of C. albicans (Table S1) and S. cerevisiae proteins revealed a predominant surface distribution in C. albicans, very distinct from the distribution observed in S. cerevisiae, where those residues are mostly buried (Fig. 1C and Fig. S1). The peculiar pattern of surface accessibility of CUG-encoded residues observed in C. albicans is a general feature of fungi belonging to the CTG clade where CUG-residues are exposed or partially exposed, particularly those in nonconserved positions (Fig. 1D).
These data show that reassignment of CUG codons in Candida (10, 11) was not random but rather specifically directed to permit ambiguity with minimal protein misfolding. This CUG-codon positional bias could explain why increasing leucine misincorporation up to 28% at CUG positions did not result in an upregulation of proteasome activity and heat shock protein response or in decreased growth rates in C. albicans (7) (Fig. S1).
Although seemingly tolerated, surface Leu-for-Ser replacements will hardly occur without structural and functional consequences. In agreement, functional differences were recently reported in C. albicans eukaryotic translation initiation factor 4E (EIF4E) upon incorporation of serine or leucine within a nonconserved site at the protein surface (12). Considering that exposed residues are often mediating macromolecular interactions, an impact of CUG ambiguity on protein activity and regulation can be expected.
Functional Impact of CUG Ambiguity in C. albicans Biology and Pathogenesis.
To understand the impact of ambiguous CUG decoding in C. albicans biology, the selected set of 680 proteins was clustered according to the relationship between their gene ontology (GO) terms. These proteins are uniformly distributed among functional classes and biological processes (Fig. S2 and Dataset S2), suggesting that CUG-codon ambiguity affects multiple cellular events simultaneously, with pleiotropic effects in C. albicans as observed upon increase of leucine-CUG misincorporation levels (7, 13). A closer analysis of the proteins containing at least one CUG-encoded residue at a position where serine is strictly conserved in other yeast orthologs (Dataset S2) revealed that many are correlated with virulence and pathogenesis (biofilm formation, morphogenesis, and mating) or associated with signal transduction, suggesting a pivotal role for CUG decoding ambiguity in pathogen–host interaction. Intriguingly, two C. albicans proteins crucial in signal transduction pathways associated with morphological switching and virulence (9–15) contain CUG-encoded residues in conserved/strictly conserved positions (Ras1 GTPase, Cek1 protein kinase), where Ser-to-Leu replacement is predicted to have a functional impact (Fig. S3). Supporting this hypothesis, increased leucine misincorporation up-regulated the expression of an adhesin (13), which is positively controlled by Ras1-dependent signaling cascades (9, 16). Ras1 is an environmental stress signal sensor that activates two morphological change-related signaling cascades (cAMP-dependent protein kinase and MAP kinase-dependent pathways) and disruption of genes encoding proteins of either pathway results in reduced virulence (9, 16, 17). Strikingly, partial Ser-to-Leu reversion of CUG identity in C. albicans also increased the expression of genes involved in cell adhesion and hyphal growth and the secretion of proteases and phospholipases (7, 13), features associated with virulence and infection (18). We have identified a number of proteins (Dataset S2) that can act as molecular switches and whose function may be affected by leucine incorporation. Therefore, determining the full extent of the structural and functional consequences of Ser-to-Leu exchange in CUG-containing proteins and their relevance for C. albicans biology and virulence must take into account its proteome variability.
Novel Structural Elements in C. albicans SerRS.
SerRS is a central player in C. albicans ambiguous CUG decoding and contains itself a CUG-encoded residue (position 197), in a position where polar amino acids are preferred in homologous enzymes (Fig. S4). The crystal structures of the two natural C. albicans SerRS isoforms were determined at 2.0 and 2.3 Å resolution, and snapshots of different states of the serine activation process were obtained for the SerRS_Ser197 isoform [apo enzyme and complexes with ATP or the nonhydrolyzable seryl-adenylate analogue 5′-O-[N-(L-seryl)-sulfamoyl]adenosine (SerSA)]. All crystals contained a SerRS monomer in the asymmetric unit, although size exclusion chromatography (19) and the nature and extent of intermolecular contacts across a crystallographic twofold axis indicated that recombinant SerRS is a dimer in solution, as observed for characterized bacterial and archaeal enzymes (20, 21). Superposition of the apo SerRS_Ser197 model (all Cα atoms) with that of SerRS_Leu197, SerRS–ATP, and SerRS–SerSA resulted in a root mean square deviation (rmsd) of 0.44, 0.25, and 0.36 Å, respectively, indicating a high degree of similarity between all the structures.
SerRS coiled-coil N-terminal domain is composed of two 62 Å long antiparallel α-helices (α3 and α4; Fig. 2A). This characteristic structural element is involved in tRNA discrimination, interacting with the long variable arm of serine-tRNAs (22). The α-β catalytic core domain is composed of a seven-stranded antiparallel β-sheet flanked by α-helices and a short parallel strand (β8), forming three conserved motifs (Fig. 2A), typical of class II aminoacyl-tRNA synthetases (aaRSs). The electron density maps for the SerRS–SerSA and SerRS–ATP complexes were unambiguous for the ligands bound to the active site pocket (Fig. S3), formed by conserved motifs 2 (residues 268–307) and 3 (residues 398–435) and the serine-binding TsE motif (residues 246–248). In the SerRS–SerSA complex, a strong positive electron density peak at the protein surface was interpreted as an additional SerSA molecule (Fig. 2B). This hydrophobic nucleotide-binding crevice is located 26 Å away from the active site, at the catalytic core domain surface. The adenine moiety of the SerSA molecule stacks perpendicularly to Tyr419 (conserved in yeast and mammalian SerRSs; Fig. S4), stabilized by polar interactions with main-chain atoms from residues forming this unique binding pocket, bordered by Pro133–Glu134 and Ala143–Ala145 (from the loop connecting strand β1 to helix α6), Gln328–Gly331 (at the end of helix α14), and Val426–Arg432 (in motif 3), adjacent to the C-terminal tail (residues 443–452), an extension unique to eukaryotic cytosolic SerRSs and associated with enzyme stability and substrate affinity (23, 24). Although structurally dissimilar, the β1-α6 loop, conserved in most SerRSs from Saccharomycetales (Figs. S4 and S5), is equivalent to the region where the archea-specific insertion is located (21), and will henceforth be referred to as Saccharomycetales-specific insertion (SSI)-loop. The relevance of this secondary, although specifically shaped, nucleotide-binding site for SerRS function and regulation is unclear, because it is very distant from the experimentally characterized tRNA-binding regions in homologous SerRSs (Fig. S5).
Fig. 2.

Novel structural elements in C. albicans SerRS_Ser197. (A) Overall structure of the SerRS_Ser197 dimer. Conserved structural elements of monomer A (gray cartoon) are colored in red (motif 1), yellow (motif 2), and purple (motif 3). The N-terminal helical coil, CUG-encoded residues (spheres) at the SerRS dimer interface, and the C-terminal extension characteristic of eukaryotic cytoplasmic SerRSs are colored blue, cyan, and pink, respectively. (B) Secondary nucleotide-binding site at the SerRS surface with bound SerSA (ball-and-stick model with carbon in magenta, oxygen in red, nitrogen in blue, sulfur in orange) and electron density map (1σ contouring, dark blue). The region altered by Ser-to-Leu197 exchange is shown in pale pink. Ser197 (cyan) and Tyr434 (purple) are shown as spheres. (C) Stereo view of the interactions between the Tyr444-centered C-terminal extension (pink) and the SerRS main core. Residues interacting with helices α6, α7, and the α16–β12 loop (sticks), water molecules (red spheres), and putative hydrogen bonds (dashed lines) are shown. The motif 3 helix-capping Gln420 is labeled red.
The C-terminal tail mediates intramonomer interactions with residues from helices α6 and α7, motif 3, and the SSI loop (Fig. 2C), and is not involved in dimer formation as seen for the C-terminal extension of bovine mitochondrial SerRS (25). The interactions between the C-terminal tail and the catalytic core are hinged at the side chain of the conserved Tyr444 (Fig. 2C and Fig. S4). This residue slots between Arg160 and the linker connecting helix α16 and strand β12 (in motif 3), participating in the intricate water-mediated hydrogen bonding network bridging helix α6, motif 3 and the C-terminal portion of the SSI loop. This network fixes (a) the position of the invariant helix α16-capping Gln420 (Fig. S4), therefore stabilizing the active site Arg410 (Fig. S3), and (b) the conformation of the SSI and α16–β12 loops, stabilizing the surface nucleotide-binding pocket. Furthermore, the C-tail Lys446 contributes to the C-terminal capping of helix α6 and to the stabilization of helix α7 via formation of both a salt bridge with Asp165 in the α6–α7 linker and water-mediated interactions with the N-terminus of helix α7. Thus, SerRS C-terminal tail stabilizes structural elements involved both in ATP and tRNA binding.
C. albicans SerRS Tolerates both Leu and Ser at the CUG Position.
The CUG-encoded residues of both SerRS monomers are facing each other (19 Å apart) on adjacent antiparallel helices α8 at the interface of the dimer, but are not directly involved in intersubunit contacts (Fig. 3), established between helix α7, helix α11, the β9–β10 hairpin, and the residue 197-containing conserved motif 1 (Figs. 2A and 3). The side chain of Ser197 establishes water-mediated hydrogen bonds with the side chains of Ser201 and Tyr434, within the same subunit (Fig. 3). In the SerRS_Leu197 crystal structure, accommodation of the bulkier leucine side chain at this position induces a local rearrangement in its immediate vicinity: the water molecules in direct contact with Ser197 disappear, the Ser201 side chain is rotated 180° away, and the side chain of Tyr434 (within helix α17, at the final segment of motif 3, stacked between Lys433 and helix α8) is shifted (0.6 Å) to accommodate the aliphatic moiety at position 197 (Fig. 3). The proximity of the dimer (and crystallographic) twofold axis, translates the concerted movement of the C-terminal Arg432-Tyr444 segment, induced by the presence of a bulkier Leu197, into a positional shift of the Tyr434–Pro436 of the opposing subunit (Fig. 3). Furthermore, the subtle structural rearrangements associated with accommodating serine or leucine in helix α8 at the SerRS dimer interface result in differences in protein stability in vitro (19), with the decreased thermal stability displayed by SerRS_Leu197 correlating well with the observed loss of polar interactions. Although the structural changes brought about by Leu197 do not affect the enzyme’s active site, an effect on its catalytic activity cannot be excluded. In fact, all observed rearrangements are adjacent to the surface adenine-binding pocket and the C tail, which stabilizes regions of the protein (e.g., helix α7; Fig. 2C) that, together with motif 2 loop and the helical arm, were shown to be involved in tRNASer binding in Thermus thermophilus and Pyrococcus horikoshii SerRSs (21, 26). Moreover, modeling studies with tRNASer show that the C-terminal tail could be directly involved in tRNA binding (Fig. S5), implying that small differences in its orientation/flexibility induced by CUG-residue identity might influence substrate affinity and thus the dynamics and catalytic activity of SerRS, modulating the aminoacylation of C. albicans unique 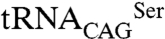 .
.
Fig. 3.

Leucine incorporation at the SerRS dimer interface induces subtle structural changes. (A) SerRS_Ser197 dimer interface, with residues involved in polar intermonomer interactions (Asn189 and Asn193) shown as sticks, and the CUG-encoded serine in cyan. Part of the mostly water (red spheres) mediated hydrogen bond network centered on Ser197 is shown as dashed black lines. (B) Superposition of the 3D structures of C. albicans SerRS natural isoforms. SerRS_Leu197 colored in mauve and SerRS_Ser197 colored as in Fig. 2A. The shifting residues in the immediate vicinity of residue 197 are represented as sticks.
Both SerRS Isoforms Are Functional.
The impact of Ser197 and Leu197 on C. albicans SerRS activity was assessed in vitro by measuring the rate of seryl-adenylate formation (27) in the presence and absence of unfractionated yeast tRNA. The results show that serine or leucine incorporation at the CUG site in SerRS does not alter the affinity for serine in the first step of the aminoacylation reaction [Km = 3.22 ± 0.33 μM (SerRS_Ser197), Km = 3.19 ± 0.29 μM (SerRS_Leu197)] and both enzymes display similar serine activation rates (Fig. 4A). However, in the presence of tRNA the SerRS_Leu197 isoform is 27% more active than the Ser197-containing enzyme (Fig. 4A).
Fig. 4.

C. albicans SerRS_Leu197 is catalytically more efficient. (A) Rates of serine activation by SerRS_Ser197 and SerRS_Leu197 in the presence and absence of tRNA. (B) Schematic representation of the C. albicans  orthogonal pair coexpression system. (C) Relative growth rate variation of E. coli transformed with the C. albicans orthogonal pairs SerRS_Ser197/tRNACAG or
orthogonal pair coexpression system. (C) Relative growth rate variation of E. coli transformed with the C. albicans orthogonal pairs SerRS_Ser197/tRNACAG or 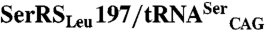 shows that both isoforms induce serine misincorporation at CUG codons. Horizontal bars represent the observed/control growth rate ratio. The growth rate with
shows that both isoforms induce serine misincorporation at CUG codons. Horizontal bars represent the observed/control growth rate ratio. The growth rate with 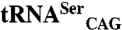 is comparable to that of the negative control, indicating that C. albicans
is comparable to that of the negative control, indicating that C. albicans 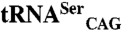 is not aminoacylated by the host SerRS. (D) Expression levels of SerRS isoforms quantified by infrared band intensities. (E) E. coli endogenous β-galactosidase activity upon coexpression of C. albicans
is not aminoacylated by the host SerRS. (D) Expression levels of SerRS isoforms quantified by infrared band intensities. (E) E. coli endogenous β-galactosidase activity upon coexpression of C. albicans  . The ratio between the β-galactosidase activity for each orthogonal pair and that of controls expressing only SerRS is represented by a horizontal bar.∗P < 1 × 10-2; ∗∗P < 1 × 10-9.
. The ratio between the β-galactosidase activity for each orthogonal pair and that of controls expressing only SerRS is represented by a horizontal bar.∗P < 1 × 10-2; ∗∗P < 1 × 10-9.
To determine if the differences observed in vitro are transposable to a cellular context, the SerRS aminoacylation activity was evaluated by coexpressing the SerRS–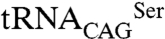 pair in Escherichia coli. Because C. albicans
pair in Escherichia coli. Because C. albicans 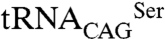 is not recognized by E. coli SerRSs, the eukaryotic SerRS–tRNA pair works independently from the endogenous bacterial serylation system, ensuring orthogonality (28, 29). The activity of Leu/Ser SerRS isoforms was therefore assessed by inducing their expression in recombinant E. coli expressing the exogenous
is not recognized by E. coli SerRSs, the eukaryotic SerRS–tRNA pair works independently from the endogenous bacterial serylation system, ensuring orthogonality (28, 29). The activity of Leu/Ser SerRS isoforms was therefore assessed by inducing their expression in recombinant E. coli expressing the exogenous 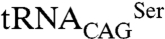 (Fig. 4B). As expected, serylation of
(Fig. 4B). As expected, serylation of 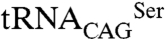 by C. albicans SerRS led to misincorporation of serine at CUG positions and had a negative impact on E. coli growth rate (Fig. 4C). Although both SerRS isoforms are catalytically active, a more significant decrease in growth rate was observed for SerRS_Leu197 (33.8 ± 2.6%) than for SerRS_Ser197 (19.4 ± 7.8%) (Fig. 4C). Because this difference was not the result of differential expression levels of the SerRS isoforms in E. coli (Fig. 4D), it likely reflects differences in serylation efficiency. Cross-validation using β-galactosidase thermostability (30) to measure serine misincorporation (31), confirmed an increased activity of the orthogonal
by C. albicans SerRS led to misincorporation of serine at CUG positions and had a negative impact on E. coli growth rate (Fig. 4C). Although both SerRS isoforms are catalytically active, a more significant decrease in growth rate was observed for SerRS_Leu197 (33.8 ± 2.6%) than for SerRS_Ser197 (19.4 ± 7.8%) (Fig. 4C). Because this difference was not the result of differential expression levels of the SerRS isoforms in E. coli (Fig. 4D), it likely reflects differences in serylation efficiency. Cross-validation using β-galactosidase thermostability (30) to measure serine misincorporation (31), confirmed an increased activity of the orthogonal 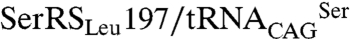 pair (Fig. 4E). Therefore, in E. coli the predominant C. albicans cytoplasmic SerRS isoform (Ser197) was less active than its minor counterpart, suggesting the existence of an elegant negative feedback mechanism for leucine misincorporation in this ambiguous yeast: upon increased
pair (Fig. 4E). Therefore, in E. coli the predominant C. albicans cytoplasmic SerRS isoform (Ser197) was less active than its minor counterpart, suggesting the existence of an elegant negative feedback mechanism for leucine misincorporation in this ambiguous yeast: upon increased 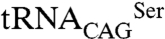 leucylation, the consequent increment of the more active SerRS_Leu197 will result in enhanced
leucylation, the consequent increment of the more active SerRS_Leu197 will result in enhanced 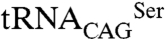 serylation, preventing overincorporation of leucine at CUG positions.
serylation, preventing overincorporation of leucine at CUG positions.
Discussion
Codon Reassignment was Constrained by Protein Structure.
Random misincorporation of leucine at CUG positions in C. albicans affects more than half of its proteome (7), producing an unique combination of protein variants in each cell. Previous studies have shown that ambiguity created a negative pressure that erased CUGs from the genome of the CTG clade ancestor (10, 11). Here we demonstrate that protein structural constraints shaped the reintroduction of CUGs in the genome of ambiguous yeasts, and that CUG-encoded residues circumvent regions where leucine misincorporation could result in protein misfolding, clustering in nonconserved solvent-exposed areas. This explains in part the high tolerance of C. albicans to increased ambiguity levels (7), as well as the homologous complementation of S. cerevisiae knockouts by C. albicans SerRS and LeuRS (32, 33). The positive pressure exerted by protein structure and folding tailored the genetic code reassignment and evolution in CTG clade species, resulting in functional proteomes under ambiguous CUG decoding. The data here presented provide a compelling example of the constraint imposed by protein folding on driving genome coding sequence evolution (34, 35).
Subtle Structural Changes in SerRS Isoforms have Functional Implications.
The three-dimensional structure of the major SerRS_Ser197 isoform disclosed two unique structural elements: the C-terminal tail and the SSI loop. The C-terminal tail is mostly involved in intramonomer interactions stabilizing the conserved structural motif 3 and the predicted tRNA-binding sites. In particular, a strictly conserved aromatic residue (Tyr444) plays a key role in the network of hydrogen bonds cross-linking the helix containing the active site Arg410 (motif 3) and the new SSI loop. The pivotal role of Tyr444 in eukaryotic SerRSs is in agreement with previous biochemical data showing that (a) C-terminal truncation mutants of S. cerevisiae/maize SerRSs are unstable, and (b) a mutant of maize SerRS lacking the conserved P[FY]X[KN] fingerprint displays decreased enzymatic activity (24). The SSI loop and residues from motif 3 delineate a remote nucleotide-binding site, occupied in one of our structures by SerSA, and might represent an allosteric site or nucleic acid binding surface (36).
The CUG-encoded residue at position 197 is located at the SerRS dimer interface and replacement of the more frequent serine at this position by leucine induces a local structural rearrangement in its immediate vicinity, affecting motif 3 and a region upstream of the C-terminal tail, which may be directly involved in interactions with the tRNA (Fig. S5). In agreement, the SerRS_Leu197 isoform, although less stable (19), is more active than SerRS_Ser197 both in vitro and in an heterologous E. coli 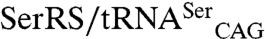 coexpression system, demonstrating that structural changes resulting from CUG ambiguity affect SerRS activity. These findings suggest that C. albicans SerRS might act as a sensor of CUG ambiguity as part of a negative feedback mechanism to maintain higher levels of serylated
coexpression system, demonstrating that structural changes resulting from CUG ambiguity affect SerRS activity. These findings suggest that C. albicans SerRS might act as a sensor of CUG ambiguity as part of a negative feedback mechanism to maintain higher levels of serylated 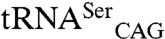 .
.
C. albicans SerRS and LeuRS: Key Regulators of CUG-Decoding Ambiguity?
A possible regulatory role of CUG ambiguity arises from the identification of a small number of proteins with CUG-encoded residues in conserved regions. From the set of proteins analyzed only two contained a CUG-encoded residue in a position where a leucine is strictly conserved—glycerol uptake protein 1 (GUP1) and CDC60 (C. albicans LeuRS). Interestingly, SerRS and LeuRS, the two enzymes competing for the single CUG-decoding tRNA, each contain a single CUG codon. Previous studies have shown that C. albicans leucine-containing LeuRS isoform is functional, because it has the ability to complement S. cerevisiae LeuRS (33). The CUG-encoded residue (position 919) is located in the C-terminal domain, which has a role both in substrate recognition, interacting with the long variable arm of tRNALeu in archaeal and bacterial LeuRSs (37, 38), and in aminoacylation activity (37, 39). The preferential incorporation of serine in C. albicans LeuRS in a position where leucine is strictly conserved in homologous fungal enzymes is thus expected to have a direct impact in 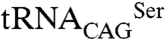 binding and leucylation activity. Thus, leucine misincorporation in this pathogen is likely regulated by a precise molecular mechanism that requires CUG ambiguity and a fine balance between the stability and aminoacylation activities of SerRS and LeuRS isoforms under different cellular conditions.
binding and leucylation activity. Thus, leucine misincorporation in this pathogen is likely regulated by a precise molecular mechanism that requires CUG ambiguity and a fine balance between the stability and aminoacylation activities of SerRS and LeuRS isoforms under different cellular conditions.
CUG Ambiguity in Candida albicans: Fine-Tuning Protein Function?
The identification of subtle structural differences in C. albicans SerRS isoforms, translating into functional changes in 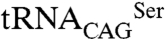 serylation activity in vivo, raised the question whether similar alterations existed in other proteins containing CUG-encoded residues. Although selected to avoid massive protein misfolding, Ser/Leu incorporation at CUG-codon positions is expected to selectively affect the function of other C. albicans proteins. In fact, mild stress conditions (decrease in pH and exposure to H2O2) increase leucine incorporation up to 5% (7) and overall SerRS protein levels decrease in response to macrophage interaction (40), hinting that the CUG-Ser/Leu levels might change with physiological conditions. Most interestingly, Ras1 and Cek1, two key effectors of signaling cascades regulating the transcription of genes associated with morphology and pathogenesis (9, 17), are expected to display functional changes upon leucine misincorporation. In the three-dimensional model of C. albicans Ras1 GTPase the CUG-encoded Ser66 is part of the conserved active site switch II region, whereas in the Cek1 protein kinase model the conserved CUG-encoded serine is facing the ATP binding-pocket (Fig. S3). The Cek1 MAPK pathway mediates mating and filamentation (41), the latter being crucial for C. albicans virulence (42). This multienzymatic signaling pathway, finely regulated by protein–protein interactions (43), was recently shown to be reprogrammed within the CTG clade where alterations occur in the scaffolding protein CaSte5 and in the interacting protein partners (44). The notable presence of CUG-encoded residues in Cek1, Hst7 (MAPKK homologue of S. cerevisiae Ste7, 4 CUGs), Ste4 (beta subunit of heterotrimeric G protein, 7 CUGs), Cst5 (scaffolding protein homologue of S. cerevisiae Ste5, 2 CUGs), Far1 (homologue of S. cerevisiae Far1—cyclin-dependent kinase inhibitor, 8 CUGs), and HOG1 (MAP kinase phosphorylated in response to H2O2, 2 CUGs) underscores the role of CUG ambiguity in modulating the protein–protein interaction dynamics required for signal transduction regulation. Here we propose that the negative impact of codon ambiguity in C. albicans can be overruled by the high adaptative potential of an expanded protein functional plasticity ultimately resulting in phenotypic diversity (7, 13) and enhanced virulence potential.
serylation activity in vivo, raised the question whether similar alterations existed in other proteins containing CUG-encoded residues. Although selected to avoid massive protein misfolding, Ser/Leu incorporation at CUG-codon positions is expected to selectively affect the function of other C. albicans proteins. In fact, mild stress conditions (decrease in pH and exposure to H2O2) increase leucine incorporation up to 5% (7) and overall SerRS protein levels decrease in response to macrophage interaction (40), hinting that the CUG-Ser/Leu levels might change with physiological conditions. Most interestingly, Ras1 and Cek1, two key effectors of signaling cascades regulating the transcription of genes associated with morphology and pathogenesis (9, 17), are expected to display functional changes upon leucine misincorporation. In the three-dimensional model of C. albicans Ras1 GTPase the CUG-encoded Ser66 is part of the conserved active site switch II region, whereas in the Cek1 protein kinase model the conserved CUG-encoded serine is facing the ATP binding-pocket (Fig. S3). The Cek1 MAPK pathway mediates mating and filamentation (41), the latter being crucial for C. albicans virulence (42). This multienzymatic signaling pathway, finely regulated by protein–protein interactions (43), was recently shown to be reprogrammed within the CTG clade where alterations occur in the scaffolding protein CaSte5 and in the interacting protein partners (44). The notable presence of CUG-encoded residues in Cek1, Hst7 (MAPKK homologue of S. cerevisiae Ste7, 4 CUGs), Ste4 (beta subunit of heterotrimeric G protein, 7 CUGs), Cst5 (scaffolding protein homologue of S. cerevisiae Ste5, 2 CUGs), Far1 (homologue of S. cerevisiae Far1—cyclin-dependent kinase inhibitor, 8 CUGs), and HOG1 (MAP kinase phosphorylated in response to H2O2, 2 CUGs) underscores the role of CUG ambiguity in modulating the protein–protein interaction dynamics required for signal transduction regulation. Here we propose that the negative impact of codon ambiguity in C. albicans can be overruled by the high adaptative potential of an expanded protein functional plasticity ultimately resulting in phenotypic diversity (7, 13) and enhanced virulence potential.
Taken together, the data here presented provide a better understanding of the molecular basis of CUG codon relocation, caused by reassignment of the universal leucine–CUG codon to a serine in the CTG clade fungi, and highlight a strategic role of natural codon ambiguity in protein multifunctionality. Considering the amazing ability of this human pathogen to assume different morphologies and its adaptability to extreme environments during infection, it is tempting to correlate CUG-induced protein plasticity with morphopathogenesis, laying the foundations for future research in Candida virulence pathways.
Methods
The experimental procedures are briefly described here and a detailed description is provided in SI Text. The distribution of CUG-encoded residues in C. albicans proteins was based on the analysis of multiple sequence alignments with orthologous proteins from yeast species. Experimental three-dimensional models for the selected CTG clade proteins were identified in the Protein Data Bank (PDB) and theoretical models generated by comparative homology modeling as described in SI Text. The C. albicans SerRS isoforms were purified and crystallized as previously described (19) and refinement is detailed in SI Text and Table S2. The C. albicans SerRS amino acid activation rate was measured using a continuous photometric assay (27), whereas the in vivo serylation activity was determined by expressing the orthogonal 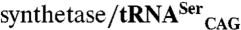 pair in E. coli and measuring the mistranslation-induced decrease in growth rate, as detailed in SI Text. The total β-galactosidase activity present in E. coli cells cotransformed with the orthogonal synthetase/tRNA pair was determined as described in SI Text and ref. 30.
pair in E. coli and measuring the mistranslation-induced decrease in growth rate, as detailed in SI Text. The total β-galactosidase activity present in E. coli cells cotransformed with the orthogonal synthetase/tRNA pair was determined as described in SI Text and ref. 30.
Supplementary Material
Acknowledgments.
We thank Frederico Silva and Joana Fraga for technical support and acknowledge the European Synchrotron Radiation Facility (ESRF) (Grenoble, beamline ID14EH1) for support during data collection. This work was funded by Fundação para a Ciência e a Tecnologia (FCT), through Grants PTDC/SAU-MII/70634/2006, PTDC/BIA-BCM/64745/2006, PTDC/SAU-GMG/098850/2008, PTDC/BIA-MIC/099826/2008, and REEQ/564/BIO/2005 (EU-FEDER and POCI 2010). R.R. is an FCT (BD/15233/2004) and Doctoral Programme on Experimental Biology and Biomedicine (University of Coimbra) PhD Fellow.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The crystallographic atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3QNE, 3QO5, 3QO7, and 3QO8).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1102835108/-/DCSupplemental.
References
- 1.Miranda I, Silva R, Santos MA. Evolution of the genetic code in yeasts. Yeast. 2006;23:203–213. doi: 10.1002/yea.1350. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Gladyshev VN. High content of proteins containing 21st and 22nd amino acids, selenocysteine and pyrrolysine, in a symbiotic deltaproteobacterium of gutless worm Olavius algarvensis. Nucleic Acids Res. 2007;35:4952–4963. doi: 10.1093/nar/gkm514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arner ES. Selenoproteins-What unique properties can arise with selenocysteine in place of cysteine? Exp Cell Res. 2010;316:1296–1303. doi: 10.1016/j.yexcr.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 4.Hao B, et al. A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science. 2002;296:1462–1466. doi: 10.1126/science.1069556. [DOI] [PubMed] [Google Scholar]
- 5.Netzer N, et al. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature. 2009;462:522–526. doi: 10.1038/nature08576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bender A, Hajieva P, Moosmann B. Adaptive antioxidant methionine accumulation in respiratory chain complexes explains the use of a deviant genetic code in mitochondria. Proc Natl Acad Sci USA. 2008;105:16496–16501. doi: 10.1073/pnas.0802779105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomes AC, et al. A genetic code alteration generates a proteome of high diversity in the human pathogen Candida albicans. Genome Biol. 2007;8:R206. doi: 10.1186/gb-2007-8-10-r206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki T, Ueda T, Watanabe K. The “polysemous” codon—a codon with multiple amino acid assignment caused by dual specificity of tRNA identity. EMBO J. 1997;16:1122–1134. doi: 10.1093/emboj/16.5.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leberer E, et al. Ras links cellular morphogenesis to virulence by regulation of the MAP kinase and cAMP signalling pathways in the pathogenic fungus Candida albicans. Mol Microbiol. 2001;42:673–687. doi: 10.1046/j.1365-2958.2001.02672.x. [DOI] [PubMed] [Google Scholar]
- 10.Butler G, et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459:657–662. doi: 10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massey SE, et al. Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG codon in Candida spp. Genome Res. 2003;13:544–557. doi: 10.1101/gr.811003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feketova Z, Masek T, Vopalensky V, Pospisek M. Ambiguous decoding of the CUG codon alters the functionality of the Candida albicans translation initiation factor 4E. FEMS Yeast Res. 2010;10:558–569. doi: 10.1111/j.1567-1364.2010.00629.x. [DOI] [PubMed] [Google Scholar]
- 13.Miranda I, et al. A genetic code alteration is a phenotype diversity generator in the human pathogen Candida albicans. PLoS ONE. 2007;2:e996. doi: 10.1371/journal.pone.0000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lengeler KB, et al. Signal transduction cascades regulating fungal development and virulence. Microbiol Mol Biol Rev. 2000;64:746–785. doi: 10.1128/mmbr.64.4.746-785.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Csank C, et al. Roles of the Candida albicans mitogen-activated protein kinase homolog, Cek1p, in hyphal development and systemic candidiasis. Infect Immun. 1998;66:2713–2721. doi: 10.1128/iai.66.6.2713-2721.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leberer E, et al. Virulence and hyphal formation of Candida albicans require the Ste20p-like protein kinase CaCla4p. Curr Biol. 1997;7:539–546. doi: 10.1016/s0960-9822(06)00252-1. [DOI] [PubMed] [Google Scholar]
- 17.Rocha CR, et al. Signaling through adenylyl cyclase is essential for hyphal growth and virulence in the pathogenic fungus Candida albicans. Mol Biol Cell. 2001;12:3631–3643. doi: 10.1091/mbc.12.11.3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shapiro RS, et al. Hsp90 orchestrates temperature-dependent Candida albicans morphogenesis via Ras1-PKA signaling. Curr Biol. 2009;19:621–629. doi: 10.1016/j.cub.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rocha R, Pereira PJB, Santos MA, Macedo-Ribeiro S. Purification, crystallization and preliminary X-ray diffraction analysis of the seryl-tRNA synthetase from Candida albicans. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;67:153–156. doi: 10.1107/S1744309110048542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vincent C, Borel F, Willison JC, Leberman R, Hartlein M. Seryl-tRNA synthetase from Escherichia coli: Functional evidence for cross-dimer tRNA binding during aminoacylation. Nucleic Acids Res. 1995;23:1113–1118. doi: 10.1093/nar/23.7.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itoh Y, et al. Crystallographic and mutational studies of seryl-tRNA synthetase from the archaeon Pyrococcus horikoshii. RNA Biol. 2008;5:169–177. doi: 10.4161/rna.5.3.6876. [DOI] [PubMed] [Google Scholar]
- 22.Borel F, Vincent C, Leberman R, Hartlein M. Seryl-tRNA synthetase from Escherichia coli: implication of its N-terminal domain in aminoacylation activity and specificity. Nucleic Acids Res. 1994;22:2963–2969. doi: 10.1093/nar/22.15.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lenhard B, Praetorius-Ibba M, Filipic S, Soll D, Weygand-Durasevic I. C-terminal truncation of yeast SerRS is toxic for Saccharomyces cerevisiae due to altered mechanism of substrate recognition. FEBS Lett. 1998;439:235–240. doi: 10.1016/s0014-5793(98)01376-3. [DOI] [PubMed] [Google Scholar]
- 24.Mocibob M, Weygand-Durasevic I. The proximal region of a noncatalytic eukaryotic seryl-tRNA synthetase extension is required for protein stability in vitro and in vivo. Arch Biochem Biophys. 2008;470:129–138. doi: 10.1016/j.abb.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Chimnaronk S, Gravers Jeppesen M, Suzuki T, Nyborg J, Watanabe K. Dual-mode recognition of noncanonical tRNAs(Ser) by seryl-tRNA synthetase in mammalian mitochondria. EMBO J. 2005;24:3369–3379. doi: 10.1038/sj.emboj.7600811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biou V, Yaremchuk A, Tukalo M, Cusack S. The 2.9 Å crystal structure of T. thermophilus seryl-tRNA synthetase complexed with tRNA(Ser) Science. 1994;263:1404–1410. doi: 10.1126/science.8128220. [DOI] [PubMed] [Google Scholar]
- 27.Lloyd AJ, Thomann HU, Ibba M, Soll D. A broadly applicable continuous spectrophotometric assay for measuring aminoacyl-tRNA synthetase activity. Nucleic Acids Res. 1995;23:2886–2892. doi: 10.1093/nar/23.15.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soma A, Himeno H. Cross-species aminoacylation of tRNA with a long variable arm between Escherichia coli and Saccharomyces cerevisiae. Nucleic Acids Res. 1998;26:4374–4381. doi: 10.1093/nar/26.19.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weygand-Durasevic I, Nalaskowska M, Soll D. Coexpression of eukaryotic tRNASer and yeast seryl-tRNA synthetase leads to functional amber suppression in Escherichia coli. J Bacteriol. 1994;176:232–239. doi: 10.1128/jb.176.1.232-239.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santos MA, Perreau VM, Tuite MF. Transfer RNA structural change is a key element in the reassignment of the CUG codon in Candida albicans. EMBO J. 1996;15:5060–5068. [PMC free article] [PubMed] [Google Scholar]
- 31.Lesjak S, Weygand-Durasevic I. Recognition between tRNASer and archaeal seryl-tRNA synthetases monitored by suppression of bacterial amber mutations. FEMS Microbiol Lett. 2009;294:111–118. doi: 10.1111/j.1574-6968.2009.01560.x. [DOI] [PubMed] [Google Scholar]
- 32.O’Sullivan JM, Mihr MJ, Santos MA, Tuite MF. The Candida albicans gene encoding the cytoplasmic leucyl-tRNA synthetase: Implications for the evolution of CUG codon reassignment. Gene. 2001;275:133–140. doi: 10.1016/s0378-1119(01)00632-1. [DOI] [PubMed] [Google Scholar]
- 33.O’Sullivan JM, Mihr MJ, Santos MA, Tuite MF. Seryl-tRNA synthetase is not responsible for the evolution of CUG codon reassignment in Candida albicans. Yeast. 2001;18:313–322. doi: 10.1002/1097-0061(20010315)18:4<313::AID-YEA673>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 34.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou T, Weems M, Wilke CO. Translationally optimal codons associate with structurally sensitive sites in proteins. Mol Biol Evol. 2009;26:1571–1580. doi: 10.1093/molbev/msp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paukstelis PJ, Chen JH, Chase E, Lambowitz AM, Golden BL. Structure of a tyrosyl-tRNA synthetase splicing factor bound to a group I intron RNA. Nature. 2008;451:94–97. doi: 10.1038/nature06413. [DOI] [PubMed] [Google Scholar]
- 37.Tukalo M, Yaremchuk A, Fukunaga R, Yokoyama S, Cusack S. The crystal structure of leucyl-tRNA synthetase complexed with tRNALeu in the post-transfer-editing conformation. Nat Struct Mol Biol. 2005;12:923–930. doi: 10.1038/nsmb986. [DOI] [PubMed] [Google Scholar]
- 38.Fukunaga R, Yokoyama S. Crystal structure of leucyl-tRNA synthetase from the archaeon Pyrococcus horikoshii reveals a novel editing domain orientation. J Mol Biol. 2005;346:57–71. doi: 10.1016/j.jmb.2004.11.060. [DOI] [PubMed] [Google Scholar]
- 39.Hsu JL, Rho SB, Vannella KM, Martinis SA. Functional divergence of a unique C-terminal domain of leucyl-tRNA synthetase to accommodate its splicing and aminoacylation roles. J Biol Chem. 2006;281:23075–23082. doi: 10.1074/jbc.M601606200. [DOI] [PubMed] [Google Scholar]
- 40.Fernandez-Arenas E, et al. Integrated proteomics and genomics strategies bring new insight into Candida albicans response upon macrophage interaction. Mol Cell Proteomics. 2007;6:460–478. doi: 10.1074/mcp.M600210-MCP200. [DOI] [PubMed] [Google Scholar]
- 41.Roman E, et al. The Cek1 MAPK is a short-lived protein regulated by quorum sensing in the fungal pathogen Candida albicans. FEMS Yeast Res. 2009;9:942–955. doi: 10.1111/j.1567-1364.2009.00545.x. [DOI] [PubMed] [Google Scholar]
- 42.Lo HJ, et al. Nonfilamentous C.albicans mutants are avirulent. Cell. 1997;90:939–949. doi: 10.1016/s0092-8674(00)80358-x. [DOI] [PubMed] [Google Scholar]
- 43.Good M, Tang G, Singleton J, Remenyi A, Lim WA. The Ste5 scaffold directs mating signaling by catalytically unlocking the Fus3 MAP kinase for activation. Cell. 2009;136:1085–1097. doi: 10.1016/j.cell.2009.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cote P, Sulea T, Dignard D, Wu C, Whiteway M. Evolutionary reshaping of fungal mating pathway scaffold proteins. MBio. 2011;2:e00230–10. doi: 10.1128/mBio.00230-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.