

Abstract
Malaria causes worldwide morbidity and mortality, and while chemotherapy remains an excellent means of malaria control, drug-resistant parasites necessitate the discovery of new antimalarials. Peptidases are a promising class of drug targets and perform several important roles during the Plasmodium falciparum erythrocytic life cycle. Herein, we report a multidisciplinary effort combining activity-based protein profiling, biochemical, and peptidomic approaches to functionally analyze two genetically essential P. falciparum metallo-aminopeptidases (MAPs), PfA-M1 and Pf-LAP. Through the synthesis of a suite of activity-based probes (ABPs) based on the general MAP inhibitor scaffold, bestatin, we generated specific ABPs for these two enzymes. Specific inhibition of PfA-M1 caused swelling of the parasite digestive vacuole and prevented proteolysis of hemoglobin (Hb)-derived oligopeptides, likely starving the parasite resulting in death. In contrast, inhibition of Pf-LAP was lethal to parasites early in the life cycle, prior to the onset of Hb degradation suggesting that Pf-LAP has an essential role outside of Hb digestion.
Keywords: protease, chemical-genetics, proteomics, small molecule, drug design
Malaria is a global disease causing at least 500 million clinical cases and more than 1 million deaths each year (1). While significant efforts to control malaria via insect vector elimination have been pursued, chemotherapy remains the principal means of malaria control. Moreover, the emergence of drug resistance in Plasmodium falciparum, the causative agent of most malaria-associated deaths, necessitates the discovery of novel antimalarials.
P. falciparum has a complex life cycle involving mosquito and human hosts. This life cycle involves both sexual and asexual stages of growth, wherein the human asexual erythrocytic phase (blood stage) is the cause of malaria-associated pathology. The erythrocytic stage begins when extracellular parasites, initially released from the liver, invade red blood cells. Once established in a specialized vacuole inside the host erythrocyte, parasites grow from the initial ring stage to the trophozoite stage, wherein much of the host hemoglobin (Hb) is proteolyzed. Parasites then replicate during the schizont stage to produce expanded populations of invasive merozoites that then rupture from the host cell approximately 48 h postinvasion and go on to recapitulate the life cycle (2). Peptidases are critical to parasite development throughout the life cycle and therefore are considered to be potential antimalarial drug targets (3–5).
One proteolytic pathway that has received significant attention is the multistep degradation of host Hb (6). While residing inside the host red blood cell, malaria parasites endocytose and proteolytically digest host Hb in a specialized lysosomal-like digestive vacuole (DV). This process liberates amino acids that the parasite can utilize for protein synthesis and general metabolism (7) and may reduce pressure on the host cell produced by the growing parasite (8). Multiple endoproteases make initial cuts in full-length Hb; however, genetic knockout studies of these enzymes, plasmepsins 1–4 and falcipain 2/2′, have revealed that all are nonessential and functionally redundant (9–11). While falcipain 3 has not been shown to be dispensable, it is expressed later in the parasite lifecycle and may have roles beyond Hb degradation. On the other hand, several exopeptidases, some of which may have roles in Hb degradation, are likely genetically essential (12, 13). Among these nonredundant enzymes are cysteine dipeptidyl aminopeptidase 1 (DPAP1) (13) and three metallo-aminopeptidases (MAPs): aminopeptidase N (PfA-M1), aminopeptidase P (PfAPP), and leucyl aminopeptidase, (Pf-LAP).
MAPs have been postulated to be important for the parasite life cycle. Early studies suggested that MAP activities were absent from the DV lumen, leading to the proposal that Hb peptides are exported to the cytosol for further degradation by MAPs (14–16). However, more recent localization and biochemical evidence suggest that PfA-M1 and PfAPP are located inside the DV (17, 18), while Pf-LAP is located in the parasite cytosol and has been proposed to act on exported globin peptides (19). A cytosolic aspartyl MAP, PfDAP, has been shown to hydrolyze substrates with an amino-terminal Asp or Glu residue (20), but its contribution to blood-stage peptide catabolism appears to be dispensable as genetic disruption causes no overt phenotype (13). Importantly, none of these studies provides any biological evidence, most importantly in live parasites, for a direct role for MAPs during Hb peptide catabolism.
Unfortunately in P. falciparum, genetic approaches to study essential genes are limited and thus direct evidence for the biological roles of these MAPs is still lacking (21). As a complementary approach, we have developed a MAP-specific chemical genetics platform that utilizes activity-based protein profiling (ABPP) based on the natural product inhibitor bestatin to study the roles of MAPs (22). ABPP is a chemical strategy that utilizes mechanism-based, tagged small molecule inhibitors to discover new enzymes, profile their activity state in complex proteomes, and identify potential functions for these enzymes during a specific biological process (23). ABPP has been utilized for a variety of enzyme classes, including serine hydrolases (24), peptidases (25), histone deacetylases (26), and kinases (27).
(-)-Bestatin is a natural product dipeptide analog of actinomycetes that potently inhibits multiple families of MAPs including the M1 and M17 families (28–31) (Fig. 1A). Importantly, bestatin has been shown to inhibit growth of P. falciparum parasites in culture and in mouse models of malaria (32–34). In addition, a recent study has indirectly implicated aminopeptidases in hemoglobin catabolism showing that parasites treated with bestatin had decreased levels of hemazoin formation, the detoxified biomineral byproduct of Hb digestion (34); likewise, isoleucine uptake was decreased in these bestatin treated parasites. However, the MAP(s) targeted by bestatin, which are responsible for these processes, were not identified.
Fig. 1.

Identification of PfA-M1 and Pf-LAP as the targets of the antiparasitic MAP inhibitor bestatin. (A) Structure of bestatin. (B) Structure of MH01. (C) Identification of PfA-M1 and Pf-LAP as the parasite targets of bestatin. Parasite lysates were prepared by freeze-thaw lysis and subsequently labeled with 5 μM MH01, UV crosslinked, and analyzed by Western blot for biotin. Four proteins were labeled by MH01. The same blot was stripped and reprobed sequentially with antibodies for PfA-M1 and Pf-LAP; three MH01 labeled proteins were accounted for by PfA-M1 and fourth by Pf-LAP. (D) A parasite line expressing a YFP-tagged PfA-M1 protein was incubated with MH01 followed by immunoprecipitation for biotin (MH01) and Western blot analysis for YFP, which confirmed that PfA-M1 is targeted by MH01 (first panel). The reciprocal experiment involving the immunoprecipitation of PfA-M1-YFP (after incubation of parasites with MH01) using a YFP-specific antibody confirmed PfA-M1-YFP is biotinylated and thus labeled by MH01 (second panel). (E) Likewise, the same analysis was performed using a parasite line expressing a YFP-tagged Pf-LAP protein; incubation of these parasites with MH01, followed by immunoprecipitation of biotin (MH01) and Western blot analysis for YFP confirmed that MH01 labels Pf-LAP. The reciprocal experiment involving immunoprecipitation of Pf-LAP-YFP and analysis by Western blot for biotin confirmed that Pf-LAP-YFP is biotinylated by MH01. Lastly, MH01 labeling of YFP-tagged MAPs, in both cases, is blocked by pretreatment with unbiotinylated bestatin as seen as a lack of labeling. Input lanes below each panel show Western blot analysis using anti-YFP of total parasite lysate just before immunoprecipitation.
Herein, we report on a multidisciplinary effort combining bestatin-based, small molecule ABPs with biochemical and peptidomic approaches to functionally analyze two essential aminopeptidases, PfA-M1 and Pf-LAP.
Results
Identification of Bestatin Targets in P. falciparum.
Because of the paucity of genetic tools for analysis of essential proteins in P. falciparum and a lack of highly specific inhibitors with which to probe the individual roles of MAPs, we chose to study the functions of these essential parasite MAPs through the development and application of a MAP-specific ABPP platform. ABPP utilizes tagged mechanism-based inhibitors, or activity-based probes (ABPs), to characterize families or individual active peptidases within complex proteomes. ABPs typically possess two main structural components that contribute to their target specificity: (i) a mechanism-based inhibitor scaffold to covalently or noncovalently target catalytic residues or the active site of peptidases and (ii) a reporter tag, such as a fluorophore or biotin, for the visualization, characterization of labeling events, and eventual affinity purification of target proteins. The mechanism-based inhibitor scaffold ensures that ABPs bind to the enzyme(s) in an activity-dependent manner.
We decided to use the natural product, bestatin, as the scaffold for the development of ABPs for MAPs because it is a general MAP inhibitor, kills Plasmodium parasites, and is synthetically tractable using both solution and solid-phase chemistry (22, 35, 36). To identify the target(s) of bestatin in P. falciparum, we utilized a previously published bestatin-based ABP, MH01 (Fig. 1B) (22). MH01 contains a biotin moiety to allow for monitoring of protein binding and affinity purification for target identification (22) and also utilizes a benzophenone for irreversible UV crosslinking to protein targets, as the inhibition of MAPs with bestatin is noncovalent. Asynchronous cultures of 3D7 parasites were treated with saponin to lyse the erythrocyte and parasitophorous vacuole membranes and isolated whole parasites were harvested by centrifugation. Crude parasite lysates, including both soluble and membrane proteins, were treated with 5 μM MH01, exposed to UV light, and analyzed by Western blot using streptavidin-HRP to detect biotinylated proteins. The observed labeling pattern consisted of four bands at approximately 100 kDa, 70 kDa, 55 kDa, and 25 kDa (Fig. 1C). Western blot analysis with PfA-M1 or Pf-LAP antibodies of parasite lysates labeled with MH01 showed that all four labeled species could be accounted for with these two antibodies: Three bands corresponded to different species of PfA-M1 and one to Pf-LAP (Fig. 1C). PfA-M1 is known to be proteolytically processed from a 120 kDa proform to a 115 kDa intermediate yielding the p96 and p68 forms (16), both of which contain the catalytic domains and are labeled by MH01. The labeled p25 band is likely a secondary proteolytic breakdown product. We further confirmed that MH01 bound PfA-M1 and Pf-LAP using individual parasite lines expressing YFP-tagged versions of these proteins (13). After incubation of each YFP-tagged transgenic line with MH01, immunoprecipitation using streptavidin and Western blotting for YFP revealed that both PfA-M1-YFP and Pf-LAP-YFP were targeted by MH01 (Fig. 1 D and E). Likewise, the reciprocal experiment involving the immunoprecipitation of YFP and Western blotting for biotin revealed that each YFP-tagged peptidase protein was biotinylated by MH01 (Fig. 1 D and E). Specificity of the interaction was confirmed by pretreatment with unlabeled bestatin, which blocked labeling. Attempted labelings using a parasite line expressing PfAPP-YFP, the other DV-localized MAP, confirmed that it was not a target of MH01 (Fig. S1B in SI Appendix). These results indicate that PfA-M1 and Pf-LAP are likely the only targets of bestatin in P. falciparum parasites. In addition, they highlight the difficulty in understanding the mechanism of bestatin toxicity, which could be due to the inhibition of either PfA-M1 or Pf-LAP or both.
MAP ABP Library Design and in Vitro Analysis Against PfA-M1 and Pf-LAP.
To investigate the individual functions of the two MAP targets of bestatin and to gain insight into the mechanism of how bestatin kills parasites, we synthesized several libraries of bestatin-based ABPs with the intention of generating specific ABPs for both PfA-M1 and Pf-LAP. Bestatin has two side chains that can be diversified, which are derived from the constituent α-hydroxy-β-amino acid and a natural α-amino acid (Fig. 1A). These side chains straddle the active sites of MAPs where the α-hydroxy-β-amino acid side chain (termed P1) fits into the S1 pocket of the enzyme (N-terminal to the scissile bond) and the adjacent natural amino acid side chain (P1′) interacts with the S1′ pocket (C-terminal to the scissile bond) (37, 38). In our initial library, the P1′ leucine residue in bestatin was replaced with a series of natural amino acids (except cysteine and methionine, which are prone to oxidation; norleucine was included as an isostere for methionine) and a limited number of nonnatural amino acids (Fig. 2A). Each library member was designed to incorporate a benzophenone to enable covalent attachment of the ABP to its targets and a terminal alkyne at the C terminus, to allow for the later addition of a variety of reporter tags using the bioorthogonal copper(I)-catalyzed [3 + 2] azide/alkyne cycloaddition (“click reaction”) (39–41). This library construction strategy increases the flexibility of downstream applications as each member has a “taggable” arm; thus, they can be used to directly treat live cells (as well as cellular lysates or recombinant enzymes) for target identification and activity profiling using a reporter tag, without any necessity for resynthesis or troubleshooting of tag placement.
Fig. 2.

Bestatin-based ABP libraries reveal distinct chemotypes produced by ABPs with increased specificity for either PfA-M1 or Pf-LAP. (A) Representative structure of the bestatin-based ABP scaffold showing the point of diversification at the P1′ position. This ABP library was screened against recombinant PfA-M1 and Pf-LAP in single fixed concentrations. Results of the assay are displayed as a heat map: Red indicates higher potency; blue indicates lower potency. The Phe-Naph ABP showed high specificity for Pf-LAP. (BES* indicates parental bestatin) (B) A library of ABPs was synthesized to identify a probe with increased specificity for PfA-M1. Representative structure of the bestatin-based ABP scaffold showing the point of diversification at the P1 position. All compounds had an Ala at the P1′ position. Results of the assay are displayed as a heat map: Red indicates higher potency; blue indicates lower potency. The (Benzyl)Tyr-Ala ABP showed high specificity for PfA-M1. (C) Synchronized parasites were treated with each compound at 1 μM and assayed for morphological changes by light microscopy of Giemsa-stained blood smears through the erythrocytic lifecycle. Scale bar, 5 μm. ABPs more specific for PfA-M1 showed swelling of the DV, while probes more specific for Pf-LAP showed an early death chemotype.
We initially screened a P1′ diverse library against both recombinant PfA-M1 and Pf-LAP via standard fluorescence protease activity assays. Results of this experiment are presented as a heat map based on percent inhibition of PfA-M1 and Pf-LAP for each ABP (Fig. 2A). The S1′ pocket of Pf-LAP tended to favor aromatic side chains such as Phe, Tyr, and Naphthyl. One probe, Phe-Naphthyl (PNAP), showed strong specificity for Pf-LAP over PfA-M1. In addition, several side chains favored binding toward PfA-M1 over Pf-LAP, which tended to be either small (Ser, Ala) or positively charged (Lys, Arg). The probes Phe-Ala, Phe-Lys, and Phe-Arg showed moderate specificity for PfA-M1; however, we decided not to pursue further studies with the positively charged probes because of potential issues with cell permeability. Although the Phe-Ala ABP was somewhat specific for PfA-M1, we felt it was not yet suitably specific for further biological studies; thus we investigated modifications to the P1 side chain to achieve higher specificity.
To increase specificity for PfA-M1, we synthesized a second bestatin-based library that diversified the α-hydroxy-β-amino acid side chain (P1 position) using a fixed alanine at the P1′ position. Given structural information indicating that the S1 pocket of the PfA-M1 enzyme was hydrophobic (38), the P1 library was synthesized with a variety of natural and nonnatural hydrophobic P1 side chains including: Ala, Leu, Diphenyl, Naphthyl, Biphenyl, and (Benzyl)Tyr (Fig. 2B). Again each library member had a clickable alkyne C-terminal to the benzophenone. This secondary ABP library was profiled against recombinant PfA-M1 and Pf-LAP and from the initial heat map analysis, the (Benzyl)Tyr-Ala ABP (BTA) gave the highest specificity of PfA-M1 over Pf-LAP.
To determine the morphological effects of inhibition using the probe libraries, parasite development was monitored throughout the entire lifecycle by Giemsa staining of thin blood smears (Fig. 2C). Three chemotypes (small molecule-induced morphological changes) were observed from this analysis: (i) no overt effect, (ii) early parasite death at the ring/trophozoite transition marked by pyknotic bodies, and (iii) swelling of the DV with parasite death occurring at the trophozoite/schizont transition. Importantly, the swollen DV as observed with these bestatin-based ABPs appeared translucent in Giemsa-stained smears, which distinguishes them from the dark swollen DV containing undigested Hb seen after treating parasites with papain family cysteine protease inhibitors such as E64 that target DV falcipains 2 and 3 (42).
Correlating the in vitro results with the live cell morphological screening results revealed that ABPs that produced the swollen DV chemotype displayed a high degree of specificity for PfA-M1 while ABPs more specific for Pf-LAP produced the early death/pyknosis chemotype. Compounds that failed to inhibit both MAPs, such as Phe-Asp (P1′-Asp) showed no chemotypes and were useful as negative control compounds (Fig. 2C). The contrasting chemotypes displayed by parasites treated with the most specific compounds, BTA or PNAP, suggested that PfA-M1 and Pf-LAP have essential yet distinct roles in the parasite.
Evaluation of BTA and PNAP Potency and Specificity.
To quantitatively assess the relative specificity of BTA and PNAP, inhibition constants against recombinant PfA-M1 and Pf-LAP were determined (Fig. 3A). All ABPs bound rapidly to PfA-M1; in contrast, binding to Pf-LAP was slow, as has been reported for bestatin (see Materials and Methods for more details). Analysis of BTA inhibition revealed that the substitution of the P1′ Leu for Ala shifted the specificity moderately toward PfA-M1. With the substitution of the P1 Phe with (Benzyl)Tyr in BTA, the affinity of the ABP was only moderately changed for PfA-M1 (Fig. 3A and Fig. S2 in SI Appendix), while the inhibition constant for Pf-LAP radically dropped nearly 100-fold, resulting in an ABP with an estimated micromolar 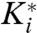 (an estimate was necessary due to insolubility at high micromolar concentrations). Thus the overall change in the ratio of inhibition constants of PfA-M1 over Pf-LAP from bestatin to BTA was approximately 75-fold and the absolute specificity difference for PfA-M1 over Pf-LAP was at least 15-fold, making BTA a useful biological tool to study PfA-M1 function. Likewise, the substitution of a naphthyl group for the P1′ leucine of PNAP increased the affinity of PNAP for Pf-LAP, resulting in a 170-fold change in the specificity of PNAP for Pf-LAP relative to BTA and approximately a 12-fold difference in absolute specificity, creating a relatively specific inhibitor for Pf-LAP.
(an estimate was necessary due to insolubility at high micromolar concentrations). Thus the overall change in the ratio of inhibition constants of PfA-M1 over Pf-LAP from bestatin to BTA was approximately 75-fold and the absolute specificity difference for PfA-M1 over Pf-LAP was at least 15-fold, making BTA a useful biological tool to study PfA-M1 function. Likewise, the substitution of a naphthyl group for the P1′ leucine of PNAP increased the affinity of PNAP for Pf-LAP, resulting in a 170-fold change in the specificity of PNAP for Pf-LAP relative to BTA and approximately a 12-fold difference in absolute specificity, creating a relatively specific inhibitor for Pf-LAP.
Fig. 3.

Biochemical and structural characterization of BTA and PNAP specificity against PfA-M1 and Pf-LAP. (A) Kinetic evaluation of inhibition for PNAP and BTA on recombinant PfA-M1 and Pf-LAP reveal over 15-fold specificity for PfA-M1 over Pf-LAP by BTA; PNAP showed greater than 10-fold specificity for Pf-LAP over PfA-M1.  for BTA for Pf-LAP was estimated due to solubility issues. (B) Activity-based protein profiling using “click” fluorescent versions of BTA or PNAP show that each probe specifically targets PfA-M1 or Pf-LAP, respectively (as indicated by an asterisk). (The superscript “C” in lane 5 of both panels indicates pretreatment with 10x of the nonfluorescent version of the respective ABP). (C) The electrostatic potential surface of the cocrystal of PfA-M1 with BTA (Left) and model of Pf-LAP with BTA bound in active site (Right). (D) The electrostatic potential surface of the model of PfA-M1 with PNAP (Left) and surface of the cocrystal of Pf-LAP with PNAP (Right). The zinc ion is shown as black sphere, the carbon atoms of inhibitors in green. Residues 755–1090 of PfA-M1 are excluded for clarity and a single monomer active site is shown for Pf-LAP. Surfaces were color coded according to electrostatic potential. Arrows show points at which either PNAP or BTA sterically clash with the enzyme.
for BTA for Pf-LAP was estimated due to solubility issues. (B) Activity-based protein profiling using “click” fluorescent versions of BTA or PNAP show that each probe specifically targets PfA-M1 or Pf-LAP, respectively (as indicated by an asterisk). (The superscript “C” in lane 5 of both panels indicates pretreatment with 10x of the nonfluorescent version of the respective ABP). (C) The electrostatic potential surface of the cocrystal of PfA-M1 with BTA (Left) and model of Pf-LAP with BTA bound in active site (Right). (D) The electrostatic potential surface of the model of PfA-M1 with PNAP (Left) and surface of the cocrystal of Pf-LAP with PNAP (Right). The zinc ion is shown as black sphere, the carbon atoms of inhibitors in green. Residues 755–1090 of PfA-M1 are excluded for clarity and a single monomer active site is shown for Pf-LAP. Surfaces were color coded according to electrostatic potential. Arrows show points at which either PNAP or BTA sterically clash with the enzyme.
Although in vitro data suggested that BTA and PNAP were quite specific for PfA-M1 and Pf-LAP, respectively, these data do not rule out the possibility that the ABPs have other targets in parasites. To confirm the specificity of each probe in parasites, we utilized the alkyne on each ABP to “click” on a fluorophore (BODIPY) tag, to identify target(s) of BTA and PNAP in crude P. falciparum proteomes. The fluorescent ABPs were incubated with parasite lysates, UV-crosslinked and targets analyzed via in-gel fluorescent scanning. As predicted from the in vitro kinetic assays, BTA exclusively labeled bands identical in migration on gels to those recognized by antibodies to PfA-M1 in this complex proteome, while PNAP was specific for a band that correlated in migration with Pf-LAP (Fig. 3B).
To investigate the structural basis for the specificity of BTA for PfA-M1 we solved the X-ray cocrystal structure of PfA-M1 bound to the BTA probe. The cocrystal structure was solved to 1.8 Å, and electron density clearly resolved the BTA probe and linker but lacked any visible density for the “clickable” alkyne C-terminal tag (Fig. 3C, Left; see Fig. S3A in SI Appendix for stereoview and Table S1 in SI Appendix for statistics). The BTA ABP bound to the essential active site zinc ion via the hydroxyl and carbonyl groups (O2/O3) and central nitrogen of the bestatin scaffold (Fig. S3A in SI Appendix). The S1 pocket showed a slight movement (approximately 1.2 Å between c-α atoms between the key S1 residue, Glu572, of the two structures) to accommodate the (Benzyl)Tyr at the P1 position. The P1′ Ala moiety did not reach far into the S1′ pocket of PfA-M1 as the remaining probe positioned itself close to the S1 pocket (Fig. 3C, Left). We also modeled BTA into the X-ray crystal structure of Pf-LAP bound to bestatin (PDB ID code 3KR4). Superposition of BTA onto the bestatin core showed that the large (Benzyl)Tyr residue at the P1 position clashed with the narrow S1 pocket of the active site in Pf-LAP (Fig. 3C, Right).
We also solved the cocrystal structure of Pf-LAP bound to PNAP (Fig. 3D, Right; see Fig. S4 in SI Appendix for hexameric structure). The 2.0-Å X-ray structure resolved the structural basis for the PNAP specificity and potency for Pf-LAP. As expected the PNAP ABP bound in a similar manner to the parent bestatin (43) dominated by coordination of two Zn2+ ions of the active site (Fig. S3B in SI Appendix). The P1-Phe ring of PNAP fit neatly into the small hydrophobic S1 pocket of Pf-LAP, and the P1′ naphthyl group also formed a series of hydrophobic interactions in the S1′ cleft. The only alteration noted to accommodate the P1′ naphthyl group was the movement of Ser550 (approximately 2.9 Å between c-α atoms of the Pf-LAP-PNAP structure versus the Pf-LAP-bestatin structure). This residue is located in a loop that lines the S1′ cleft, and the movement noted in the Pf-LAP-PNAP structure effectively flips the serine residue away from the naphthyl group, dragging the loop and preventing any close contacts with the P1′ residue (Fig. S3B in SI Appendix). It was also possible to model PNAP into the X-ray crystal structure of Pf-LAP bound to bestatin (PDB ID code 3EBH). Superposition of PNAP onto the bestatin core showed that the naphthyl side chain at the P1′ position clashed with the wall of the S1′ pocket of the active site in Pf-LAP (Fig. 3D, Left).
Inhibition of PfA-M1 Kills Parasites via Disruption of Hb Digestion Whereas Inhibition of Pf-LAP Kills via a Distinct Mechanism.
After confirming the specificities of BTA and PNAP, we next wanted to more fully characterize the effects of these ABPs on parasites through their life cycle. To do this, synchronized parasites were treated at the ring stage and followed by light microscopy evaluation of Giemsa-stained thin blood smears (Fig. 4A). We found that treating parasites with BTA at its IC99 caused a delay in the life cycle and swelling of the DV at the trophozoite stage with eventual parasite death around 60 hr posttreatment. As a comparison, parasites treated with E-64d, a cysteine protease inhibitor that blocks DV falcipains (and initial endoproteolytic cleavage of Hb) had a similar delay and swollen DV (darkly stained rather than translucent) but remained alive at the 60-hr time point. In contrast, PNAP-treated parasites were arrested at the transition to the trophozoite stage; therefore it appeared that PNAP exerted its effect on parasites significantly earlier than the time of major Hb digestion. PNAP treatment caused no prominent morphological features (other than death), thus complicating hypothesis generation as to its mechanism of action. To our knowledge the only other inhibitor that kills rings is artesunate and the other members of the artemisinin family (also shown in Fig. 4A for comparative purposes); although the mechanism of action for artesunate may be different than PNAP it is intriguing that there could be overlap among these two structurally divergent inhibitor classes (44).
Fig. 4.

Inhibition of PfA-M1 kills parasites via disruption of Hb digestion whereas inhibition of Pf-LAP kills via a distinct mechanism. (A) Parasites were treated with BTA (10 μM), E-64d (10 μM), PNAP (3 μM), and artesunate (10 nM) at concentrations roughly equivalent to their IC99, and followed by Giemsa staining and light microscopy throughout the lifecycle. Scale bar, 5 μm. (B) DV swelling was confirmed by the dose-dependent enlargement of the average DV area. Parasites expressing YFP-tagged plasmepsin II (PMII-YFP), which localizes to the DV, were treated with increasing concentrations of BTA (and 250 nM PNAP) and imaged by fluorescence microscopy. (C) DV swelling was determined to be saturable and quantified by treating the PMII-YFP parasites with increasing concentrations of BTA (PNAP was also used at 250 nM) at midring stage and measuring fluorescent DV area 20 hrs later using a minimum of 10 parasites (+/- standard deviation). (D) Treatment of parasites with BTA in media lacking exogenous amino acids, except for isoleucine, results in a more than twofold decrease in the IC50 of BTA, while parasites treated with PNAP or artesunate show a nonsignificant difference. Shown are representative IC50 plots for each compound in both I-Media (lacking exogenous amino acids except for isoleucine), and AA-Media (containing all natural amino acids). The inlay bar graphs show differences of the mean IC50 of three experiments carried out in triplicate (*P < 0.05, Student’s t test).
Inhibition of PfA-M1 by BTA treatment of parasites caused a novel swollen DV chemotype. To investigate this phenomenon more closely we visualized the swelling of the DV in live parasites using a parasite line expressing YFP-tagged plasmepsin II that serves as a DV marker (45). Transgenic parasites were treated with increasing concentrations of BTA and evaluated by fluorescent microscopy (Fig. 4B). From these images, we estimated the relative average DV size (measuring 10 DVs) after each treatment. Parasites treated with as little as 250 nM BTA (the Ki of BTA for PfA-M1) showed a statistically significant increase in DV size relative to untreated parasites, with saturation of this swelling at 1 μM (Fig. 4 B and C). The observation that the degree of DV swelling is dose-dependent and saturable is consistent with the hypothesis that, within this concentration range, PfA-M1 is likely the sole target and performs a key function in the DV. In contrast, parasites treated with PNAP at a concentration over eightfold greater than its Ki against Pf-LAP did not show any significant DV swelling (Fig. 4B and Fig. S5 in SI Appendix). Although we saw no evidence of labeling of any other peptidase in parasite proteomes (Figs. 1 and 3), we wanted to further confirm that the BTA-derived DV chemotype was not caused by inhibition of other proteases; thus we assayed for inhibition of other DV peptidases: DPAP1, PfAPP and falcipain 2. No inhibition of any enzyme was seen at a concentration up to 30 μM BTA, indicating that there is likely no cross-reactivity of BTA with these enzymes in live parasites and that the DV swelling is caused solely by inhibition of PfA-M1 (Fig. S6 in SI Appendix).
Because disruption in the endocytosis or subsequent catabolic breakdown of Hb is thought to be lethal to parasites (9), we hypothesized that PfA-M1 inhibition by BTA leads to starvation of the parasite via blockage of proteolysis of Hb peptides. To test this idea, we assayed whether parasites forced to rely only on Hb catabolism are more sensitive to BTA than parasites cultured with exogenous amino acids, by assaying the potency of BTA on parasites cultured in media lacking all amino acids except isoleucine (the only amino acid not present in Hb). Indeed, parasites were sensitized by approximately 2.4-fold to inhibition by BTA in media with only isoleucine (Fig. 4D). In contrast, parasites treated with PNAP or the antimalarial artesunate, which kills ring stage parasites prior to initiation of large-scale Hb degradation and thus acts as a negative control, showed statistically insignificant differences in sensitization to either compound in the isoleucine media. This evidence suggests a role for PfA-M1 in the Hb digestion pathway and also provides further evidence that the primary role of Pf-LAP is not within the Hb digestion pathway.
Initial proteolytic events are thought to be carried out by the redundant endopeptidases falcipains 2/2′/3, plasmepsins I, II, IV, and HAP (46). Hb-derived oligopeptides are then broken down by exopeptidases. Considering that PfA-M1 is an aminopeptidase, its likely role in the DV would occur after initial Hb proteolysis by the endopeptidases. To confirm this, we treated synchronous cultures of parasites during the trophozoite stage, in which the majority of Hb degradation takes place. Fig. S7 in SI Appendix shows that parasites treated with E-64d leads to an accumulation of full-length Hb and causes the swelling of the DV with undigested Hb (42). Conversely, parasites treated with BTA showed no inhibition of proteolysis of full-length Hb but still caused the DV to swell. Treatment of parasites with PNAP was similar to DMSO.
Inhibition of PfA-M1 Blocks Proteolysis of Specific Hb-Derived Oligopeptides.
To obtain direct evidence for the role of PfA-M1 in the proteolysis of Hb-derived oligopeptides, we endeavored to find peptide substrates for this enzyme. We used a mass spectrometry-based peptidomics approach to assay the relative abundance of peptides (< 10 kDa) in parasites either untreated or treated with the specific PfA-M1 inhibitor BTA. To do this, trophozoite stage parasites were treated with BTA or DMSO for 24 hr. Whole parasite extracts were prepared and peptides were enriched by an acid extraction followed by a filtration through a 10 kDa filter column. Resulting peptide extracts were analyzed by nano LC-MS/MS. Mass spectrometry analysis of the peptide peaks against both Hb α and β sequences revealed that the great majority of these oligopeptides showed no difference between the treated and untreated samples; however, several oligopeptides, from both the α and β chains of Hb, appeared to accumulate after treatment of parasites with BTA (Fig. 5 A and B).
Fig. 5.

Inhibition of PfA-M1 blocks proteolysis of specific Hb oligopeptides. (A) Global peptide profiling of treated parasites identifies a subset of peptides that accumulate in BTA-treated parasites relative to DMSO. Peptides were extracted from either DMSO or BTA-treated parasites and peptides analyzed by LC-MS/MS. Peptides identified are displayed according to elution time, intensity, and M/Z. Area of the peptide peak corresponds to relative abundance. (B) LC-MS/MS sequencing of the peptides reveals accumulated peptides are derived from Hb. Ratio of peptide abundance was calculated by determining the area of peptide intensity of BTA-treated vs untreated. (C) A synthesized version of an abundantly accumulated peptide, identified in the prior LC-MS/MS analysis, was efficiently proteolyzed at the N-terminal valine by PfA-M1, while DPAP1, the other likely essential DV aminopeptidase, failed to catalyze any proteolysis. Shown are HPLC traces displaying the synthetic peptide without enzyme (top trace), with PfA-M1 (middle trace), or with DPAP1 (lower trace). Insets show peptide sequences corresponding to HPLC peaks.
Further sequence analysis of the accumulated Hb-derived oligopeptides revealed that the majority were likely poor substrates for DPAP1, the other essential aminopeptidase with broad substrate specificity found in the DV (47). We thus hypothesized that PfA-M1 was necessary for proteolysis of these oligopeptides. To test this idea, a highly enriched peptide that was identified from the peptidomics study of BTA-treated samples was resynthesized and shown to be resistant to cleavage by DPAP1, yet efficiently cleaved by PfA-M1 (Fig. 5C). This data provides direct evidence of a role for the genetically essential enzyme, PfA-M1, in the digestion of small Hb-derived oligopeptides in P. falciparum.
Discussion
Peptidases likely have many essential functions in P. falciparum, yet the biological roles of the majority of putative proteases encoded by the parasite genome remain to be characterized. One reason for this rests in the difficulty in genetically manipulating essential parasite genes. To circumvent this deficit in genetic tools, a small molecule approach may be used to perturb and thus investigate essential protein functions in the parasite. Several issues arise from the use of small molecule probes, including (i) target identification, (ii) specificity, and (iii) permeability in live cells. To address some of these issues, we generated a library of MAP-specific, “clickable” ABPs. Replacement of a bulky reporter tag with an alkyne group resulted in smaller, more versatile ABPs that allowed for their use in live cells for phenotypic analysis along with more tradition lysate-based ABPP.
Using this set of chemical tools, we first identified the targets of bestatin in P. falciparum to be PfA-M1 and Pf-LAP; although there are limitations to this ABPP approach—i.e., it is hard to determine with absolute certainty that there are no low abundance targets—further diversification of the MAP inhibitor scaffold to generate structure-activity relationship (SAR) trends can strengthen functional conclusions. Thus, through diversification of the bestatin scaffold, we were able to create a suite of ABPs with increasing specificity for both PfA-M1 and Pf-LAP and used these ABPs to further understand the functions of these essential enzymes.
The aggregate data using the BTA ABP strongly suggests that PfA-M1 plays a key role in oliogopeptide proteolysis in the DV. The most prominent morphological feature of PfA-M1 inhibition was the novel swollen, translucent DV chemotype, which was likely due to the accumulation of oligopeptides that created hyperosmotic conditions. DV swelling also occurs after E-64d inhibition of DV falcipains; yet these parasites do not die until they attempt to egress, which suggests that swelling alone is not lethal to parasites, and that there must be enough amino acids generated (perhaps by plasmepsins) or obtained from the medium in the presence of this inhibitor to allow life cycle progression. We therefore propose that the likely cause of death upon PfA-M1 inhibition is a severe deprivation of the DV production of amino acids in the parasite. (Although we cannot rule out that killing may be due to indirect effects distinct to PfA-M1 inhibition, such as the accumulation of small peptides in the DV, which could potentially be toxic.) To support the hypothesis that inhibition of PfA-M1 disrupts Hb degradation, we demonstrated that the effects of BTA were enhanced when parasites were forced to rely solely on Hb degradation. Why the amino acids present in complete media do not allow for the parasite to completely complement for the genetic or pharmacological loss of PfA-M1 via the utilization of exogenous amino acids is an interesting question. This sensitivity may be explained by the fact that the parasite is particularly dependent on leucine generated in the DV from Hb, which is thought to be exchanged for isoleucine via an antiport mechanism (48).
From our peptidomics experiments we showed that Hb oligopeptides were substrates for PfA-M1 proteolysis. One issue with this analysis is that small peptides were not found using our mass spectrometry method, which was limited to the identification of peptides greater than four amino acids in length. It is likely that the concerted action of DV endo- and exoproteases also produced smaller tri- and dipeptides, which are substrates for PfA-M1 in the DV. In support of this idea, we also attempted to identify small peptide species using a Single Quad LC/MS (which allows for profiling peptides between 200 and 400 kDa) that accumulated in BTA-treated parasites (Fig. S8 in SI Appendix). These low molecular weight species all matched to predicted dipeptide molecular weights, and more than half were the molecular weight of dipeptides found in Hb. However, this method precluded the definitive identification of these molecules as peptides (as opposed to metabolites) and their origin (i.e., Hb). However, we believe these data are suggestive that several dipeptides, in addition to oligopeptides, are likely important substrates for the PfA-M1 enzyme.
Our data using the PNAP ABP for Pf-LAP indicated that this enzyme has an important role quite early in the intraerythrocytic life cycle rather than during the major period of Hb digestion. Formulating a testable hypothesis about a specific role for Pf-LAP is complicated by the fact that its inhibition did not yield any overt morphological change in the parasite other than death. However, we suspect Pf-LAP may have an essential housekeeping function in the cytosolic turnover of dipeptides (49) and perhaps acts in concert with the parasite proteasome, as has been shown for other neutral cytosolic leucine aminopeptidases pathways (50). Like PNAP, lethal amounts of proteasome inhibitors exert their effect in the ring-trophozoite transition and parasites do not progress into the later trophozoite stage (51). Our data does not completely rule out the possibility of a minor role for Pf-LAP in the Hb degradation pathway via proteolysis of Hb-derived dipeptides that have been transported from the DV into the cytoplasm. However, the fact that PNAP-treated parasites die prior to the major period of Hb degradation suggests that the essential role for Pf-LAP is not within the Hb digestion pathway.
Our collective data suggest that these two MAPs are both potential antiparasitic drug targets. In fact, PNAP is, to our knowledge, the most potent parasite MAP inhibitor with an IC50 in the 200 nM range, which gives us hope that these types of inhibitors could be further developed into more drug-like therapeutics. In addition, P. falciparum MAPs share little homology with their human counterparts; less than 35% in the case of the M1 family proteases. It is therefore reasonable to suggest that potent, specific inhibitors of P. falciparum MAPs can be designed over human MAPs. In addition, information gleaned from our preliminary SAR and crystallography efforts may provide a jumping off point for future medicinal chemistry efforts against both enzymes. Our data here suggest that combination therapy involving endopeptidase inhibitors, such as those for falcipains, and PfA-M1-specific inhibitors might provide an opportunity for a synergistic drug combination (52). Ultimately, this strategy may represent a good way to reduce the chance of parasite resistance.
Materials and Methods
General Methods.
See SI Appendix for additional chemical and experimental protocols. A summary of the methods is given below.
Parasite Culture and IC50 Determination for Bestatin-Based ABPs.
Briefly, 3D7 parasites were cultured in RPMI 1640 (Invitrogen) supplemented with Albumax II (Invitrogen). For synchronization, schizont stage parasites were magnet purified using a SuperMACS™ II Cell Separation Unit (Miltenyi Biotech). For IC50 determinations, synchronized parasites were plated at 1% parasitemia and 6% hematocrit in 96-well plates at a total volume of 50 μL. Serial dilutions of 2x concentration of the respective compound were added to the wells to bring the total volume up to 100 μL and 0.5% parasitemia and 3% hematocrit. Compounds were assayed for a 72 h period, after which 2x Vybrant DyeCycle Green DNA (Invitrogen) in PBS was added for a final concentration of 10 μM and incubated at 37 °C for 30 min. DNA content, as an indicator of parasitemia, was analyzed on an Accuri C6 Flow Cytometer with C-Sampler. IC50 curves were generated using GraphPad Prism (GraphPad Software).
Labeling of Parasite MAPs with Activity-Based Probes.
For parasite labeling, mixed stage parasites were harvested and released from erythrocytes with 1% saponin followed by centrifugation at 1,500 × g for 5 min and 3 washes in cold PBS. Parasite lysates were prepared by freeze-thaw in 50 mM Tris-HCl pH 7.0, 50 mM NaCl, 10 μM ZnCl, and protease inhibitor cocktail (EDTA-free) (Roche) and extracts were clarified by centrifugation at 1,100 × g for 10 min at 4 °C. Labeling was performed with indicated concentrations of the ABP for 1 hr at 37 °C followed by UV crosslinking (365 nm) for 1 hr on ice. Competition of labeling was carried out by preincubating lysates for 1 hr at 37 °C. For immunoprecipitation, lysates were passed through 7 K MWCO desalting columns (Pierce) after UV crosslinking then incubated overnight with streptavidin Ultralink Resin (Pierce). Proteins were visualized by standard Western blotting and VECTASTAIN ABC kit (Vector Labs) or rabbit anti-GFP (ab6556, Abcam). For fluorescent probes, labeled proteins were visualized in-gel using a Typhoon flatbed scanner (GE Healthcare).
Synthesis of Bestatin-Based ABP Libraries.
A detailed description of the synthesis and characterization of these compounds may be found in SI Appendix.
Recombinant Proteins.
Details of the expression in Escherichia coli and purification of recombinant PfA-M1 (residues 192 to 1,085) will be described separately (53). Pf-LAP lacking the N-terminal Asn-rich region (residues 79–605) was expressed with a C-terminal hexahistidine tag in E. coli and purified as previously described (43).The estimated molecular mass of the purified species from size exclusion chromatography (343 kDa) was in good agreement with the predicted mass for the hexameric enzyme (357 kDa). The purification of recombinant DPAP1 has been published (47).
X-ray Crystallography.
PfA-M1 and Pf_LAP enzymes were purified and crystallized as previously described (38). Crystals of the PfA-M1-BTA complex were obtained by cocrystallization of BTA with PfA-M1 in mother liquor containing 1 mM ligand. Crystals of the Pf-LAP-PNAP complex were obtained by cocrystallisation of PNAP with Pf-LAP in mother liquor containing 1 mM ligand. Prior to data collection, Pf-LAP-PNAP cocrystals were soaked in mother liquor containing 1 mM ligand and 1 mM ZnSO4. Data were collected at 100 K using synchrotron radiation at the Australian synchrotron micro crystallography beamline 3ID1. A summary of statistics is provided in Table S1 in SI Appendix. Raw data and images will be available from TARDIS (54).
The inhibitor complex was initially solved and refined against the unbound PfA-M1 and Pf-LAP structure (protein atoms only) as described previously (38) and clearly showed unbiased features in the active site for both structures. After placement of inhibitors into unbiased density, CNS composite omit maps were calculated using all atoms. Fig. S3 in SI Appendix shows inhibitor density contoured at 1.0σ. Fig. S3 in SI Appendix was generated using MacPymol and uses a 2.0 carve value around each inhibitor and zinc ion for clarity. Superposition of BTA into the Pf-LAP active site was performed using the X-ray crystal structure of Pf-LAP-bestatin (3KR4) where the bestatin scaffold was used to superpose BTA.
Anti-PfA-M1 Sera.
Details of the production of rabbit antisera against recombinant PfA-M1 have been previously described (53).
Screening of P1 and P1′ ABP Libraries.
Screens of relative ABP potencies were conducted at single fixed ABP concentrations for the P1 (PfA-M1- 250 nM; Pf-LAP- 750 nM), P1′ natural (PfA-M1- 1 μM; Pf-LAP- 250 nM) P1′ nonnatural (0.37 μM for both enzymes) libraries. PfA-M1 assays contained 50 mM HEPES pH 7.5, 100 mM NaCl, 25 mM leucyl-7-amido-4-methylcoumarin (Leu-AMC) and 0.1% Triton X-100; Pf-LAP assays contained 50 mM HEPES pH 7.5, 100 μM ZnCl2, 50–250 μM Leu-AMC and 0.1% Triton X-100. For Pf-LAP, steady-state rates were approximated by linear fits to the progress curves after a 1 hr equilibration period in the presence of substrate and inhibitor.
Determination of Ki and  Values.
Values.
Ki values for inhibition of PfA-M1 were determined by Dixon plots and a detailed protocol for determining Pf-LAP K1∗ values may be found in SI Appendix.
Mass Spectrometry-Based Peptide Profiling.
Briefly, parasites were treated with 2 μM BTA or DMSO at the midring stage. Parasites were treated for 24 hr at which point they were harvested by saponin treatment, centrifuged, and stored at -80 °C in the presence of protease inhibitors. To isolate peptides, parasite samples were boiled in water for 10 min and then centrifuged for 10 min at 18,000 × g. The supernatant was saved, and the pellet was resuspended in 0.25% acetic acid and disrupted by freeze-thaw and microsonicated. All fractions were combined and centrifuged at 20,000 × g at 4 °C for 20 min. The supernatant was passed through a 10 kDa molecular weight cutoff filter (Millipore).
The retention times and m/z values of the peptides identified were used to map corresponding peptide peaks in the chromatograms generated from nano-HPLC–ESI-MS (LCQ-DecaXP Plus). These peptide peaks were manually aligned and then for semiquantitative assessment of the abundances of individual peptides, the total peak areas were determined using the Bioworks algorithm PepQuan (the Area/Height Calculation) with parameters set to area, mass tolerance of 1.5, minimum threshold of 5,000, five smoothing points, and including all proteins. The alignment was based on retention times, m/z values, and patterns of peaks in close proximity.
Supplementary Material
Acknowledgments.
We thank the Australian Synchrotron for beamtime and Tom Caradoc-Davis in particular for technical assistance. We thank John Dalton, PhD, McGill University, for the Pf-LAP antisera. Authors acknowledge the support by Penn Genome Frontiers Institute (D.C.G), Ritter Foundation (D.C.G), R01-AI-076342-01 (D.B.), R01-AI-077638 (M.K.), NIH5T32AI007532 (M.B.H.), and 1R56-AI-081770-01A2 (D.C.G.). S.M. is an Australian Research Council (ARC) Future Fellow, J.C.W. is an ARC Federation Fellow and a National Health and Medical Research Council (NHMRC) Principal Research Fellow. We thank the NHMRC and the ARC for funding support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. B.F.C. is a guest editor invited by the Editorial Board.
See Author Summary on page 13885.
Data deposition: The crystal coordinates have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 3T8V (PfA-M1-BTA) and 3T8W (Pf-LAP-PNAP)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1105601108/-/DCSupplemental.
References
- 1.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 3.O’Donnell RA, et al. Intramembrane proteolysis mediates shedding of a key adhesin during erythrocyte invasion by the malaria parasite. J Cell Biol. 2006;174:1023–1033. doi: 10.1083/jcb.200604136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arastu-Kapur S, et al. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat Chem Biol. 2008;4:203–213. doi: 10.1038/nchembio.70. [DOI] [PubMed] [Google Scholar]
- 5.Skinner-Adams TS, et al. Plasmodium falciparum neutral aminopeptidases: new targets for anti-malarials. Trends Biochem Sci. 2010;35:53–61. doi: 10.1016/j.tibs.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Francis SE, Sullivan DJ, Jr, Goldberg DE. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol. 1997;51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 7.Sherman IW. Amino acid metabolism and protein synthesis in malarial parasites. \Bull World Health Organ. 1977;55:265–276. [PMC free article] [PubMed] [Google Scholar]
- 8.Lew VL, Tiffert T, Ginsburg H. Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum-infected red blood cells. Blood. 2003;101:4189–4194. doi: 10.1182/blood-2002-08-2654. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Istvan ES, Gluzman IY, Gross J, Goldberg DE. Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc Natl Acad Sci USA. 2006;103:8840–8845. doi: 10.1073/pnas.0601876103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sijwali PS, Koo J, Singh N, Rosenthal PJ. Gene disruptions demonstrate independent roles for the four falcipain cysteine proteases of Plasmodium falciparum. Mol Biochem Parasitol. 2006;150:96–106. doi: 10.1016/j.molbiopara.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 11.Omara-Opyene AL, et al. Genetic disruption of the Plasmodium falciparum digestive vacuole plasmepsins demonstrates their functional redundancy. J Biol Chem. 2004;279:54088–54096. doi: 10.1074/jbc.M409605200. [DOI] [PubMed] [Google Scholar]
- 12.Klemba M, Gluzman I, Goldberg DE. A Plasmodium falciparum dipeptidyl aminopeptidase I participates in vacuolar hemoglobin degradation. J Biol Chem. 2004;279:43000–43007. doi: 10.1074/jbc.M408123200. [DOI] [PubMed] [Google Scholar]
- 13.Dalal S, Klemba M. Roles for two aminopeptidases in vacuolar hemoglobin catabolism in Plasmodium falciparum. J Biol Chem. 2007;282:35978–35987. doi: 10.1074/jbc.M703643200. [DOI] [PubMed] [Google Scholar]
- 14.Curley GP, et al. Aminopeptidases from Plasmodium falciparum, Plasmodium chabaudi chabaudi and Plasmodium berghei. J Eukaryot Microbiol. 1994;41:119–123. doi: 10.1111/j.1550-7408.1994.tb01483.x. [DOI] [PubMed] [Google Scholar]
- 15.Kolakovich KA, Gluzman IY, Duffin KL, Goldberg DE. Generation of hemoglobin peptides in the acidic digestive vacuole of Plasmodium falciparum implicates peptide transport in amino acid production. Mol Biochem Parasitol. 1997;87:123–135. doi: 10.1016/s0166-6851(97)00062-5. [DOI] [PubMed] [Google Scholar]
- 16.Allary M, Schrevel J, Florent I. Properties, stage-dependent expression and localization of Plasmodium falciparum M1 family zinc-aminopeptidase. Parasitology. 2002;125:1–10. doi: 10.1017/s0031182002001828. [DOI] [PubMed] [Google Scholar]
- 17.Ragheb D, Bompiani K, Dalal S, Klemba M. Evidence for catalytic roles for Plasmodium falciparum aminopeptidase P in the food vacuole and cytosol. J Biol Chem. 2009;284:24806–24815. doi: 10.1074/jbc.M109.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azimzadeh O, Sow C, Geze M, Nyalwidhe J, Florent I. Plasmodium falciparum PfA-M1 aminopeptidase is trafficked via the parasitophorous vacuole and marginally delivered to the food vacuole. Malar J. 2010;9:189–205. doi: 10.1186/1475-2875-9-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stack CM, et al. Characterization of the Plasmodium falciparum M17 leucyl aminopeptidase. A protease involved in amino acid regulation with potential for antimalarial drug development. J Biol Chem. 2007;282:2069–2080. doi: 10.1074/jbc.M609251200. [DOI] [PubMed] [Google Scholar]
- 20.Teuscher F, et al. The M18 aspartyl aminopeptidase of the human malaria parasite Plasmodium falciparum. J Biol Chem. 2007;282:30817–30826. doi: 10.1074/jbc.M704938200. [DOI] [PubMed] [Google Scholar]
- 21.Crabb BS, et al. Transfection of the human malaria parasite Plasmodium falciparum. Methods Mol Biol. 2004;270:263–276. doi: 10.1385/1-59259-793-9:263. [DOI] [PubMed] [Google Scholar]
- 22.Harbut MB, Velmourougane G, Reiss G, Chandramohanadas R, Greenbaum DC. Development of bestatin-based activity-based probes for metallo-aminopeptidases. Bioorg Med Chem Lett. 2008;18:5932–5936. doi: 10.1016/j.bmcl.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barglow KT, Cravatt BF. Activity-based protein profiling for the functional annotation of enzymes. Nat Methods. 2007;4:822–827. doi: 10.1038/nmeth1092. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: The serine hydrolases. Proc Natl Acad Sci USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenbaum D, Medzihradszky KF, Burlingame A, Bogyo M. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem Biol. 2000;7:569–581. doi: 10.1016/s1074-5521(00)00014-4. [DOI] [PubMed] [Google Scholar]
- 26.Salisbury CM, Cravatt BF. Activity-based probes for proteomic profiling of histone deacetylase complexes. Proc Natl Acad Sci USA. 2007;104:1171–1176. doi: 10.1073/pnas.0608659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat Chem Biol. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suda H, Aoyagi T, Takeuchi T, Umezawa H. Inhibition of aminopeptidase B and leucine aminopeptidase by bestatin and its stereoisomer. Arch Biochem Biophys. 1976;177:196–200. doi: 10.1016/0003-9861(76)90429-x. [DOI] [PubMed] [Google Scholar]
- 29.Burley SK, David PR, Lipscomb WN. Leucine aminopeptidase: Bestatin inhibition and a model for enzyme-catalyzed peptide hydrolysis. Proc Natl Acad Sci USA. 1991;88:6916–6920. doi: 10.1073/pnas.88.16.6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsuge H, et al. Crystallization and preliminary X-ray crystallographic studies of recombinant human leukotriene A4 hydrolase complexed with bestatin. J Mol Biol. 1994;238:854–856. doi: 10.1006/jmbi.1994.1341. [DOI] [PubMed] [Google Scholar]
- 31.Taylor A. Aminopeptidases: Structure and function. FASEB J. 1993;7:290–298. doi: 10.1096/fasebj.7.2.8440407. [DOI] [PubMed] [Google Scholar]
- 32.Nankya-Kitaka MF, Curley GP, Gavigan CS, Bell A, Dalton JP. Plasmodium chabaudi chabaudi and P.falciparum: Inhibition of aminopeptidase and parasite growth by bestatin and nitrobestatin. Parasitol Res. 1998;84:552–558. doi: 10.1007/s004360050447. [DOI] [PubMed] [Google Scholar]
- 33.Gavigan CS, Dalton JP, Bell A. The role of aminopeptidases in haemoglobin degradation in Plasmodium falciparum-infected erythrocytes. Mol Biochem Parasitol. 2001;117:37–48. doi: 10.1016/s0166-6851(01)00327-9. [DOI] [PubMed] [Google Scholar]
- 34.Naughton JA, Nasizadeh S, Bell A. Downstream effects of haemoglobinase inhibition in Plasmodium falciparum-infected erythrocytes. Mol Biochem Parasitol. 2010;173:81–87. doi: 10.1016/j.molbiopara.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Nishizawa R, Saino T, Takita T, Suda H, Aoyagi T. Synthesis and structure-activity relationships of bestatin analogues, inhibitors of aminopeptidase B. J Med Chem. 1977;20:510–515. doi: 10.1021/jm00214a010. [DOI] [PubMed] [Google Scholar]
- 36.Rich DH, Moon BJ, Harbeson S. Inhibition of aminopeptidases by amastatin and bestatin derivatives. Effect of inhibitor structure on slow-binding processes. J Med Chem. 1984;27:417–422. doi: 10.1021/jm00370a001. [DOI] [PubMed] [Google Scholar]
- 37.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 38.McGowan S, et al. Structural basis for the inhibition of the essential Plasmodium falciparum M1 neutral aminopeptidase. Proc Natl Acad Sci USA. 2009;106:2537–2542. doi: 10.1073/pnas.0807398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 40.Wang Q, et al. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 41.Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 42.Rosenthal PJ, McKerrow JH, Aikawa M, Nagasawa H, Leech JH. A malarial cysteine proteinase is necessary for hemoglobin degradation by Plasmodium falciparum. J Clin Invest. 1988;82:1560–1566. doi: 10.1172/JCI113766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGowan S, et al. Structure of the Plasmodium falciparum M17 aminopeptidase and significance for the design of drugs targeting the neutral exopeptidases. Proc Natl Acad Sci USA. 2010;107:2449–2454. doi: 10.1073/pnas.0911813107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skinner TS, Manning LS, Johnston WA, Davis TM. In vitro stage-specific sensitivity of Plasmodium falciparum to quinine and artemisinin drugs. Int J Parasitol. 1996;26:519–525. doi: 10.1016/0020-7519(96)89380-5. [DOI] [PubMed] [Google Scholar]
- 45.Klemba M, Beatty W, Gluzman I, Goldberg DE. Trafficking of plasmepsin II to the food vacuole of the malaria parasite Plasmodium falciparum. J Cell Biol. 2004;164:47–56. doi: 10.1083/jcb200307147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goldberg DE. Hemoglobin degradation. Curr Top Microbiol Immunol. 2005;295:275–291. doi: 10.1007/3-540-29088-5_11. [DOI] [PubMed] [Google Scholar]
- 47.Wang F, et al. Biochemical characterization of Plasmodium falciparum dipeptidyl aminopeptidase 1. Mol Biochem Parasitol. 2011;175:10–20. doi: 10.1016/j.molbiopara.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin RE, Kirk K. Transport of the essential nutrient isoleucine in human erythrocytes infected with the malaria parasite Plasmodium falciparum. Blood. 2007;109:2217–2224. doi: 10.1182/blood-2005-11-026963. [DOI] [PubMed] [Google Scholar]
- 49.Botbol V, Scornik OA. Degradation of abnormal proteins in intact mouse reticulocytes: Accumulation of intermediates in the presence of bestatin. Proc Natl Acad Sci USA. 1979;76:710–713. doi: 10.1073/pnas.76.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saric T, Graef CI, Goldberg AL. Pathway for degradation of peptides generated by proteasomes: A key role for thimet oligopeptidase and other metallopeptidases. J Biol Chem. 2004;279:46723–46732. doi: 10.1074/jbc.M406537200. [DOI] [PubMed] [Google Scholar]
- 51.Reynolds JM, et al. Antimalarial activity of the anticancer and proteasome inhibitor bortezomib and its analog ZL3B. BMC Clin Pharmacol. 2007;7:13–19. doi: 10.1186/1472-6904-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gavigan CS, Machado SG, Dalton JP, Bell A. Analysis of antimalarial synergy between bestatin and endoprotease inhibitors using statistical response-surface modelling. Antimicrob Agents Chemother. 2001;45:3175–3181. doi: 10.1128/AAC.45.11.3175-3181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ragheb D, Dalal S, Bompiani KM, Ray WK, Klemba M. Distribution and biochemical properties of an M1-family aminopeptidase in Plasmodium falciparum indicate a role in vacuolar hemoglobin catabolism. J Biol Chem. 2011;206:27255–27265. doi: 10.1074/jbc.M111.225318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Androulakis S, et al. Federated repositories of X-ray diffraction images. Acta Crystallogr D Biol Crystallogr D. 2008;64:810–814. doi: 10.1107/S0907444908015540. [DOI] [PubMed] [Google Scholar]
