

Abstract
Background
Siraitia grosvenorii (Luohanguo) is an herbaceous perennial plant native to southern China and most prevalent in Guilin city. Its fruit contains a sweet, fleshy, edible pulp that is widely used in traditional Chinese medicine. The major bioactive constituents in the fruit extract are the cucurbitane-type triterpene saponins known as mogrosides. Among them, mogroside V is nearly 300 times sweeter than sucrose. However, little is known about mogrosides biosynthesis in S. grosvenorii, especially the late steps of the pathway.
Results
In this study, a cDNA library generated from of equal amount of RNA taken from S. grosvenorii fruit at 50 days after flowering (DAF) and 70 DAF were sequenced using Illumina/Solexa platform. More than 48,755,516 high-quality reads from a cDNA library were generated that was assembled into 43,891 unigenes. De novo assembly and gap-filling generated 43,891 unigenes with an average sequence length of 668 base pairs. A total of 26,308 (59.9%) unique sequences were annotated and 11,476 of the unique sequences were assigned to specific metabolic pathways by the Kyoto Encyclopedia of Genes and Genomes. cDNA sequences for all of the known enzymes involved in mogrosides backbone synthesis were identified from our library. Additionally, a total of eighty-five cytochrome P450 (CYP450) and ninety UDP-glucosyltransferase (UDPG) unigenes were identified, some of which appear to encode enzymes responsible for the conversion of the mogroside backbone into the various mogrosides. Digital gene expression profile (DGE) analysis using Solexa sequencing was performed on three important stages of fruit development, and based on their expression pattern, seven CYP450s and five UDPGs were selected as the candidates most likely to be involved in mogrosides biosynthesis.
Conclusion
A combination of RNA-seq and DGE analysis based on the next generation sequencing technology was shown to be a powerful method for identifying candidate genes encoding enzymes responsible for the biosynthesis of novel secondary metabolites in a non-model plant. Seven CYP450s and five UDPGs were selected as potential candidates involved in mogrosides biosynthesis. The transcriptome data from this study provides an important resource for understanding the formation of major bioactive constituents in the fruit extract from S. grosvenorii.
Background
Siraitia grosvenorii, which belongs to the Cucurbitaceae family, has long been used in traditional Chinese medicine as a natural sweetener and as a folk medicine for the treatment of lung congestion, colds and sore throats. In recent years, important pharmacological characteristics, such as anti-cancer and anti-hyperglycemic effects and inhibition of oxidative modification of low-density lipoprotein, have been reported [1-4]. Many cucurbitane-type triterpene glycosides have been isolated and characterized from the fruits [5-10]. The mixed mogrosides have been estimated to be about 300 times as sweet as sucrose so that an 80% extract was nearly 250 times sweeter than sugar [7]. Among them, mogroside V which accounts to 20% of mogrosides is extremely sweet. The purified, sweet principle, mogroside V, has been approved as a high-intensity sweetening agent in Japan [11] and the non-caloric sweet taste extract is a generally recognized as safe (GRAS) non-nutritive sweetener, flavor enhancer, and food ingredient in the USA [12].
The active components responsible for the sweetness are mogrosides, which are members of the family of triterpene glycosides. In studies the relationships of the structure and the taste, the number of glucose units at the 3 and 24-position of the aglycone moiety are thought responsible for the perception of taste [13]. Among them, mogroside IV, V and mogroside VI which have more glucose units and are extremely sweet, but the fruit also contains some tasteless glycosides, as well as bitter-tasting glycosides such as mogroside III and mogroside II E which possess less glucose units (shown in Figure 1). Previous research has indicated that the bitter mogroside II E and tasteless mogroside III are the main products in the fruit at the early growing stage, but these decrease as the fruit matures [13]. The content of mogroside V increases rapidly from 50 to 70 DAF, and levels off after 85 DAF.
Figure 1.

Putative mogrosides biosynthesis pathway in Siraitia grosvenorii. AACT: acetyl-CoA acetyltransferase, EC:2.3.1.9; HMGS: hydroxymethylglutaryl-CoA synthase, EC:2.3.3.10; HMGR: 3-hydroxy-3-methylglutaryl-coenzyme A reductase, EC:1.1.1.34; MK: mevalonate kinase, EC:2.7.1.36; PMK: phosphomevalonate kinase, EC:2.7.4.2; MVD: diphosphomevalonate decarboxylase, EC:4.1.1.33; DXS: 1-deoxy-D-xylulose-5-phosphate synthase, EC:2.2.1.7; DXR: 1-deoxy-D-xylulose-5-phosphate reductoisomerase, EC:1.1.1.267; MCT: 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase, EC:2.7.7.60; CMK: 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase, EC:2.7.1.148; MCS: 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase, EC:4.6.1.12; HDS: 4-hydroxy-3-methylbut-2-enyl diphosphate synthase, EC:1.17.7.1; IDS: 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (isopentenyl/dimethylallyl diphosphate synthase), EC:1.17.1.2; IPI: isopentenyl-diphosphate delta-isomerase, EC:5.3.3.2; GPS: geranyl diphosphate synthase, EC:2.5.1.1; FPS: farnesyl diphosphate synthase/farnesyl pyrophosphate synthetase, EC:2.5.1.10; SQS: squalene synthetase; CAS: cycloartenol synthase, EC:2.5.1.21; SQE: squalene epoxidase, EC:1.14.99.7; CS: cucurbitadienol synthase, EC:5.4.99.8; P450: cytochrome P450, EC:1.14.-.-; and UDPG: UDP-glucosyltransferase, EC:2.4.1.-.
Although the various chemical and pharmacological properties of mogrosides in S. grosvenorii have been extensively studied, the biosynthetic pathway of the mogrosides remains poorly understood. Triterpene saponins are synthesized via the isoprenoid pathway by cyclization of 2, 3-oxidosqualene to give primarily cucurbitane skeleton. The triterpenoid backbone then undergoes various modifications (oxidation, substitution and glycosylation) mediated by cytochrome P450-dependent monooxygenases, glycosyltransferases and other enzymes [14]. Mogrosides, cucurbitane-type triterpene glycosides, are synthesized by the isoprenoid pathway and share the same precursor, 2, 3-oxidosqualene, with sterol. In the early stage of active isoprene unit formation, both the cytosolic mevalonate pathway (MVA pathway) and the plastidial 2-Cmethyl-D-erythritol-4-phosphate pathway (MEP pathway) can produce isopentenyl diphosphate (IPP), which is then converted to its allylic isomer, isopentenyl/dimethylallyl diphosphate (DMAPP), through the action of IPP isomerase [15]. Triterpenoids are known to be formed by the MVA pathway because they are cytosolic products. However, there are examples that two pathways can act cooperatively to create a molecule [16]. Little progress has been made toward determining the precise source of isoprene units in mogrosides biosynthesis. Due to the biological importance of sterol and diterpenoid, the previous steps in its conversion from acetyl-CoA (MVA pathway) and 1-deoxy-D-xylulose-5-phosphate (MEP pathway) to IPP have been widely studied in many plant species, but the next steps are still unclear, especially the late steps of the pathway. The cyclization of oxidosqualene is the branch point for the biosynthesis of mogrosides and sterol. However, the portion of the pathway that lies downstream of cyclization remains largely unknown. According to the proposed pathway (shown in Figure 1), some specific CYP450s and UDP-glycosyltransferases (UGTs) may catalyze the conversion of cucurbitadienol to various mogrosides. To date, no genes involved in mogroside biosynthesis have been identified from S. grosvenorii or other mogrosides-producing plants.
Next generation sequencing technology (NGS) has emerged as a cost effective approach for high-throughput sequence determination that has dramatically improved the efficiency and speed of gene discovery [17,18]. RNA-Seq refers to whole transcriptome shotgun sequencing wherein mRNA or cDNA is mechanically fragmented, resulting in overlapping short fragments that cover the entire transcriptome. DGE is a tag-based transcriptome sequencing approach where short raw tags are generated by endonuclease. The expression level of genes in the sample is measured by counting the number of individual mRNA molecules produced from each gene. A combination of RNA-Seq and DGE approaches is powerful, as it leverages the advantages of each, enabling both large scale functional assignment of genes via the assembly of large sequenced transcriptome library (RNA-Seq), and the ability to easily perform quantitative gene expression comparisons without potential bias, allowing for a more sensitive and accurate profiling of the transcriptome that more closely resembles the biology of the cell (DGE) [19]. Despite the commercial and medicinal importance of S. grosvenorii, RNA-Seq and DGE have not yet been applied to S. grosvenorii genomic research.
In this study a cDNA library generated from of equal amount of RNA taken from S. grosvenorii fruit at 50 DAF and 70 DAF were sequenced using Illumina/Solexa platform. More than 48,755,516 HQ reads from a cDNA library generated that was assembled into 43,891 unigenes. Bioinformatic analysis indicated that cDNA sequences could be identified that matched all known enzymes involved in the biosynthesis of the mogroside backbone. DGE of S. grosvenorii at 3, 50 and 70 DAF identified seven CYP450s and five UDPGs out of a total of eighty-five CYP450 and ninety UDPG unigenes, as potential candidate genes responsible for mogroside backbone modifications. These results demonstrated the powerful ability of high-throughput sequencing to identify candidate genes involved in novel metabolic pathways in non-model plant systems.
Results
Illumina Sequencing and de novo assembly
To obtain an overview of the S. grosvenorii fruit transcriptome, a cDNA library was generated from an equal mixture of RNA isolated from 50 DAF and 70 DAF fruits, and pair end sequenced using the Illumina platform. After cleaning and quality checks, 48 million of 75 bp reads were assembled into 98,510 contigs (shown in Table 1). The mean contig size was 313 bp with a length ranging from as small as 75 bp to as large as 7,056 bp. Using paired-end joining and gap-filling, the contigs were further assembled into 67,061 scaffolds with a mean size of 484 bp including 7,929 scaffolds larger than 1,000 bp (Table 1). After clustering using TGICL software [20], the 67,061 scaffolds generated 43,891 unigenes (a scaffold that matches no other scaffold) with a mean size of 668 bp. The size distribution of these contigs, scaffolds and unigenes is shown in Additional file 1.
Table 1.
Summary for the Siraitia grosvenorii transcriptome
| Total number of reads | 48,755,516 |
| Total base pairs (bp) | 3,656,663,700 |
| Average read length | 75 bp |
| Total number of contigs | 98,510 |
| Mean length of contigs | 323 bp |
| Total number of scaffolds | 67,061 |
| Mean length of scaffolds | 484 bp |
| Total number of unigenes | 43,891 |
| Mean length of unigenes | 668 bp |
| Sequences with E-value < 10-5 | 26,307 |
Annotation of predicted proteins
For annotation, distinct gene sequences were first searched using BLASTx against the non-redundant (nr) NCBI nucleotide database using a cut-off E-value of 10-5. Using this approach, 26,307 unigenes (59.9% of all unigenes) returned a significant BLAST result (shown in Additional file 2). Because of the relatively long length of unigenes (mean size of 668 bp) and the addition of 75 Uni-ESTs from a suppression subtractive hybridization cDNA library of S. grosvernorii as a reference (data not shown), most of the 43,891 assembled sequences have been matched to known genes (59.9%).
Gene ontology (GO) classification
GO assignments were used to classify the functions of the predicted S. grosvenorii genes. Based on sequence homology, 3,117 sequences can be categorized into 41 functional groups (shown in Figure 2). In each of the three main categories (biological process, cellular component and molecular function) of the GO classification, 'metabolic process', 'cell'&'cell part' and 'Binding' terms are dominant respectively. We also noticed a high-percentage of genes from categories of 'cellular process', 'organelle' and 'catalytic' and only a few genes from terms of 'biological adhesion', 'extracellular region part' and 'nutrient reservoir' (shown in Figure 2). The GO analysis showed that the functions of the identified genes are involved various biological processes. 1,209 sequences were annotated as 'metabolic process' category, which suggests that our study may allow for the identification of novel genes involved in the secondary metabolite synthesis pathways.
Figure 2.

Histogram of gene ontology classification. The results are summarized in three main categories: biological process, cellular component and molecular function. The right y-axis indicates the number of genes in a category. The left y-axis indicates the percentage of a specific category of genes in that main category.
Functional classification by KEGG
Functional classification and pathway assignment was performed by the Kyoto Encyclopedia of Genes and Genomes (KEGG) [21]. First, the 43,891 unique sequences were compared using BLASTX with an E-value cutoff of < 10-5 against the KEGG database. To identify the biological pathways that are active in the S. grosvenorii, the 26,307 annotated sequences were mapped to the reference canonical pathways in KEGG. In total, 11,475 sequences were assigned to 217 KEGG pathways. The pathways with most representation by the unigenes were starch and sucrose metabolism (287 members), purine metabolism (287 members) and pyrimidine metabolism (251 members). These annotations provide a valuable resource for investigating specific processes, functions and pathways during S. grosvenorii research. Interestingly, 739 unigenes involved in biosynthesis of secondary metabolites were found (shown in Table 2). Among them, the cluster for 'Phenylpropanoid biosynthesis [PATH: ko00940]' represents the largest group (134, 17.4%) followed by 'Limonene and pinene degradation [PATH: ko00903]' (119, 16.1%) and 'Stilbenoid, diarylheptanoid and gingerol biosynthesis [PATH: ko00945]' (85, 11.5%). Mogrosides belong to the terpenoid saponins, which share a common pathway from acetyl-CoA to 2, 3-oxidosqualene with sterol. Most of the enzymes identified in the fruit cDNA library were mapped into the terpenoid backbone [PATH: ko00900] and sterol biosynthesis [PATH: ko00100] groups by KEGG. All of the genes encoding enzymes involved in the biosynthesis of the mogroside backbone were present in our transcriptome of the S. grosvenorii fruit (shown in Table 3). In most cases, more than one unigene was annotated as the same enzyme. Such unigenes may represent different fragments of a single transcript, different members of a gene family, or both. Additionally, eighty-five unigenes for CYP450 gene and ninety unigenes for UDPG were also discovered (shown in Additional file 3). All these unigenes are important resources for S. grosvenorii genetic engineering work in future. These results also demonstrated the powerful ability of high-throughput sequencing to identify genes in metabolic pathways.
Table 2.
The unigenes related to secondary metabolites
| Biosynthesis of Secondary Metabolites | Unigene Numbers |
|---|---|
| Anthocyanin biosynthesis | 6 |
| Betalain biosynthesis | 4 |
| Brassinosteroid biosynthesis | 7 |
| Caffeine metabolism | 2 |
| Carotenoid biosynthesis | 53 |
| Diterpenoid biosynthesis | 37 |
| Flavone and flavonol biosynthesis | 11 |
| Flavonoid biosynthesis | 34 |
| Glucosinolate biosynthesis | 7 |
| Indole alkaloid biosynthesis | 27 |
| Isoquinoline alkaloid biosynthesis | 29 |
| Limonene and pinene degradation | 119 |
| Novobiocin biosynthesis | 24 |
| Phenylpropanoid biosynthesis | 134 |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | 85 |
| Streptomycin biosynthesis | 37 |
| Terpenoid backbone biosynthesis | 63 |
| Tetracycline biosynthesis | 12 |
| Tropane, piperidine and pyridine alkaloid biosynthesis | 22 |
| Zeatin biosynthesis | 26 |
| Total | 739 |
Table 3.
The numbers of Unigene involved in mogrosides biosynthesis
| Gene | Enzyme No. | Numbers |
|---|---|---|
| 1-deoxy-D-xylulose-5-phosphate synthase (DXS) | [EC:2.2.1.7] | 11 |
| 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) | [EC:1.1.1.267] | 4 |
| 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (MCT) | [EC:2.7.7.60] | 1 |
| 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase (CMK) | [EC:2.7.1.148] | 2 |
| 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MCS) | [EC:4.6.1.12] | 2 |
| 4-hydroxy-3-methylbut-2-enyl diphosphate synthase (HDS) | [EC:1.17.7.1] | 1 |
| 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (HDR/IDS) | [EC:1.17.1.2] | 1 |
| acetyl-CoA acetyltransferase (AACT) | [EC:2.3.1.9] | 5 |
| hydroxymethylglutaryl-CoA synthase (HMGS) | [EC:2.3.3.10] | 1 |
| 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGR) | [EC:1.1.1.34] | 3 |
| mevalonate kinase (MK) | [EC:2.7.1.36] | 4 |
| phosphomevalonate kinase (PMK) | [EC:2.7.4.2] | 3 |
| diphosphomevalonate decarboxylase (MVD) | [EC:4.1.1.33] | 2 |
| isopentenyl-diphosphate delta-isomerase (IPI) | [EC:5.3.3.2] | 5 |
| geranyl diphosphate synthase (GPS) | [EC:2.5.1.1] | 1 |
| farnesyl diphosphate synthase (FPS) | [EC:2.5.1.10] | 2 |
| hexaprenyl pyrophosphate synthetase (HPS) | [EC:2.5.1.33] | 1 |
| geranylgeranyl pyrophosphate synthetase (GGPS) | [EC:2.5.1.29] | 1 |
| squalene synthetase (SQS) | [EC:2.5.1.21] | 3 |
| squalene epoxidase (SQE) | [EC:1.14.99.7] | 1 |
| cycloartenol synthase (CAS) | [EC:5.4.99.8] | 5 |
| cucurbitadienol synthase (CS) | 1 | |
| Total | 60 | |
Changes in gene expression profiles during different developmental stages
DGE generates absolute rather than relative gene expression measurements and avoids many of the inherent limitations of microarray analysis. DGE was used to analyze the gene expression during three stages of S. grovenorii fruit development. Three DGE libraries: 3, 50 and 70 DAF, were sequenced that generated between 3.4 to 3.7 million high quality tags and the number of tag entities with unique nucleotide sequences ranged from 85,737 to 91,130 (shown in Table 4). The distributions of total tags and distinct tags over different tag abundance categories are shown in Additional file 4. The tag sequences were mapped to S. grosvenorii reference transcriptome database. Between 46,700 to 51,320 distinct tags were mapped to a gene in the reference database and up to 49.58% (21,761) of the sequences in transcriptome reference tag database could be unequivocally identified by unique tag (shown in Table 4).
Table 4.
Statistics of DGE sequencing
| Summary | 3 DAF | 50 DAF | 70 DAF | |
|---|---|---|---|---|
| Raw Data | Total | 3510563 | 3738193 | 3784929 |
| Raw Data | Distinct Tag | 207988 | 202720 | 184923 |
| Clean Tag | Total number | 3382707 | 3612715 | 3682563 |
| Clean Tag | Distinct Tag number | 87140 | 85737 | 91130 |
| All Tag Mapping to Gene | Total number | 1573645 | 1237119 | 1379598 |
| All Tag Mapping to Gene | Total % of clean tag | 46.52% | 34.24% | 37.46% |
| All Tag Mapping to Gene | Distinct Tag number | 46700 | 46865 | 51320 |
| All Tag Mapping to Gene | Distinct Tag % of clean tag | 53.59% | 54.66% | 56.32% |
| Unambiguous Tag Mapping to Gene | Total number | 1566698 | 1233379 | 1376138 |
| Unambiguous Tag Mapping to Gene | Total % of clean tag | 46.31% | 34.14% | 37.37% |
| Unambiguous Tag Mapping to Gene | Distinct Tag number | 46520 | 46689 | 51138 |
| Unambiguous Tag Mapping to Gene | Distinct Tag % of clean tag | 53.39% | 54.46% | 56.12% |
| All Tag-mapped Genes | number | 19607 | 21534 | 21865 |
| All Tag-mapped Genes | % of ref genes | 44.67% | 49.06% | 49.82% |
| Unambiguous Tag-mapped Genes | number | 19492 | 21406 | 21761 |
| Unambiguous Tag-mapped Genes | % of ref genes | 44.41% | 48.77% | 49.58% |
| Unknown Tag | Total number | 1809062 | 2375596 | 2302965 |
| Unknown Tag | Total % of clean tag | 53.48% | 65.76% | 62.54% |
| Unknown Tag | Distinct Tag number | 40440 | 38872 | 39810 |
| Unknown Tag | Distinct Tag % of clean tag | 46.41% | 45.34% | 43.68% |
Differentially expressed tags between samples were identified by an algorithm developed by Audic et al [22]. As expected the majority of gene expression changes occurred between the DAF 3 and DAF 50/DAF 70 (Figure 3 & Additional file 5, 6, 7) with slightly more down-regulated genes observed. More than 40%-45% of the highly regulated genes were found to be orphan sequences - no homologues found in the NCBI database. This may indicate that S. grosvenorii fruit development contains many unique processes and pathways.
Figure 3.

Changes in gene expression profile among the different developmental stages. The number of up-regulated and down-regulated genes between 3 DAF and 50 DAF; 3 DAF and 70 DAF; 50 DAF and 70 DAF are summarized. DEGs: Differentially Expressed Genes.
Expression levels of putative genes involved in mogrosides biosynthesis
As shown in Figure 4, to examine the expression of the 21 genes putatively involved in mogrosides backbone biosynthesis revealed very low expression levels of AACT, MVD, CMK, HDS, SQS genes. The 16 remaining genes had significant expression levels that showed changes in gene expression during fruit development (shown in Figure 4). Between 3 DAF and 50 DAF, DXS, DXR, MCS, IDS are down-regulated, whereas the remaining 12 genes are up-regulated. Between 50 DAF and 70 DAF, HMGR, IDS, IPI-I, IPI-II, GPS, FPS and CAS were found to be down-regulated, whereas, the other 9 genes were up-regulated. Overall the trend between 3 DAF and 70 DAF was that DXR, MCS, IDS, IPI -I showed decreased expression over time, while HMGS, HMGR, MK, PMK, MCT, IPI-II, GPS, FPS, SQS, CAS, and CS genes showed degrees of up-regulation. SQE and CS genes were found to change the most in this period, (8.9 times and 9.7 times respectively). The rapid accumulation of mogroside V from 50 DAF to 70 DAF may result from the increase in expression of SQE and CS. SQE plays a key role in the cyclical transformation of squalene to 2, 3-oxidosqualene. The cyclization of 2, 3-oxidosqualene is the branch point of sterol and mogrosides biosynthesis and controls the carbon flux through the branched biosynthetic pathways. The next important step in the pathway is via CS and CAS, which catalysis 2, 3-oxidosqualene formed cucurbitadenol and sterol respectively [23]. Cucurbitadenol is the precursor of different mogrosides. To date, no genes involved in mogroside biosynthesis have been cloned from S. grosvenorii or other mogrosides-producing plants. The correlation between the expression of SQE and CS and the rapid formation of mogrol during fruit development presents a way of finding possible CYP450 and UDPG genes involved in mogrosides biosynthesis via identification of candidate genes with similar expression profiles. This method of finding candidate genes has successfully identified a gene involved in saponin biosynthesis [24].
Figure 4.
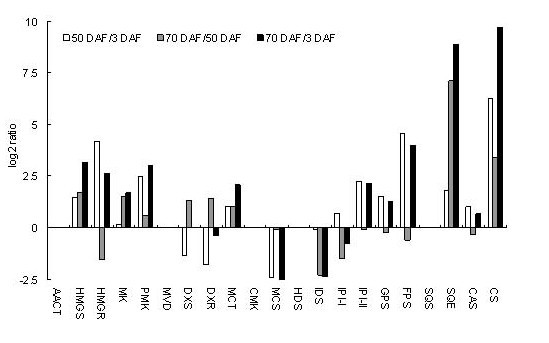
Expression pattern of genes involved in mogrosides biosynthesis at different fruit developmental stages by DGE. 21 genes expression in 3 DAF, 50 DAF and 70 DAF were analyzed by DGE.
Candidate CYP450 enzymes involved in mogrosides biosynthesis
CYP450 proteins are the largest family of plant proteins and catalyze most of the oxidation steps in plant secondary metabolism [25,26]. In the biosynthetic pathway of mogrosides, the conversion from cucurbitadenol to mogrol is catalyzed by one or more CYP450s. A total of eighty-five unigenes were annotated as CYP450s in our S. grosvenorii transcriptome (shown in Additional file 8). To find the CYP450s putatively involved in mogrosides biosynthesis, these unigenes expression profiles were compared to SQE and CS by DGE (shown in Figure 5). From 3 DAF to 70 DAF, the SQE and CS genes are up-regulated, and so only CYP450s that displayed some up-regulation during this developmental period were considered candidates for mogrosides biosynthesis. A total of seventeen CYP450s that were found to be differentially expressed between at least one of the three developmental time points were chosen, and their expression profiles hierarchically clustered with that of SQE and CS (shown in Figure 5). Seven candidate CYP450s were found to cluster closely with either SQE or CS (correlation > 0.99): Unigene5315 (transcriptome short reads assembly (TSA) accession number JL554661), Unigene23541 (JL554662), Unigene24189 (JL554663), Unigene26598 (JL554664), Unigene34139 (JL554665), Unigene36601 (JL554666) and Unigene43109 (JL554667). These genes are therefore promising candidates that may catalyze the oxidation of cucurbitadinol and the formation of mogrol.
Figure 5.
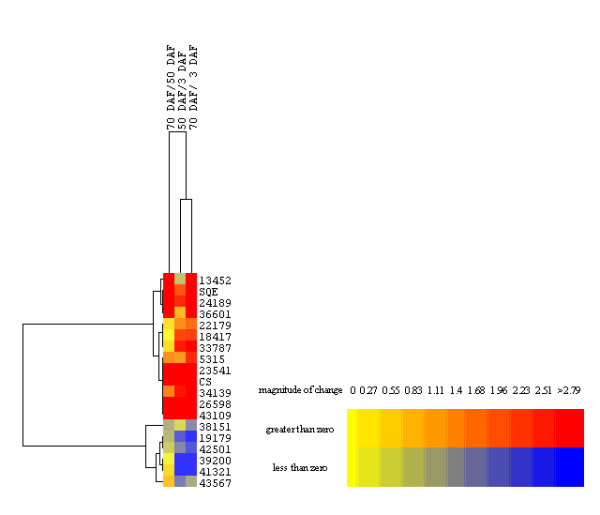
Clustering of CYP450 genes expression profiles at three different fruit developmental stages. Hierarchical clustering of expression data for 17 candidate CYP450 genes using CS and SQE as reference profiles. Expression ratios are expressed as Log 2 values.
Candidate UDPG enzymes involved in mogrosides biosynthesis
Glucosyltransferases (UDPG) are another large multi-gene family in plants. In general, glycosylation is the last step in the biosynthesis of secondary metabolites and sugar conjugation results in both increased stability and water solubility [27-29]. The S. grosovenorii cDNA library contained seventy-two glycosyltransferase and ninety glucosyltransferase unigenes (shown in Additional file 9). Only glucosyltransferases not glycosyltransferases, which used glucose as the donor and mogrol as the acceptor, catalyzed the formation of mogrosides. Similar to the candidate CYP450s, 16 differentially expressed UDPGs that showed up-regulation of gene expression during fruit development were selected and their expression profiles hierarchically clustered with that of SQE and CS (shown in Figure 6). The expression pattern of five UDPGs showed strong correlation to that of SQE and CS (> 0.99, Figure 6) and appear the best UDPGs candidates for encoding the enzymes responsible for mogrosiedes biosynthesis. These UDPGs consisted of Unigene2346 (JL554668), Unigene4016 (JL554669), Unigene8672 (JL554670), Unigene15400 (JL554671), and Unigene37589 (JL554672).
Figure 6.
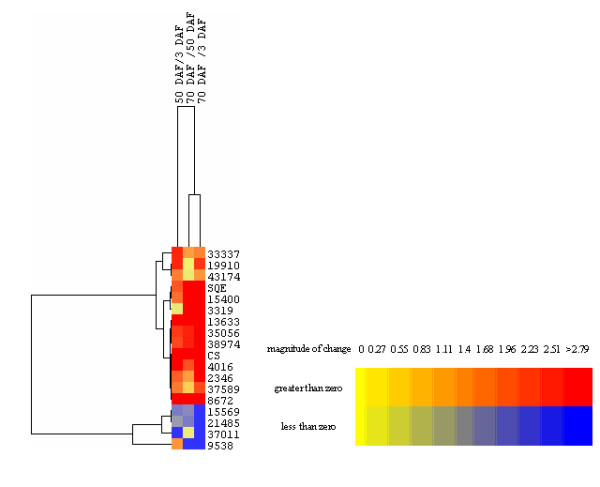
Clustering of UDPG genes expression profiles at three different fruit developmental stages. Hierarchical clustering of expression data for 16 candidate UDPG genes using CS and SQE as reference profiles. Expression ratios are expressed as Log 2 values.
Discussion
EST analysis is one of the most valuable tools for gene discovery. However, until the advent of NGS, significant coverage of the transcriptome was labor intensive and expensive. Therefore, EST analyses tended to identified only a few candidate genes involved in complex biosynthetic pathways such as that observed from studies in Panax quinquefolius and Panax ginseng [30-32]. Even in combination with subtracted cDNA libraries strategies in S. grosvenorii, traditional EST sequencing only identified one CYP450 and two Glycosyltransferases (1, 200 ESTs) and none potentially involved in mogrosides biosynthesis (data not shown). The development of NGS has removed these limitations, and a number of successful identifications of secondary metabolism genes from plants with little genomics sequence information including cotton [33], American ginseng [34], Glycyrrhiza uralensis [35], Medicago truncatula [24,36], Huperzia serrata and Phlegmariurus carinatus [37] have been performed.
Despite the commercial and medicinal importance of S. grosvenorii, molecular information on this species is lacking preventing analysis of the mogrosides biosysnthetic pathway. Creating a cDNA library from the S. grosvenorii fruit and performing RNA-seq and DGE provided a very efficient means for identifying the genes associated with known enzymes involved in the biosynthesis of secondary metabolites and for providing candidate genes that could be associated with currently unknown steps in the pathway. Thus, candidates for all of the known genes encoding enzymes involved in mogrosides backbone biosynthesis, including two 2, 3-oxidosqualene cyclases: CAS and CS were identified in this study. Very little is known about the late stages of mogrosides biosynthesis. This part of the pathway includes multiple oxidation and glycosylation steps catalyzed by enzymes from the CYP450 and glucosyltransferase superfamilies, respectively. These families of enzymes display a wide range of substrate specificities and are responsible for the diversity of many plant secondary metabolites. For example, approximately one hundred and twenty UGT genes and two hundreds and seventy-two CYP450 genes were identified in the model plant Arabidopsis thaliana. Because of the biological, pharmacological, and agricultural importance of secondary metabolites, UGTs and CYP450s have attracted considerable interest for decades, but only a few have been characterized by traditional biochemistry and genetics. To date, only two CYP450s involved in triterpene saponin biosynthesis are functionally characterized: a β-amyrin 11-oxidase from Glycyrrhiza uralensis [38] and a β-amyrin and sophoradiol 24-hydroxylase from Glycine max [39]. Also, five UGTs in triterpene saponin biosynthesis have been identified: soybean [40], UGT73K1, UGT71G1 and UGT73F3 from Medicago truncatula [24,41] and UGT74M1 from Saponaria vaccaria [42]. Genomic and co-expression analyses by DNA microarray provide a basis for future studies to define genetically the roles of multiple cytochromes P450 and glycosyltransferases in triterpene saponin biosynthesis in model plant Medicago truncatula. UGT73F3 have been identified for glucosylating hederagenin at the C28 position [24]. However, all of the aforementioned enzymes are involved in soyasapogenol and oleanane-type ginsenoside biosynthesis. No CYP450s or UGTs in the cucurbitane-type mogrosides biosynthetic pathway have been previously functionally characterized. Therefore, this study focused on the discovery of CYP450s and UGTs involved in the biosynthesis of cucurbitane-type mogrosides, which are the major mogrosides type in the S. grosvenorii fruit.
In the cucurbitane-type mogorisdes biosynthetic pathway, CYP450s catalyze the C6 and C12 hydroxylation of cucurbitadienol, while glycosylation generally occurs on C3 and C24 of the aglycones. In total, eighty-five CYP450 unigenes and ninety UDPG unigenes were found in the S. grosvenorii cDNA library. However, the large number of candidate genes produced by NGS leads to difficulty in the characterization of the enzymes that are actually involved in this pathway. One CYP450 and four UDPG were selected as the candidates most likely to be involved in ginsenoside biosynthesis through a methyl jasmonate (MeJA) inducibility experiment and tissue-specific expression pattern analysis based on NGS and real-time PCR assay [34]. Three CYP450 enzymes and six UGTS were selected as the candidates most likely to be involved in glycyrrhizin biosynthesis by the same approach [35]. The real-time PCR method is time-consuming and labor intensive, as it can only study a few genes at the same time. DNA microarrays are mainly used in model plants as they generally require either extensive gene sequence information or large scale amplification of cDNA libraries. Unfortunately, there is little information about gene cloning and EST in NCBI database in S. grosvenorii. In contrast, the DGE method based on high-throughput sequencing technology in combination with NGS transcriptome information, is more suitable and affordable for tens of thousands gene expressions studies at once. Therefore, in this study, the DGE of three different development stages were carried out to try to select those CYP450s and UGTs most likely to be involved in mogrosides biosynthesis. The expression levels of 20 genes involved in mogrosides backbone biosynthesis all showed some variation in expression pattern through DGE analysis. Among these, SQE and CS genes in late steps changed the most. They increased 7.11 times and 3.40 times respectively from 50 DAF to 70 DAF, increased 8.89 times and 9.69 times respectively in 70 DAF vs. 3 DAF. Mogroside V is the main constituent of ripe fruits, while mogroside III and II E are the leading components in unripe fruits [13]. At the early growing stage (from 3 DAF to 50 DAF), mogroside V increases from 0.057% to 0.083%, then increases rapidly to 2.150% (25.9 times) from 50 DAF to 70 DAF and gets stability at 2.700% after 85 DAF. Therefore levels of SQE and CS gene expression levels are coincident with mogroside V content. Selecting SQE and CS as reference expression patterns for CYP450s and UDPGs that would be functional at the same development periods, similar to the work on saponin biosynthesis [24], identified only seven CYP450s and five UDPGs candidates. The function of these candidate CYP450s and UDPGs will be confirmed by heterologous expression in Escherichia coli or yeast, and in vitro enzymatic assay.
Conclusion
This study demonstrates the feasibility of using combination of RNA-Seq and DGE to provide a method for identifying and studying the genes involved in secondary metabolism not only in S. grosvenorii but also other non-model medicinal plants. Candidate genes encoding enzymes potentially involved in mogrosides backbone biosynthesis as well as CYP450 and UDPG genes likely to be involved in mogrosides biosynthesis were rapidly identified by this approach.
Methods
Sample collection and preparation
Routinely, the S. grosvenorii fruits are harvested between 70 DAF to 85 DAF for medicinal use. The mogroside V appears in 50 DAF and peaks after 70 DAF. Therefore, 50 DAF and 70 DAF S. grosvenorii were collected for transcriptome sequencing from the field in Xing-an County, Guilin, Guangxi autonomous region, China. 3 DAF, 50 DAF and 70 DAF fruits were using for DGE analysis. The plant tissues were then cut into small pieces and were immediately frozen in liquid nitrogen. All materials were stored at -80°C until further processing.
RNA isolation and library preparation for transcriptome analysis
Total RNA was isolated using the Trizol reagent (Invitrogen). RNA samples were treated with Dnase I (Promega) at a concentration of 1 unit/μg of total RNA. Total RNA purity and degradation were checked on 1% agarose gels before proceeding. The samples for transcriptome analysis were prepared using the Illumina's kit following the manufacturer's recommendations. Briefly, mRNA was purified from 6 μg of total RNA using oligo (dT) magnetic beads. Following purification, the mRNA was fragmented into small pieces using divalent cations under elevated temperature and the cleaved RNA fragments were used for first strand cDNA synthesis using reverse transcriptase and random primers. This was followed by second strand cDNA synthesis using DNA polymerase I and RNaseH. These cDNA fragments then went through an end repair process and ligation of adapters. These products were purified and enriched with PCR to create the final cDNA library.
Analysis of Illumina sequencing results
The cDNA library was sequenced on the Illumina sequencing platform (GAII). The size of the library is approximately 200 bp and both ends of the libraries are sequenced. Sequences from the Illumia sequencing were deposited in the GenBank Short Read Archive (Accession SRP006841). Image deconvolution and quality value calculations were performed using the Illumina GA pipeline 1.3. The raw reads were cleaned by removing adaptor sequences, empty reads and low quality sequences (reads with unknown sequences 'N'). The reads obtained were randomly clipped into 21 bp K-mers for assembly using de Bruijn graph and SOAP denovo software [43]. After assessing different K-mer sizes, we found that the 21-mer provided the best result for transcriptome assembly. Small K-mers make the graph very complex; while large K-mers can have poor overlap in regions with low sequencing depth. After sequence assembly, the resultant contigs were joined into scaffolds using the read mate pairs. To obtained distinct gene sequences, the scaffolds were clustered using TGI Clustering tools [44]. Distinct sequences were used for blast search and annotation against an NCBI nr database using an E-value cut-off of 10-5. Functional annotation by gene ontology terms (GO, http://www.geneontology.org) was analyzed by Blast2go software. The KEGG pathways annotation was performed using Blastall software against the KEGG database. The assembled sequences can be searched using the Gene-ID listed in Additional file 3.
DGE library preparation and sequencing
Tag library preparation for the three different growth period samples (3 DAF, 50 DAF, and 70 DAF) was performed in parallel using the Illumina gene expression sample preparation kit. Briefly, total RNA from the three samples was used for mRNA capture with magnetic oligo (dT) beads. First and second strand cDNA were synthesized and beadbound cDNA was subsequently digested with NlaIII. The cDNA fragments with 3' ends were then purified with magnetic beads and Illumina adapter 1 was added to their 5' ends. The junction of the Illumina adapter 1 and CATG site is the recognition site of MmeI, which cuts 17 bp downstream of the CATG site, producing tags with adapter 1. After removing 3' fragments with magnetic beads precipitation, Illumina adapter 2 was introduced at 3' ends of tags, acquiring tags with different adapters at both ends to form a tag library. After 15 cycles of linear PCR amplification, 85 base strips were purified by PAGE gel electrophoresis. These strips were then digested, and the single-chain molecules were fixed onto the Illumina sequencing chip for sequencing. The reproducibility of DGE were > 0.99 [45]. The sequences from the DGE analysis were deposited in the GenBank Short Read Archive (Accession SRP006090).
Analysis and mapping of DGE tags
Sequencing-received raw image data was transformed by base calling into sequence data. Prior to mapping reads to the reference database, we filtered all sequences to remove adaptor sequence, low quality sequences (tags with unknown sequences 'N'), empty tags (sequence with only adaptor sequences but no tags); low complexity, and tags with a copy number of 1 (probably sequencing error). A preprocessed database of all possible CATG+17 nucleotide tag sequences was created using our transcriptome reference database. For annotation, all tags were mapped to the reference sequences and only allowed 1 or fewer nucleotide mismatches. All the tags mapped to reference sequences from multiple genes were filtered and the remaining tags were designed as unambiguous tags. For gene expression analysis, the number of expressed tags was calculated and then normalized to TPM (number of transcripts per million tags); and the differentially expressed tags were used for mapping and annotation. The complete lists of differentially expressed genes are shown in Additional file 5, 6 and 7.
Evaluation of DGE libraries
A statistical analysis of the frequency of each tag in the different cDNA libraries was performed to compare gene-expression in different developmental stages. Statistical comparison was performed using the method described by Audic et al [22]. FDR (false discovery rate) was used to determine the threshold of P-value in multiple test and analysis. We used FDR < 0.001 as the threshold to judge the significance of gene expression difference. For pathway enrichment analysis, we mapped all differentially expressed genes to terms in the KEGG database and looked for significantly enriched KEGG terms compared to the genome background.
Clustering of CYP450 and UDPG gene expression profiles
Hierarchical clustering of log-transformed expression data was carried out using the Cluster 3.0 and Treeview programs [46]. Correlations between gene clusters were determined using Pearson's correlation. Heat maps were constructed using the University of Toronto BAR Heatmapper tool http://www.bar.utoronto.ca/ntools/cgi-bin/ntools_heatmapper.cgi.
List of abbreviations
DGE: Digital Gene Expression; DAF: days after flowering; MVA: mevalonate pathway; MEP: 2-Cmethyl-D-erythritol-4-phosphate pathway; IPP: isopentenyl diphosphate; DMAPP: isopentenyl/dimethylallyl diphosphate; NGS: Next generation sequencing technology; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; AACT: acetyl-CoA acetyltransferase; HMGS: hydroxymethylglutaryl-CoA synthase; HMGR: 3-hydroxy-3-methylglutaryl-coenzyme A reductase; MK: mevalonate kinase; PMK: phosphomevalonate kinase; MVD: diphosphomevalonate decarboxylase; DXS: 1-deoxy-D-xylulose-5-phosphate synthase; DXR: 1-deoxy-D-xylulose-5-phosphate reductoisomerase; MCT: 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase; CMK: 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase; MCS: 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase; HDS: 4-hydroxy-3-methylbut-2-enyl diphosphate synthase; IDS: 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (isopentenyl/dimethylallyl diphosphate synthase); IPI: isopentenyl-diphosphate delta-isomerase; GPS: geranyl diphosphate synthase; FPS: farnesyl diphosphate synthase/farnesyl pyrophosphate synthetase; SQS: squalene synthetase; SQE: squalene epoxidase; CAS: cycloartenol synthase; CS: cucurbitadienol synthase; CYP450: cytochrome P450; UDPG: UDP-glucosyltransferase.
Authors' contributions
QT conceived the study, designed and built the cDNA library, participated in data analysis, and drafted the manuscript. XJM initiated the project, helped to conceive the study, and participated in the design and coordination. DYQ designed the study and data analysis. IW participated in clustering of gene expression profiles. CS participated in manuscript formatting and editing. CMM participated in experiment materials preparation. YFY, WF and HZ participated in RNA extraction and RACE experiments. All authors read and approved the final manuscript.
Supplementary Material
Overview of Siraitia grosvenorii transcriptome sequencing and assembly. (A) Size distribution of Illumina sequencing contigs. (B) Size distribution of Illumina sequencing scaffolds and which after paired-end and gap filling. (C) Size distribution of Illumina sequencing unigenes and which after paired-end and gap filling.
Top BLAST hits from NCBI nr database. BLAST results against the NCBI nr database for all the distinct sequences with a cut-off E value above 10-5 are shown.
Top BLAST hits from NCBI Swissprot database. BLAST results against the NCBI Swissprot database for all the distinct sequences with a cut-off E value above 10-5 are shown.
Distribution of total tags and distinct tags over different tag abundance categories. (A) Distribution of total clean tags. Numbers in the square brackets indicate the range of copy numbers for a specific category of tags. For example, [2,5] means all the tags in this category has 2 to 5 copies. Numbers in the parentheses show the total tag copy number and ratio for all the tags in that category. (B) Distribution of distinct clean tags. Numbers in the square brackets indicate the range of copy numbers for a specific category of tags. Numbers in the parentheses show the total types of tags in that category.
Differentially expressed genes between 3 DAF and 50 DAF. TPM: transcript copies per million tags. Raw intensity: the total number of tags sequenced for each gene. FDR: false discovery rate. We used FDR < 0.001 and the absolute value of log2Ratio ≤ 1 as the threshold to judge the significance of gene expression difference. In order to calculate the log2Ratio and FDR, we used TPM value of 0.001 instead of 0 for genes that do not express in one sample.
Differentially expressed genes between 3 DAF and 70 DAF
Differentially expressed genes between 50 DAF and 70 DAF
Putative CYP450 genes in the Solexa cDNA library
Putative UDPG genes in the Solexa cDNA library
Contributor Information
Qi Tang, Email: tangqi423@sina.com.
Xiaojun Ma, Email: xjma@public.bta.net.cn.
Changming Mo, Email: mochming@126.com.
Iain W Wilson, Email: Iain.wilson@csiro.au.
Cai Song, Email: cai.song2011@gmail.com.
Huan Zhao, Email: 53522722@163.com.
Yanfang Yang, Email: echojln@yahoo.com.cn.
Wei Fu, Email: fuwei0212@163.com.
Deyou Qiu, Email: qiudy@caf.ac.cn.
Acknowledgements
We acknowledge the Beijing Genomics Institute at Shenzhen for its assistance in original data processing and related bioinformatics analysis. This work was supported by the National Natural Science Foundation of China (grant No. 30860379 & 30960500).
References
- Takasaki M, Konoshima T, Murata Y, Sugiura M, Nishino H, Tokuda H, Matsumoto K, Kasai R, Yamasaki K. Anticarcinogenic activity of natural sweeteners, cucurbitane glycosides, from Momordica grosvenori. Cancer Letters. 2003;198:37–42. doi: 10.1016/S0304-3835(03)00285-4. [DOI] [PubMed] [Google Scholar]
- Suzuki YA, Murata Y, Inui H, Sugiura M, Nakano Y. Triterpene glycosides of Siraitia grosvenori inhibit rat intestinal maltase and suppress the rise in blood glucose level after a single oral administration of maltose in rats. J Agric Food Chem. 2005;53(8):2941–2946. doi: 10.1021/jf0478105. [DOI] [PubMed] [Google Scholar]
- Takeo E, Yoshida S, Tada S, Shingu T, Matsuura H, Murata Y, Yoshikawa S, Ishikawa T, Nakamura H, Ohsuzu F, Kohda H. Sweet elements of Siraitia grosvenori inhibit oxidative modification of low-density lipoprotein. J Atheroscler Thromb. 2002;9(2):114–120. doi: 10.5551/jat.9.114. [DOI] [PubMed] [Google Scholar]
- Lin GP, Jiang T, Hu XB, Qiao XH, Tuo QH. Effect of Siraitia grosvenorii Polysaccharide on Glucose and Lipid of Diabetic Rabbits Induced by Feeding High Fat/High Sucrose Chow. Exp Diabetes Res. 2007;2007:67435. doi: 10.1155/2007/67435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. Intense sweetener from Lo Han Kuo. Experientia. 1975;31(5):533–534. doi: 10.1007/BF01932442. [DOI] [PubMed] [Google Scholar]
- Takemoto T, Arihara S, Nakajima T, Okuhira M. Studies on the constituents of Fructus momordicae. II. Structure of sapogenin. Yakugaku Zasshi. 1983;103(11):1155–1166. doi: 10.1248/yakushi1947.103.11_1155. [DOI] [PubMed] [Google Scholar]
- Kasai R, Nie RL, Nashi K, Ohtani K, Zhou J, Tao GD, Tanaka O. Sweet cucurbitane glycosides from fruits of Siraitha siamensis (chi-zi luo-han-guo), a Chinese folk medicine. Agric Biol Chem. 1989;53(12):3347–3349. [Google Scholar]
- Matsumoto K, Kasai R, Ohtani K, Tanaka O. Minor cucurbitane-glycosides from fruits of Siraitia grosvenorii (Cucurbitaceae) Chem Pharm Bull. 1990;38(7):2030–2032. [Google Scholar]
- Li DP, El-Aasr M, Ikeda T, Ogata M, Miyashita H, Yoshimitsu H, Nohara T. Two new cucurbitane-type glycosides obtained from roots of Siraitia grosvenori SWINGLE. Chem Pharm Bull (Tokyo) 2009;57(8):870–872. doi: 10.1248/cpb.57.870. [DOI] [PubMed] [Google Scholar]
- Jia Z, Yang X. A minor, sweet cucurbitane glycoside from Siraitia grosvenorii. Nat Prod Commun. 2009;4(6):769–772. [PubMed] [Google Scholar]
- Jakinovich W Jr, Moon C, Choi YH, Kinghorn AD. Evaluation of plant extracts for sweetness using the Mongolian gerbil. J Nat Prod. 1990;53(1):190–195. doi: 10.1021/np50067a030. [DOI] [PubMed] [Google Scholar]
- Xia Y, Rivero-Huguet ME, Hughes BH, Marshall WD. Isolation of the sweet components from Siraitia grosvenorii. Food Chemistry. 2008;107(3):1022–1028. doi: 10.1016/j.foodchem.2007.09.014. [DOI] [Google Scholar]
- Li D, Ikeda T, Huang Y, Liu J, Nohara T, Sakamoto T, Nonaka G-I. Seasonal variation of mogrosides in Lo Han Kuo (Siraitia grosvenori) fruits. J Nat Med. 2007;61(3):307–312. doi: 10.1007/s11418-006-0130-7. [DOI] [Google Scholar]
- Haralampidis K, Trojanowska M, Osbourn AE. Biosynthesis of triterpenoid saponins in plants. Adv Biochem Eng Biotechno. 2002;75:31–49. doi: 10.1007/3-540-44604-4_2. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler HK. The 1-deoxy-D-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu Rev Plant Physiol Plant Mol Biol. 1999;50:47–65. doi: 10.1146/annurev.arplant.50.1.47. [DOI] [PubMed] [Google Scholar]
- Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. Wiley; 2009. [Google Scholar]
- Schuster SC. Next-generation sequencing transforms today's biology. Nat Methods. 2008;5(1):16–18. doi: 10.1038/nmeth1156. [DOI] [PubMed] [Google Scholar]
- Ansorge WJ. Next-generation DNA sequencing techniques. N Biotechnol. 2009;25(4):195–203. doi: 10.1016/j.nbt.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Xiang LX, He D, Dong WR, Zhang YW, Shao JZ. Deep sequencing-based transcriptome profiling analysis of bacteria-challenged Lateolabrax japonicus reveals insight into the immune-relevant genes in marine fish. BMC Genomics. 2010;11:472. doi: 10.1186/1471-2164-11-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B. et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19(5):651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Shibuya M, Adachi S, Ebizuka Y. Cucurbitadienol synthase, the first committed enzyme for cucurbitacin biosynthesis, is a distinct enzyme from cycloartenol synthase for phytosterol biosynthesis. Tetrahedron. 2004;60(33):6995–7003. doi: 10.1016/j.tet.2004.04.088. [DOI] [Google Scholar]
- Naoumkina MA, Modolo LV, Huhman DV, Urbanczyk-Wochniak E, Tang Y, Sumner LW, Dixon RA. Genomic and coexpression analyses predict multiple genes involved in triterpene saponin biosynthesis in Medicago truncatula. Plant Cell. 2010;22(3):850–866. doi: 10.1105/tpc.109.073270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon MJ. Cytochrome P450: nature's most versatile biological catalyst. Annu Rev Pharmacol Toxicol. 2005;45:1–25. doi: 10.1146/annurev.pharmtox.45.120403.100030. [DOI] [PubMed] [Google Scholar]
- Morant M, Bak S, Møller BL, Werck-Reichhart D. Plant cytochromes P450: tools for pharmacology, plant protection and phytoremediation. Curr Opin Biotechnol. 2003;14(2):151–162. doi: 10.1016/S0958-1669(03)00024-7. [DOI] [PubMed] [Google Scholar]
- Bowles D, Lim EK, Poppenberger B, Vaistij FE. Glycosyltransferases of lipophilic small molecules. Annu Rev Plant Biol. 2006;57:567–597. doi: 10.1146/annurev.arplant.57.032905.105429. [DOI] [PubMed] [Google Scholar]
- Gachon CM, Langlois-Meurinne M, Saindrenan P. Plant secondary metabolism glycosyltransferases: the emerging functional analysis. Trends Plant Sci. 2005;10(11):542–549. doi: 10.1016/j.tplants.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- Wu Q, Song J, Sun Y, Suo F, Li C, Luo H, Liu Y, Li Y, Zhang X, Yao H. et al. Transcript profiles of Panax quinquefolius from flower, leaf and root bring new insights into genes related to ginsenosides biosynthesis and transcriptional regulation. Genome Res. 2010;20(2):265–272. doi: 10.1101/gr.097261.109. [DOI] [PubMed] [Google Scholar]
- Kim MK, Lee BS, In JG, Sun H, Yoon JH, Yang DC. Comparative analysis of expressed sequence tags (ESTs) of ginseng leaf. Plant Cell Rep. 2006;25(6):599–606. doi: 10.1007/s00299-005-0095-0. [DOI] [PubMed] [Google Scholar]
- Jung JD, Park HW, Hahn Y, Hur CG, In DS, Chung HJ, Liu JR, Choi DW. Discovery of genes for ginsenoside biosynthesis by analysis of ginseng expressed sequence tags. Plant Cell Rep. 2003;22(3):224–230. doi: 10.1007/s00299-003-0678-6. [DOI] [PubMed] [Google Scholar]
- Wang QQ, Liu F, Chen XS, Ma XJ, Zeng HQ, Yang ZM. Transcriptome profiling of early developing cotton fiber by deep-sequencing reveals significantly differential expression of genes in a fuzzless/lintless mutant. Genomics. 2010;96(6):369–376. doi: 10.1016/j.ygeno.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Sun C, Li Y, Wu Q, Luo H, Sun Y, Song J, Lui EM, Chen S. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genomics. 2010;11:262. doi: 10.1186/1471-2164-11-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Luo HM, Sun C, Song JY, Sun YZ, Wu Q, Wang N, Yao H, Steinmetz A, Chen SL. EST analysis reveals putative genes involved in glycyrrhizin biosynthesis. BMC Genomics. 2010;11:268. doi: 10.1186/1471-2164-11-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung F, Haas BJ, Goldberg SM, May GD, Xiao Y, Town CD. Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology. BMC Genomics. 2006;7:272. doi: 10.1186/1471-2164-7-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo HM, Li Y, Sun C, Wu Q, Song JY, Sun YZ, Steinmetz A, Chen SL. Comparison of 454-ESTs from Huperzia serrata and Phlegmariurus carinatus reveals putative genes involved in lycopodium alkaloid biosynthesis and developmental regulation. BMC Plant Biology. 2010;10:209. doi: 10.1186/1471-2229-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki H, Ohyama K, Sawai S, Mizutani M, Ohnishi T, Sudo H, Akashi T, Aoki T, Saito K, Muranaka T. Licorice beta-amyrin 11-oxidase, a cytochrome P450 with a key role in the biosynthesis of the triterpene sweetener glycyrrhizin. Proc Natl Acad Sci USA. 2008;105(37):14204–14209. doi: 10.1073/pnas.0803876105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M, Hoshino M, Katsube Y, Hayashi H, Kushiro T, Ebizuka Y. Identification of beta-amyrin and sophoradiol 24-hydroxylase by expressed sequence tag mining and functional expression assay. FEBS J. 2006;273(5):948–959. doi: 10.1111/j.1742-4658.2006.05120.x. [DOI] [PubMed] [Google Scholar]
- Kurosawa Y, Takahara H, Shiraiwa M. UDP-glucuronic acid: soyasapogenol glucuronosyltransferase involved in saponin biosynthesis in germinating soybean seeds. Planta. 2002;215(4):620–629. doi: 10.1007/s00425-002-0781-x. [DOI] [PubMed] [Google Scholar]
- Achnine L, Huhman DV, Farag MA, Sumner LW, Blount JW, Dixon RA. Genomics-based selection and functional characterization of triterpene glycosyltransferases from the model legume Medicago truncatula. Plant. 2005;41(6):875–887. doi: 10.1111/j.1365-313X.2005.02344.x. [DOI] [PubMed] [Google Scholar]
- Meesapyodsuk D, Balsevich J, Reed DW, Covello PS. Saponin biosynthesis in Saponaria vaccaria. cDNAs encoding beta-amyrin synthase and a triterpene carboxylic acid glucosyltransferase. Plant Physiol. 2007;143(2):959–969. doi: 10.1104/pp.106.088484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B. et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19(5):651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 't Hoen PA, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RH, de Menezes RX, Boer JM, van Ommen GJ, den Dunnen JT. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008;36(21):e141. doi: 10.1093/nar/gkn705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95(25):14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Overview of Siraitia grosvenorii transcriptome sequencing and assembly. (A) Size distribution of Illumina sequencing contigs. (B) Size distribution of Illumina sequencing scaffolds and which after paired-end and gap filling. (C) Size distribution of Illumina sequencing unigenes and which after paired-end and gap filling.
Top BLAST hits from NCBI nr database. BLAST results against the NCBI nr database for all the distinct sequences with a cut-off E value above 10-5 are shown.
Top BLAST hits from NCBI Swissprot database. BLAST results against the NCBI Swissprot database for all the distinct sequences with a cut-off E value above 10-5 are shown.
Distribution of total tags and distinct tags over different tag abundance categories. (A) Distribution of total clean tags. Numbers in the square brackets indicate the range of copy numbers for a specific category of tags. For example, [2,5] means all the tags in this category has 2 to 5 copies. Numbers in the parentheses show the total tag copy number and ratio for all the tags in that category. (B) Distribution of distinct clean tags. Numbers in the square brackets indicate the range of copy numbers for a specific category of tags. Numbers in the parentheses show the total types of tags in that category.
Differentially expressed genes between 3 DAF and 50 DAF. TPM: transcript copies per million tags. Raw intensity: the total number of tags sequenced for each gene. FDR: false discovery rate. We used FDR < 0.001 and the absolute value of log2Ratio ≤ 1 as the threshold to judge the significance of gene expression difference. In order to calculate the log2Ratio and FDR, we used TPM value of 0.001 instead of 0 for genes that do not express in one sample.
Differentially expressed genes between 3 DAF and 70 DAF
Differentially expressed genes between 50 DAF and 70 DAF
Putative CYP450 genes in the Solexa cDNA library
Putative UDPG genes in the Solexa cDNA library