

Abstract
We present complete sequences of the mitochondrial genomes for two important mosquitoes, Aedes aegypti and Culex quinquefasciatus, that are major vectors of dengue virus and lymphatic filariasis, respectively. The A. aegypti mitochondrial genome is 16,655 bp in length and that of Culex quinquefasciatus is 15,587 bp, yet both contain 13 protein coding genes, 22 transfer RNA (tRNA) genes, one 12S ribosomal RNA (rRNA) gene, one 16S rRNA gene and a control region (CR) in the same order. The difference in the genome size is due to the difference in the length of the control region. We also analyzed insertions of nuclear copies of mtDNA-like sequences (NUMTs) in a comparative manner between the two mosquitoes. The NUMT sequences occupy ~0.008% of the A. aegypti genome and ~ 0.001% of the C. quinquefasciatus genome. Several NUMTs were found localized in the introns of predicted protein coding genes in both genomes (32 genes in A. aegypti but only four in C. quinquefasciatus). None of these NUMT-containing genes had an ortholog between the two species or had paralogous copies within a genome that was also NUMT-containing. It was further observed that the NUMT-containing genes were relatively longer but had lower GC content compared to the NUMT-less paralogous copies. Moreover, stretches of homologies are present among the genic and non-genic NUMTs that may play important roles in genomic rearrangement of NUMTs in these genomes. Our study provides new insights on understanding the roles of nuclear mtDNA sequences in genome complexities of these mosquitoes.
Keywords: mitochondrial DNA, NUMTs, pseudogene, Culicidae, phylogeny, genome organization
1. Introduction
Much of our knowledge on Culicidae disease vectors has resulted from studies on Aedes and Culex mosquitoes. Aedes aegypti and Culex quinquefasciatus are major vectors of dengue virus and lymphatic filariasis, respectively. Complete genome sequences are available for both species (Nene et al. 2007, Arensburger et al. 2010). Along with nuclear genome sequencing projects, mitochondria genome sequences are regarded as important additional resources. Mitochondrial sequences are useful in inferring phylogenetic relationships, population genetics and molecular evolution in general and have been widely exploited in studying insect ecology, in particular, using them as molecular markers (Behura 2006). Mitochondrial DNA has a simple structure, it is maternally inherited, with no recombination, and it replicates independently from the nuclear genome. In addition, it has a different rate of evolution, usually much faster than that of its nuclear counterpart, facilitating our ability to infer phylogenetic relationships among closely related organisms (Arctander 1995; Lopez 1996; Krzywinski, Grushko, Besansky 2006).
It is believed, according to the endosymbiotic theory, that mitochondria in animals may be derived from α-proteobacteria. While the genome size of proteobacteria is ~4000–6000 kb, the present-day eukaryotic mitochondrial DNA is only ~15 to 40 kb. This has been linked to reduction of mitochondria genome size over evolutionary periods possibly by gradual transfer of mitochondrial genes to the nuclear genome (Thorsness, Weber 1996; Timmis et al. 2004). Transferred copies of mitochondrial sequences in the nuclear genome have been discovered in many eukaryotes (Bensasson et al. 2001; Richly and Leister 2004). They are commonly referred to as nuclear mitochondrial sequences (NUMTs). The presence of mitochondrial sequences in nuclear genomes was first reported in mice (du Buy and Riley, 1967). Identification of NUMTs is important for many reasons. Because the transferred mtDNA sequences remain as “molecular fossils” in the nuclear genome, their identification can help us better understand the confounding effects in phylogenetic inferences of species (Bensasson et al. 2001). Moreover, NUMT insertion loci may be used to predict common ancestry for a particular lineage and to determine phylogenetic branching orders of various species (Zischler et al. 1995). Also, within species variation of NUMT copy number can be used as a population genetics tool as well (Bensasson et al. 2001).
The integration of mtDNA sequences into nuclear DNA is generally accomplished by non-homologous end joining mechanisms (Blanchard et al. 1996). They are commonly inserted in the intergenic regions and, to some extent, in the intron regions of coding genes as has been observed in humans as well as in yeast (Blanchard et al. 1996). The insert size varies drastically from small fragments (<100 bp) to several kilobases of mtDNA (Bensasson et al. 2001). Moreover, NUMTs are not equally abundant in all species (Richly and Leister 2004). In insects, the presence of NUMTs has been demonstrated in tiger beetles (Pons and Vogler, 2005), grasshopper (Bensasson et al. 2000), Sitobion aphids (Sunnucks and Hales, 1996), honey bee (Pamilo et al. 2007; Behura 2007) and Nasonia vitripennis (Viljakainen et al. 2010) among others. The genome sequence of Anopheles gambiae showed no inserts of mitochondrial sequences and that of Drosophila melanogaster showed only a few (6 to 8) copies (Richly and Leister 2004). NUMTs in the nuclear genome of the A. aegypti genome have been identified (Hlaing et al. 2009; Black et al. 2009). However, no detailed study has been performed on the genomic rearrangement patterns of these sequences or their presence in the protein coding genes of this mosquito.
In this study, we determined complete sequences of mtDNA for A. aegypti and C. quinquefasciatus; then performed comparative analysis of mtDNA-like sequences between their nuclear genomes. Our results show that nuclear mtDNA sequences represent an important component of genomic complexity of these mosquitoes. The information obtained from this investigation has important implications to further our ability to use mtDNA as a tool to study these important mosquito vectors.
2. Materials and Methods
2.1. Assembly, annotation and sequence validation of A. aegypti mtDNA
We initially identified and extracted A. aegypti mtDNA sequences by BLAST analysis with the Aedes albopictus mtDNA genome (accession no. AY072044) against the A. aegypti whole genome sequence (WGS) trace files at NCBI (http://www.ncbi.nlm.nih.gov). The traces were assembled into a single contig using the ‘AMOS’ package (http://www.amos.sourceforge.net) and a circularized molecule was predicted using the ‘slice tools’ package (http://sourceforge.net/projects/slicetools/). Mate pair relationships in the circular contig were confirmed numerically and visually using the ‘HAWKEYE’ tool of AMOS. From this assembly, a total of 23 primer pairs (Table S1) were designed to generate overlapping amplicons representing the complete mtDNA genome and were used for PCR amplification from total DNA extracted from A. aegypti (LVPIB12 strain). PCR was performed in a 25 μl reaction volume containing 20 pmole each of forward primer and reverse primer, 200 μM dNTP mix, 1ul (~20 ng) of template DNA and 0.25 unit of Taq DNA polymerase with thermocycler conditions of 94°C for 15 sec, 55°C for 1 min and 72°C for 2 min for 30 cycles. The amplified fragments were column purified (Qiagen QIAquick PCR Purification Kit) and direct sequenced with the same primers using BigDye™ on a 3730XL DNA Analyzer (Applied Biosystems). The sequences were then assembled using Seqman (DNASTAR) into a circular molecule that showed complete sequence identity with the initial putative assembly derived from the genome trace files. The tRNA genes were identified using tRNAscan-SE (Lowe and Eddy, 1997), while the protein coding genes, ribosomal RNA genes and the control region were manually annotated by comparisons to the A. gambiae (accession no. L20934) and A. albopictus (accession no. AY072044) mitochondrial genomes. The A. aegypti mtDNA sequence has been submitted to GenBank (accession no. EU352212).
2.2. Assembly and annotation of C. quinquefasciatus mtDNA
Similar bioinformatics and experimental methods as described above were adopted to to identify the mtDNA sequences from the C. quinquefasciatus trace file database at NCBI and to assemble the complete C. quinquefasciatus mitochondrial genome (accession no. GU188856).
2.3. Analysis of mtDNA-like sequences in nuclear genomes
The mtDNA genome sequences were used as the query in local BLASTN (Altschul et al. 1990) searches (with E-value < 0.00001) to identify mtDNA-like sequences in the assembled A. aegypti and C. quinquefasciatus nuclear genomes (AaegL1 and CpipJ1, respectively). The ‘hits’ were obtained in tabular form and the number, size and distributions of NUMT fragments were calculated using Excel (Table S2). The locations of NUMTs in relation to the predicted protein coding genes were determined from the start and end coordinates of the predicted gene sets (AaegL1.1 and CpipJ1.2) and those of the hits. The gene lists for orthologous relationships of A. aegypti genes with C. quinquefasciatus genes and the paralogous copies of the NUMT-containing genes of A. aegypti were downloaded from http://www.vectorbase.org/ in October, 2008. NUMT fragmentations were determined by comparing the distance between neighboring NUMTs with the corresponding homologous sequences in the mtDNA. If the distance between two NUMTs was ‘similar’ (± 100 bp) to the distance between the corresponding regions in mtDNA, then these NUMTs were regarded as fragmentation products of a larger original insert. This approach has been used to identify fragmented NUMTs in previous work (Behura 2007).
Mitochondrial DNA specific primer pairs (Table S1) were used for PCR-screening of mtDNA-like sequences in an A. aegypti BAC (bacterial artificial chromosome) library. The library has been described earlier (Jiménez et al. 2004). The clones of each 384-well plate were pooled to generate 131 plate-pools (representing 50,304 BAC clones) of this library. The purified lysates of each plate-pool were used as template with the mtDNA specific primers to screen for possible mtDNA insertions in the BAC DNAs. A total of 15 plate-pools were identified as positive by PCR screening (Table S3). PCR was performed under similar conditions as previously described (Jiménez et al. 2004). As PCR with primer pair #13 was most often positive among these BAC pools, the corresponding amplicon was used as a radiolabeled probe to identify individual BAC clones for further analysis. Probe preparation and membrane hybridizations were performed as described earlier (Jiménez et al. 2004). Individual BAC clones verified as NUMT-positive from library hybridizations were subjected to PCR using primer pair #13 to confirm the presence of NUMT sequences. Authenticity of NUMT-positive clones was confirmed by direct sequencing of the PCR products.
We observed that the near complete C. quinquefasicatus mtDNA sequence (the sequence contained frame-shift mutations in three coding genes) was misassembled into supercontig 3.816 by the genome assembly pipelines. To test the validity of the supercontig 3.816 assembly, we performed PCR assays across the putative mtDNA-nuclear DNA boundary. One of the primers (primer #1; supercont3.816:51-70: 5′-AAGTGTTGCCAGTCACCTCA-3′) was anchored to the nuclear DNA and the other primer (primer #2; supercont3.816:592-573: 5′-TGGGGTATGAACCCAATAGC-3′) was specific to the mtDNA-like sequence. A positive control was included in the PCR assay in which primer #1 was used along with primer #3 (supercont3.816:214-195: 5′-TGGGGTATGAACCCAATAGC-3′); these primer pairs targeted a 164 bp product within the nuclear DNA only, immediately flanking the 15.587 kb mtDNA-like sequence. The reaction mixtures for the negative controls were identical to test and positive control, but minus primers. PCR reactions were conducted as above except that the annealing temperature was kept at 59°C and the template was total DNA (~20 ng) of C. quinquefasciatus (JHB strain). PCR results were visualized in 1.2% ethidium bromide agarose gels.
3. Results
3.1. Assembly of the Aedes aegypti mitochondrial genome sequence
To identify mtDNA-like sequences in the sequenced genome of A. aegypti, we first determined the complete nucleotide sequence of the A. aegypti mtDNA. The A. aegypti whole genome sequence (WGS) reads were used, guided by the A. albopictus mitochondrial genome (AY072044) as the template, to construct an assembly (see methods). The resulting contig was 16,655 bp in length. It was composed of 728 traces with 37.29-fold mean sequence coverage (compared with 7.63-fold mean coverage for the WGS genome assembly). The assembly was verified by PCR using primer pairs designed from the assembly (Table S1) and also by sequencing the amplified products. The genome was annotated for coding and structural genes and submitted to the GenBank as the A. aegypti mtDNA under the accession# EU352212. The 16,655 bp A. aegypti mitochondrial genome contains 13 protein coding genes, 22 transfer RNA (tRNA) genes, one 12S ribosomal RNA (rRNA) gene, one 16S rRNA gene and a control region (Figure 1). Each of the tRNA genes has only one copy for each amino acid, except for leucine and serine which have two copies each. The control region (CR) consists of two repeats; each is repeated three times within the region (data not shown). The A+T content of A. aegypti mtDNA is close to that observed in other insect mitochondrial genomes, with a total A+T content of 78.9 % for the entire genome and 77.1% for coding regions (Stewart and Beckenbach, 2006).
Figure 1.
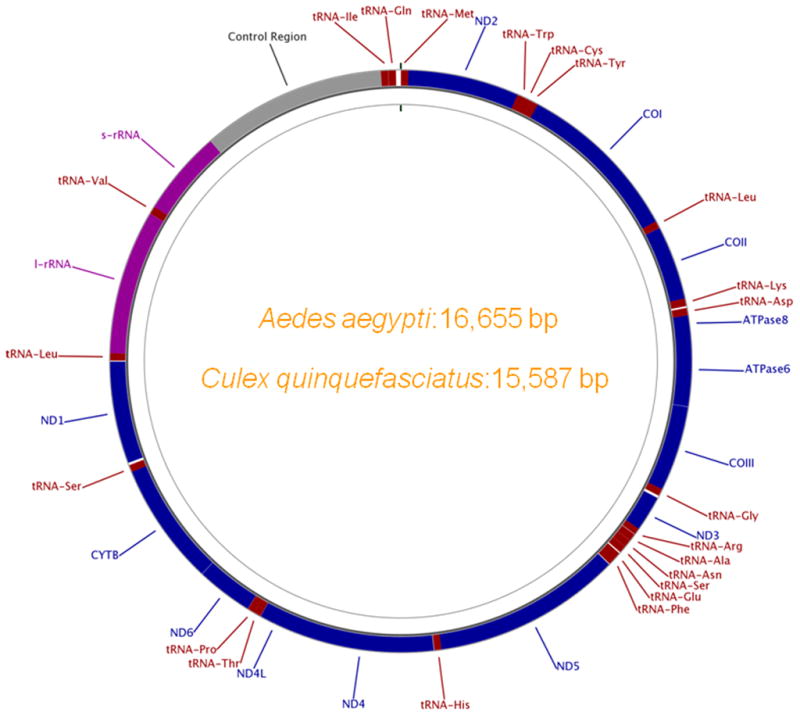
The circular map of Aedes aegypti and Culex quinquefasciatus mtDNA. The predicted genes are labeled and color-coded; protein coding genes: blue, tRNA genes: red, rRNA genes: purple and control region (CR): grey. The gene names are labeled outside the circle.
3.2. Assembly of Culex quinquefasciatus mitochondrial genome sequence
We used the A. aegypti mtDNA sequence as a query to explore sequence homologies with the C. quinquefasciatus trace files at NCBI and genome assembly (CpipJ1). In this process, we identified a ‘hit’ to a 14.96 kb region in supercontig3.816 (position: 413 to 15272 of this supercontig) that showed high homology (E-value = 0) to the complete mtDNA of A. aegypti except for the variable CR region. The immediate flanking sequences of this hit (742 bp: from position 15257 to 15999 of this supercontig) also showed high homology (just three bp differences) with the CR region (accession no. U69572) previously identified in Culex pipiens (Guillemaud et al. 1997). We performed PCR using genomic DNA template with primers specific to this mtDNA-like sequence (specific to the CR-end) and the flanking non-mtDNA region in order to test the continuity of the junction. We were unable to amplify the region across this junction (data not shown). This indicates that supercontig3.816 most likely represents a misassembly, but contains the near-complete mtDNA genome sequence. The whole genome sequences (WGS) generated from the C. quinquefasciatus genome project were searched for correct trace sequences and these were used to assemble and annotate the complete mtDNA genome sequence of C. quinquefasciatus using a similar approach to that we adopted to annotate the A. aegypti mtDNA. The sequence is 15,587 bp and has been submitted to GenBank (accession no. GU188856). It contains all the coding and non-coding genes in the same order as that observed for A. aegypti (Figure 1). However, the length of control region is considerably shorter (710 bp) compared to 1,771 bp and 1,708 bp for A. albopictus and A. aegypti (respectively), but similar to that observed in anopheline mtDNAs (~500–700 bp).
3.3. Comparison of mtDNA-like sequences between Aedes and Culex nuclear genomes
Significant ‘hits’ from our BLASTN searches of both nuclear genomes included 250 mtDNA-like sequences scattered over 142 supercontigs in A. aegypti and 22 mtDNA-like sequences scattered over 15 supercontigs (excluding supercontig 3.816) in C. quinquefasciatus (Table S2). The sum totals of these inserts are approximately 111.8 kb for A. aegypti and only 7.6 kb for C. quinquefasciatus. This represents 0.0085% (~8 bp NUMT per 100 kb) of the genome assembly of A. aegypti (AaegL1) and 0.0013% (~1 bp NUMT per 100 kb) of the genome assembly of C. quinquefasciatus. This further indicates that though these species reflect ~52–54 million years of divergence (Arensburger et al. 2010), transfer of mtDNA to the nuclear genome in A. aegypti is significantly higher (~8 fold) than in C. quinquefasciatus (Figure 2). Of note, A. gambiae and D. melanogaster, with estimated divergence times of ~145–200 and ~260 million years from Aedes and Culex respectively, contain no or only a few nuclear copies of mitochondrial DNAs in their genomes (Richly and Leister, 2004). However, the frequency of NUMTs in the A. aegypti and C. quinquefasciatus genomes is significantly lower compared to that of the honey bee (Apis mellifera) nuclear genome which contains ~1 bp NUMT per 1 kb of nuclear DNA (Pamilo et al. 2007).
Figure 2.
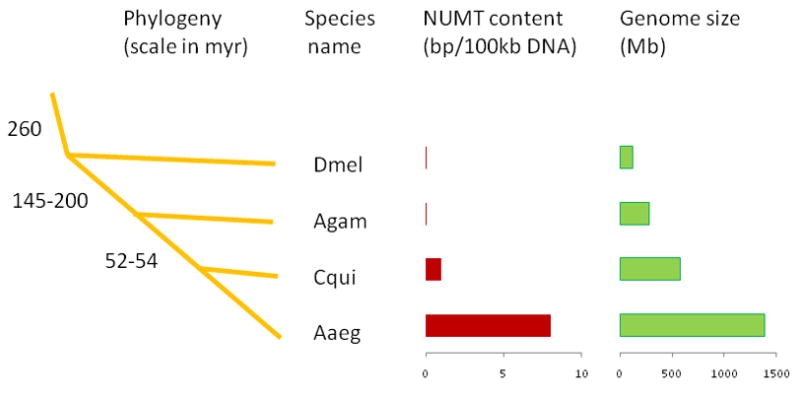
Amount of NUMTs (in bp per 100 kb of genomic DNA) in the genome of the three mosquitoes and D. melanogaster and its comparison to species phylogeny and genome size. The estimated times of divergence are shown on the common ancestral nodes in millions of years. The tree is not shown to scale; it only shows the phylogenetic relationships. The species names are shown corresponding to each branch. The amount of NUMTs (in bp per 100 kb of genomic DNA) as well as the estimated genome size (in Mb) are shown on the right to each species.
The NUMT insertions vary in sizes from 57 bp to 13,590 bp in A. aegypti and from 43 bp to 2,789 bp in C. quinquefasciatus. The large-size NUMTs (>2 kb) are few in number in both genomes (eight in A. aegypti and just one in C. quinquefasciatus) (Table S2). A total of 95 insertions were identified in A. aegypti genome and only four in C. quinquefasciatus genome that represented medium-size (200 bp to 2 kb) NUMTs. On the other hand, 147 insertions in A. aegypti and 17 insertions in C. quinquefasciatus genome were identified that represented small-size (< 200 bp) NUMTs. Excess numbers of small inserts are, however, common features of NUMTs because larger inserts are most often fragmented after transfer to the nuclear genome (Behura 2007).
3.4. PCR-based validation of mtDNA insertions in Aedes aegypti
PCR assays were used to experimentally verify mtDNA inserts in A. aegypti nuclear DNA. An A. aegypti BAC library was screened by PCR as previously described (Jiménez et al. 2004) with primer pairs spanning throughout the complete A. aegypti mtDNA sequence (Table S1). We used a PCR-based plate-pool DNA screening strategy in order to identify individual BAC pools that contained mtDNA-like sequences. Of the 131 pools representing the complete BAC library, 15 pools showed amplicons of the expected size (Table S3). For each pool, however, only a subset of primer pairs showed amplifications of the expected size band indicating that fragments of the mtDNA sequence were present in BAC pools. Also, some primer pairs produced stronger PCR bands compared to other primer pairs. These could suggest possible sequence variations within the primer binding sites. We also noticed that some primer pairs amplified a product from more than one BAC DNA pool (Table S3). For example, primer pair #13 amplified a product from seven BAC pools. This indicates that the individual BACs within each of these pools likely contain particular (mtDNA-like) sequences in multiple locations in the nuclear genome. We further analyzed two individual BAC clones BL46J23 (from the BL46 pool) and BL93K5 (from the BL93 pool) that were identified as NUMT positive by BAC library hybridizations. Primer pairs (# 5 through # 13) produced amplicons of the expected sizes from BAC BL46J23. This indicates the presence of a continuous NUMT of at least 7.93 kb in this clone (based on positions of primer 5F and 13R in mtDNA). Similarly, with BAC BL93K5, primer-pairs #1 to #13 (with the exception of # 3, 4 and 11) produced amplicons of the expected sizes. These results validate the presence of large NUMTs in the A. aegypti genome. Our experimental results thus agree with an earlier report suggesting the presence of large NUMTs in the A. aegypti genome (Hlaing et al. 2009).
3.5. Intronic NUMTs in predicted protein coding genes
We identified NUMTs within predicted protein coding genes of both mosquitoes. In A. aegypti, the genic NUMTs account for a total of 9,680 bp, whereas in the C. quinquefasciatus genome, the genic NUMTs sum to only 524 bp. In A. aegypti, a total of 32 genes were identified that contained NUMT sequences (Table S4). In C. quinquefasciatus, only four such genes were identified. None of these NUMT-containing genes are orthologous between the two species. Some of these genes contain multiple NUMTs; for example, nine NUMTs are located in gene AAEL002648 and three NUMTs in gene AAEL001547. Most of the NUMTs that are found within the predicted genes are small to medium size (< 500 bp) (except for three genes: AAEL009076, AAEL001547 and CPIJ018968). In all of the NUMT-containing genes except two, the NUMT sequences are located within intron regions. The two genes where coding sequences show homology to mtDNA (NADH dehydrogenase 2) are AAEL009076 of A. aegypti and CPIJ018968 of C. quinquefasciatus. However, it remains to be established if these genes are functional.
We also compared the NUMT-containing A. aegypti genes with NUMT-less, otherwise putative duplicate genes (predicted paralogous copies). Results show that the majority of the NUMT-less paralogous copies have relatively higher GC content than that of the NUMT-containing gene (Table S5). Our data is in agreement with an earlier observation that NUMTs preferentially insert into regions of low GC content (Lascaro et al. 2008). Also, the majority of the NUMT-less paralogous genes are relatively smaller than the NUMT-containing gene (Table S5). It is not known if gene length and GC content are generally negatively related in A. aegypti, for example as indicated in Drosophila and other species (Moriyama and Powell, 1998). However, for the select paralogous genes of A. aegypti analyzed in this study, our results indicate that NUMTs may have a role on variation of gene length and GC content of these genes. We also identified stretches of sequence similarities among intronic- and intergenic-NUMTs in these mosquitoes (Table 1). Sequence similarities were also observed in the intronic NUMTs of different genes in a pair-wise manner (Table S6). Though these common sequences may possibly play a role in recombination among NUMT-containing genes, it is not known if such sequences may have any role in rearrangement of NUMTs in the chromosomes. This is because NUMTs are generally integrated in the chromosomal DNA by a non-homologous end joining mechanism (Blanchard et al. 1996; Hazkani-Covo and Covo, 2008).
Table 1.
Sequence homologies among genic and non-genic NUMTs of the A. aegypti and C. quinquefasciatus genomes. The origins of the mtDNA as well as of the nuclear positions of NUMT sequences in the genic or non-genic region are shown.
| mtDNA | Genic*/Non-genic | Supercontig | NUMT position |
|---|---|---|---|
| A. aegypti | |||
| 1276-1550 | AAEL002648 | supercont1.62 | 2333418-2333698 |
| Non-genic | supercont1.287 | 519121-519402 | |
| Non-genic | supercont1.34 | 812505-812785 | |
| 4941-5239 | AAEL006582 | supercont1.211 | 1186826-1187123 |
| Non-genic | supercont1.198 | 12074-12372 | |
| 9116-9552 | AAEL002648 | supercont1.62 | 2331091-2331517 |
| Non-genic | supercont1.34 | 814603-815029 | |
| 13079-13311 | AAEL002648 | supercont1.62 | 2332600-2332827 |
| Non-genic | supercont1.287 | 518101-518330 | |
| Non-genic | supercont1.522 | 77641-77868 | |
| 13659-13750 | AAEL002648 | supercont1.62 | 2332850-2332941 |
| Non-genic | supercont1.287 | 518354-518445 | |
| Non-genic | supercont1.23 | 1309462-1309553 | |
| Non-genic | supercont1.857 | 28423-28514 | |
| 14288-14397 | AAEL010739 | supercont1.504 | 547213-547322 |
| Non-genic | supercont1.118 | 1545080-1545189 | |
| 14670-14743 | AAEL014296 | supercont1.1061 | 81559-81632 |
| Non-genic | supercont1.522 | 533879-533952 | |
| C. quinquefasciatus | |||
| 11355-11450 | CPIJ012100 | supercont3.408 | 100145-100240 |
| Non-genic | supercont3.415 | 41838-42276 | |
| 9670-9741 | CPIJ018968 | supercont3.1381 | 37-2825 |
| CPIJ006219 | supercont3.118 | 743910-744023 | |
| Non-genic | supercont3.258 | 256287-256350 | |
| Non-genic | supercont3.909 | 17757-17799 | |
| Non-genic | supercont3.118 | 739586-739656 | |
| Non-genic | supercont3.118 | 742449-742519 | |
| 1064-1232 | CPIJ009068 | supercont3.228 | 5857-6025 |
| Non-genic | supercont3.507 | 356204-357611 | |
All of these genic regions are intronic regions. The gene ID is shown.
3.6. Genomic rearrangements of NUMTs
Upon insertion, large mtDNA sequences are often fragmented during the assimilation process into the nuclear genome (Behura 2007). The fragmented NUMTs are identifiable as clusters of NUMTs where the inter-NUMT distance resembles the distance between their corresponding sequences in mtDNA. In A. aegypti, we identified several clusters of NUMTs that showed such characteristics (Figure S1). In C. quinquefasciatus, two such NUMT clusters were found: one in supercontig3.398 (140 bp and 183 bp NUMTs) and another in supercontig3.181 (173 and 257 bp NUMTs) (Table S2).
In A. aegypti, we also observed NUMT clusters wherein the inter-NUMT distance is less than the distance between their corresponding homologues in the mtDNA genome possibly because of sequence deletions from the original inserts (Table 2). Often some of these NUMTs are located in multiple locations in the genome as shown in Figure 3. Two specific NUMTs (one originating from mtDNA positions 383/384–2076 and the other from mtDNA positions 12295/12406–13206) are located at four locations in the A. aegypti genome with very similar distances (254–260 bp) between them at each location (Figure 3). The intervening DNA sequence between the two NUMTs is highly conserved and does not show homology to any known gene (data not shown). The distance between these NUMTs is considerably shorter than that in the mtDNA. In some cases, a portion of DNA was deleted and then the left and right side sequences were joined (Figure 4). Specific repeat motifs at the termini appear to mediate such joining. In contrast with A. aegypti, we did not find such NUMT clusters in C. quinquefasciatus. However, we did observe one example of a duplicated NUMT that was found in supercont3.118 (Table S2).
Table 2.
NUMT pairs that show deletion of the intervening sequences compared to the mtDNA. The inter-NUMT gap is less than the distance between their corresponding sequences in the mtDNA. The shorter gap between these NUMTs is most likely due to deletion of sequences from the mtDNA insert followed by end joining by specific termini motifs (shown). A negative value in the NUMT gap indicates overlapping NUMTs that originated from two distinct regions of mtDNA.
| mtDNA position | mtDNA gaps (bp) | Supercontig position | NUMT gap (bp) | End motif |
|---|---|---|---|---|
| 2014-2076 | 1420 | 1.240: 1309107-1309169 | 44 | AAA |
| 383-594 | 1.240: 1309213-1309426 | |||
| 13079-13206 | 3832 | 1.287: 518101-518220 | 260 | AAA |
| 384-2076 | 1.287: 518480-518742 | |||
| 1460-1550 | 414 | 1.287: 519314-519402 | 87 | AAA |
| 1964-2076 | 1.287: 519489-519600 | |||
| 384-2076 | 3832 | 1.348: 813167-813428 | 257 | AAA |
| 13116-13206 | 1.348: 813685-813775 | |||
| 13600-13641 | 4884 | 1.36: 2227040-2227081 | 970 | TTT |
| 1871-2153 | 1.36: 2228051-2228333 | |||
| 1320-1628 | 4712 | 1.454: 604405-604713 | 78 | TAT |
| 6340-6466 | 1.454: 604791-604914 | |||
| 13079-13206 | 3831 | 1.554: 77641-77760 | 254 | AAA |
| 383-2076 | 1.554: 78014-78276 | |||
| 13079-13206 | 3832 | 1.62: 2332600-2332719 | 257 | AAA |
| 384-2076 | 1.62: 2332976-2333237 | |||
| 383-647 | 3817 | 1.894: 28324-28386 | 245 | AAA |
| 13116-13220 | 1.894: 28631-28735 | |||
| 16470-16601 | 91 | 1.90: 2645439-2645570 | 47 | AAA |
| 38-72 | 1.90: 2645617-2645651 | |||
| 1633-1701 | 5267 | 1.569: 203384-203452 | 4 | AAG |
| 6968-7034 | 1.569: 203456-203522 | |||
| 1201-1398 | 641 | 1.929: 647-844 | −6 | ATTT |
| 2039-2644 | 1.929: 838-1442 | |||
| 1993-2058 | 9 | 1.495: 486627-486692 | −3 | CTCA |
| 2067-2141 | 1.495: 486689-486763 |
Figure 3.
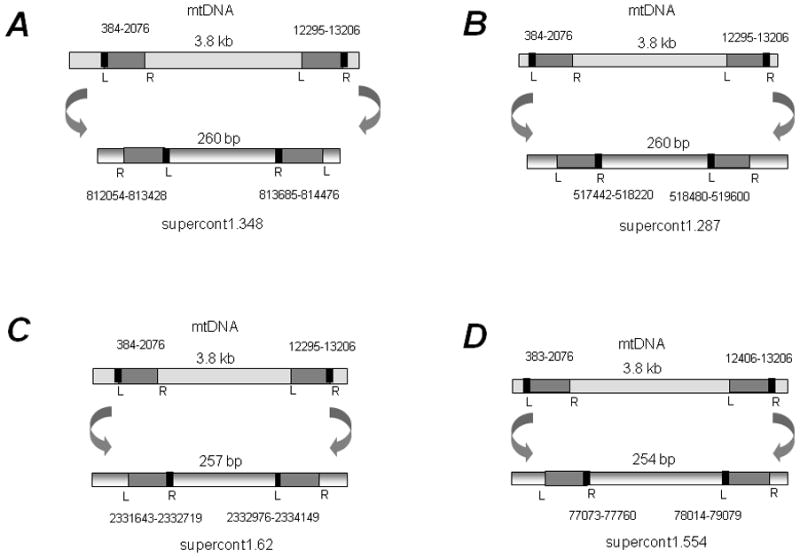
Duplication of NUMT pairs in the genome of Aedes aegypti. Four NUMT pairs are shown (A-D). In each, the mtDNA sequences are shown on the top and the corresponding nuclear sequences are shown below (with the supercontig ID). The distance between their homolog sequences in the mtDNA is shown in each case. The arrows indicate their insertion in different supercontigs and their orientations based on the relative positions of one of the termini sequences (AAA) that is highlighted in black (also see Table 2).
Figure 4.
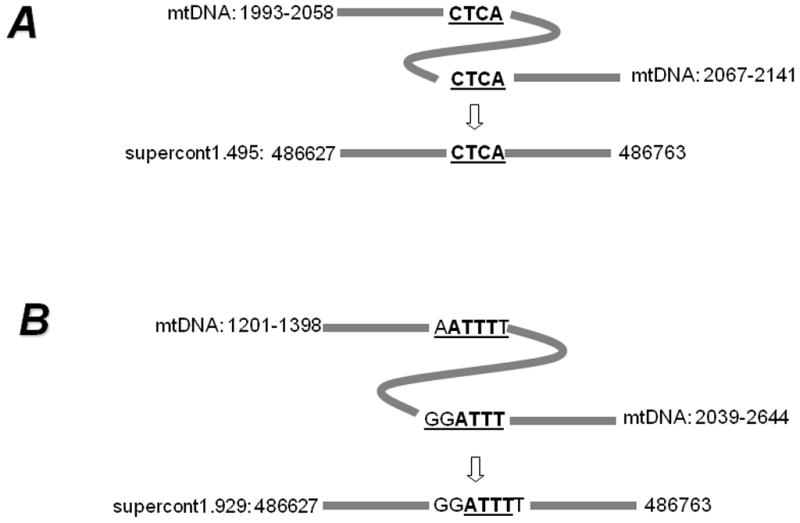
Sequence deletion and end-joining of NUMT sequences. A) A 9-bp sequence (shown by a curve) was deleted as the left- and right- sequences were joined at the direct termini motif ‘CTCA’. B) A portion (641 bp, shown by curve) was deleted as the left- and right- sequences were joined at the direct termini motif ‘ATTT’.
The chromosomal rearrangements of NUMTs do not seem to be independent events. Multiple evolutionary events may have shaped the present day NUMT structures in these mosquitoes. An example is illustrated in Figure 5 that provides evidence for these multiple events. Figure 5 shows the orientation of three NUMTs identified in A. aegypti supercontig1.495 and their corresponding homologues in the mtDNA. In the mtDNA, sequences corresponding to NUMTs 1 and 2 are overlapping with each other, while NUMT 3 is separated from them by nine bp. But in the nuclear genome, NUMTs 2 and 3 overlap with each other, whereas NUMT 1 is separated from NUMT 2 by 728 bp. This indicates that multiple rearrangement events (fragmentation of NUMT 1 and 2 along with recombination between NUMT 2 and 3) have occurred among these NUMTs, and reflects the complex nature of rearrangements of mtDNA inserts in the nuclear genome.
Figure 5.
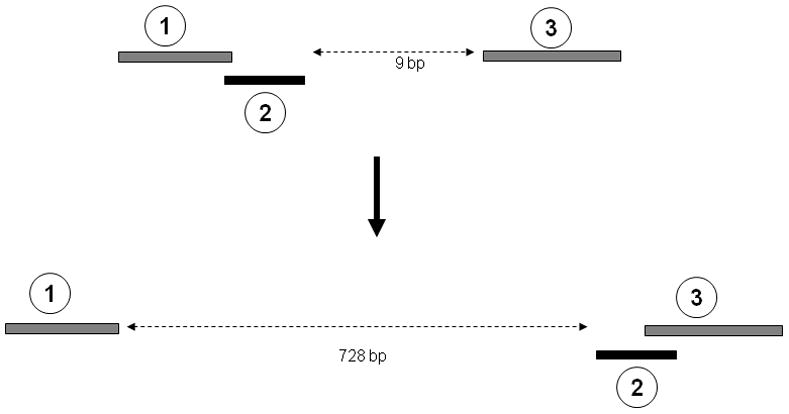
Example of multiple events associated with NUMT rearrangements. The top panel shows three regions within positions 1946-2141 of the mtDNA of A. aegypti. Sequence-1 and -2 are overlapping (1 bp overlap), while sequence-3 is 9 bp from sequence-2. The bottom panel shows the structure of the NUMTs corresponding to these three sequences. They are located in a cluster in supercontig1.495. NUMT-2 and -3 are overlapping (by 3 bp) in the supercontig (position: 486627-486763). NUMT 1 is located 728 bp from NUMT 2 (position: 485851-486692).
4. Discussion
In this study, we report the sequence of the complete mitochondrial genomes of A. aegypti and C. quinquefasciatus mosquitoes and determine insertion patterns of mtDNA sequences into their nuclear genomes. To determine the A. aegypti mtDNA sequence, we first constructed a DNA assembly using mtDNA containing trace sequences from the A. aegypti whole genome sequenceing (WGS) project (Nene et al. 2007). The assembled A. aegypti complete mtDNA sequence was then verified by PCR and direct amplicon sequencing. In C. quinquefasciatus, we initially identified what appears to be the near-complete mtDNA genome misassembled within a genome supercontig (3.816). We reassembled this sequence by similar approaches as described for A. aegypti mtDNA using mtDNA-containing trace files from the C. quinquefasciatus WGS project (Arensburger et al. 2010) to obtain the complete mtDNA sequence.
The A. aegypti and C. quinquefasciatus mtDNA shows high sequence homologies throughout with mtDNA genomes of other mosquitoes and fruit flies (data not shown). Also, the genes are packaged in the Ae. aegypti and C. quinquefasciatus mitochondrial genomes in the usual manner that reflects compactness of the mitochondrial genomes. This is reflected from small intergenic distances between consecutive genes of mitochondrial genomes of insects (< 23 bp in A. aegypti, < 24 bp in C. quinquefasciatus, < 21 bp in A. albopictus, < 16 bp in A. gambiae, < 18 bp in Anopheles quadrimaculatus, < 30 bp in Drosophila yakuba and < 30 bp in D. melanogaster). The A. aegypti and C. quinquefasciatus mtDNA genes, however, show slight variation in length (in the range of 25–33 bp) for the nd3, nd5 and nd1 genes unlike other mosquito and fruit fly species mentioned above. The tRNA-Gln, tRNA-Met, nd2 and co2 genes, on the other hand, have exactly the same length in all these species. Also, the rates of transition/transversion mutations of A. aegypti and C. quinquefasciatus are comparable to most of the mitochondrial genes of Anopheles mosquitoes or Drosophila except for nd3, nd4, nd4l and nd5 (data not shown). Overall, our data indicate that the mitochondrial genome structures of these two mosquitoes are similar to other closely related insect mitochondria.
We identified 250 NUMTs in the nuclear genome of A. aegypti and only 22 NUMTs in the nuclear genome of C. quinquefasciatus. In this regard, it is interesting to note that no NUMTs are detectable in the genome of A. gambiae (Richly and Leister 2004). There are theoretical considerations that predict factors responsible for differential rates of mtDNA transfer in different species. One of the factors is the mutation rate of the nuclear genome (Yamauchi, 2005; Lopez et al. 1994; Arctander 1995). Comparative analysis of orthologous genes reveals a higher rate of mutation of A. aegypti nuclear genes than in the same genes of A. gambiae (Nene et al. 2007). It is possible that such differences in mutation rate may have contributed to differences in NUMT frequencies between the Aedes and Anopheles genomes. NUMT survival in the chromosomal DNA is also affected by recombination frequencies in the nuclear genome (Hazkani-Covo et al. 2003; Ricchetti et al. 2004). The average genomic recombination rates in A. aegypti are less than that of A. gambiae (Severson et al. 2002; Wilfert et al. 2007) and that may also have contributed to the differences in NUMT density. The NUMT insertions may also utilize repeat sequences especially transposons. It has been shown that NUMT insertion sites are associated with transposable elements that influence the ongoing integration of mtDNA sequences and their subsequent duplication within the nuclear DNA (Pamilo et al. 2007). The A. aegypti genome contains large numbers of repetitive elements and transposable elements (Nene et al. 2007). Thus, it is highly possible that they may have effects on abundance of NUMTs integrations in A. aegypti genome. Apart from these speculative causes, other mechanisms such as intensity of intracellular competition, mtDNA leakage and effective population size are also considered to affect differential mtDNA transfer in different species (Yamauchi 2005).
Although we have not extended our study to compare NUMTs within different populations of these mosquitoes, it may be anticipated that population level variation exists among NUMT sequences, and due to the relatively large numbers of NUMTs, is likely to be the most variable in A. aegypti. We focused on the Liverpool-IB12 strain of A. aegypti as its genome has been sequenced and provided the trace files screened for mtDNA sequences as well as our basis for identifying NUMTs. The computational as well as the direct PCR amplicon sequencing methods showed no ambiguity in the A. aegypti mtDNA sequence. The use of WGS trace files in annotating mtDNA genomes was supported with a high coverage (>37 fold). However, further research is needed to determine how the mtDNA and the NUMT sequences vary among different populations of both mosquitoes. It has been shown that because of the high mutation rate of mtDNA, mtDNA sequences and the corresponding NUMT sequences do not reveal similar patterns of sequence evolution (Behura 2007).
The role of genome size may have relevancy on frequency of NUMTs as comparisons of 85 species showed a strong correlation between NUMT content and genome size (Hazkani-Covo et al. 2010). Given the relatively large genome size of A. aegypti (~1310 Mb) and C. quinquefasciatus (~ 579 Mb) compared to the A. gambiae genome (~278 Mb) (Figure 2), the relative abundance of NUMTs in these species is in agreement with observations by Hazkani-Covo et al. (2010). Such a relationship between NUMT density and genome size is also consistent with the assumption that transfers of mtDNA fragments into expressed regions of the genome is counter-selected. Under this assumption, NUMT numbers should increase in species with more noncoding nuclear DNA which could explain the abundance of NUMTs, for example, in Homo sapiens but not their rarity or absence in species such as Caenorhabditis and Anopheles (Richly and Leister, 2004). Thus, genome size is likely not the only factor determining NUMT density in genomes. Assuming that co-adaptive selection does exist between genome size and mtDNA transfer, it is not known if that selection is limited to phylogenetically related species or such a selection force is universally applicable even to unrelated species. However, the exceptionally high density of NUMTs in some species with small nuclear genomes, for example in honey bee (Pamilo et al. 2007 and Behura 2007), argues against a universal process. Its genome size (~236 Mb) is significantly smaller than the three mosquitoes, but contains relatively large numbers of NUMTs compared to these mosquitoes. Thus, though genome size may explain taxonomic variation in NUMT density (Bensasson et al. 2001, Hazkani-Covo et al. 2010), it does not explain the differences in NUMT density among related species, particularly between the honeybee and other holometabolous insects studied so far (Pamilo et al. 2007) including mosquitoes.
In the A. aegypti genome, we observed several large size NUMTs including two in excess of 12 kb. Large NUMTs have been reported only in a few animal species: a ~12.5 kb NUMT in the domestic cat (Felis catus) (Kim et al. 2006), a ~7.6 kb NUMT in the Italian wall lizard (Podarcis sicula) (Podnar et al. 2007), a 3.0 kb NUMT in the arvicoline rodent (Microtus rossiaemeridionalisin) (Triant and DeWoody, 2007) and a 8.8 kb NUMT in the human genome (Mishmar et al. 2004). This suggests that upon transfer to the nuclear genome, the mtDNAs undergo extensive fragmentation events that lead to large number of smaller NUMTs. We have also described some of these events in A. aegypti in the current study.
In conclusion, identification and characterization of NUMTs in the genome assemblies enhances our understanding of the genome sequences of both A. aegypti and C. quinquefasciatus. Particularly, identification of NUMTs in these genome sequences will provide us better opportunities for comparative analysis of these inserts within and between populations of these mosquitoes. Moreover, our data also provides a precautionary note about the presence of nuclear copies of mtDNA sequence that should be taken into consideration while using PCR-based mtDNA marker data for population studies of these mosquitoes. These have potential to interfere with PCR data due to sequence similarities between mtDNA and nuclear copies (Behura 2007). We also presented a comparative analysis of intronic NUMTs between A. aegypti and C. quinquefasciatus that will set a direction for further studies on the role of NUMTs in intron evolution of coding genes. To the best of our knowledge, no such studies have been reported in connection to NUMTs in these species. Furthermore, our results also suggested that NUMT rearrangement in A. aegypti chromosomes may be a continuous and complex evolutionary process. These chromosomal rearrangements might have shaped the current status of NUMTs in A. aegypti. Thus, our findings open up new directions on understanding the roles of nuclear mtDNA sequences on genome complexities of Culicidae mosquitoes.
Supplementary Material
Acknowledgments
This work was supported by grants RO1-AI059342, RO1-AI079125, UO1-AI50936, and contracts HHSN266200309D266030071, HHSN266200400001C and HHSN266200400039C from the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
This paper contains 1 figure and 6 tables in the form of supplementary data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Susanta K Behura, Email: sbehura@nd.edu.
Neil F Lobo, Email: nlobo@nd.edu.
Brian Haas, Email: bhaas@broad.mit.edu.
Becky deBruyn, Email: debruyn.1@nd.edu.
Diane D Lovin, Email: lovin.1@nd.edu.
Martin F Shumway, Email: shumwaym@ncbi.nlm.nih.gov.
Daniela Puiu, Email: dpuiu@umiacs.umd.edu.
Jeanne Romero-Severson, Email: jromeros@nd.edu.
Vishvanath Nene, Email: v.nene@cgiar.org.
David W Severson, Email: severson.1@nd.edu.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Arctander P. Comparison of a mitochondrial gene and a corresponding nuclear pseudogene. Proc Biol Sci. 1995;262:13–29. doi: 10.1098/rspb.1995.0170. [DOI] [PubMed] [Google Scholar]
- Arensburger P, Megy K, Waterhouse RM, Abrudan J, Amedeo P, Antelo B, Bartholomay L, Bidwell S, Caler E, Camara F, Campbell CL, Campbell KS, Casola C, Castro MT, Chandramouliswaran I, Chapman SB, Christley S, Costas J, Eisenstadt E, Feschotte C, Fraser-Liggett C, Guigo R, Haas B, Hammond M, Hansson BS, Hemingway J, Hill SR, Howarth C, Ignell R, Kennedy RC, Kodira CD, Lobo NF, Mao C, Mayhew G, Michel K, Mori A, Liu N, Naveira H, Nene V, Nguyen N, Pearson MD, Pritham EJ, Puiu D, Qi Y, Ranson H, Ribeiro JM, Roberston HM, Severson DW, Shumway M, Stanke M, Strausberg RL, Sun C, Sutton G, Tu ZJ, Tubio JM, Unger MF, Vanlandingham DL, Vilella AJ, White O, White JR, Wondji CS, Wortman J, Zdobnov EM, Birren B, Christensen BM, Collins FH, Cornel A, Dimopoulos G, Hannick LI, Higgs S, Lanzaro GC, Lawson D, Lee NH, Muskavitch MA, Raikhel AS, Atkinson PW. Sequencing of Culex quinquefasciatus establishes a platform for mosquito comparative genomics. Science. 2010;330:86–80. doi: 10.1126/science.1191864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behura SK. Molecular marker systems in insects: current trends and future avenues. Mol Ecol. 2006;15:3087–3113. doi: 10.1111/j.1365-294X.2006.03014.x. [DOI] [PubMed] [Google Scholar]
- Behura SK. Analysis of nuclear copies of mitochondrial sequences in honeybee (Apis mellifera), genome, Mol. Biol Evol. 2007;24:1492–1505. doi: 10.1093/molbev/msm068. [DOI] [PubMed] [Google Scholar]
- Bensasson D, Zhang D, Hartl DL, Hewitt GM. Mitochondrial pseudogenes: evolution’s misplaced witnesses. Trends Ecol Evol. 2001;2:314–321. doi: 10.1016/s0169-5347(01)02151-6. [DOI] [PubMed] [Google Scholar]
- Bensasson D, Zhang DX, Hewitt GM. Frequent assimilation of mitochondrial DNA by grasshopper nuclear genomes. Mol Biol Evol. 2000;17:406–415. doi: 10.1093/oxfordjournals.molbev.a026320. [DOI] [PubMed] [Google Scholar]
- Black WC, IV, Bernhardt SA. Abundant nuclear copies of mitochondrial origin (NUMTs) in the Aedes aegypti genome. Insect Mol Biol. 2009;18:705–713. doi: 10.1111/j.1365-2583.2009.00925.x. [DOI] [PubMed] [Google Scholar]
- Blanchard JL, Schmidt GW. Mitochondrial DNA migration events in yeast and humans: integration by a common end-joining mechanism and alternative perspectives on nucleotide substitution patterns. Mol Biol Evol. 1996;13:537–548. doi: 10.1093/oxfordjournals.molbev.a025614. [DOI] [PubMed] [Google Scholar]
- du Buy HG, Riley FL. Hybridization between the nuclear and kinetoplast DNA’s of Leishmania enriettii and between nuclear and mitochondrial DNA’s of mouse liver. Proc Natl Acad Sci U S A. 1967;57:790–797. doi: 10.1073/pnas.57.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemaud T, Pasteur N, Rousset F. Contrasting levels of variability between cytoplasmic genomes and incompatibility types in the mosquito Culex pipiens. Proc Biol Sci. 1997;264:245–251. doi: 10.1098/rspb.1997.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazkani-Covo E, Covo S. Numt-mediated double-strand break repair mitigates deletions during primate genome evolution. PLoS Genet. 2008;4:e1000237. doi: 10.1371/journal.pgen.1000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazkani-Covo E, Sorek R, Graur D. Evolutionary dynamics of large numts in the human genome: rarity of independent insertions and abundance of post-insertion duplications. J Mol Evol. 2003;56:169–174. doi: 10.1007/s00239-002-2390-5. [DOI] [PubMed] [Google Scholar]
- Hazkani-Covo E, Zeller RM, Martin W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010;6:e1000834. doi: 10.1371/journal.pgen.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlaing T, Tun-Lin W, Somboon P, Socheat D, Setha T, Min S, Chang MS, Walton C. Mitochondrial pseudogenes in the nuclear genome of Aedes aegypti mosquitoes: implications for past and future population genetic studies. BMC Genet. 2009;10:11. doi: 10.1186/1471-2156-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez LV, Kang BK, deBruyn B, Lovin DD, Severson DW. Characterization of an Aedes aegypti bacterial artificial chromosome (BAC), library and chromosomal assignment of BAC clones for physical mapping quantitative trait loci that influence Plasmodium susceptibility. Insect Mol Biol. 2004;13:37–44. doi: 10.1046/j.0962-1075.2004.00456.x. [DOI] [PubMed] [Google Scholar]
- Kim JH, Antunes A, Luo SJ, Menninger J, Nash WG, O’Brien SJ, Johnson WE. Evolutionary analysis of a large mtDNA translocation (numt) into the nuclear genome of the Panthera genus species. Gene. 2006;366:292–302. doi: 10.1016/j.gene.2005.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lascaro D, Castellana S, Gasparre G, Romeo G, Saccone C, Attimonelli M. The RHNumtS compilation: features and bioinformatics approaches to locate and quantify Human NumtS. BMC Genomics. 2008;9:267. doi: 10.1186/1471-2164-9-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez JV, Yuhki N, Masuda R, Modi W, O’Brien SJ. Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J Mol Evol. 1994;39:174–190. doi: 10.1007/BF00163806. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishmar D, Ruiz-Pesini E, Brandon M, Wallace DC. Mitochondrial DNA-like sequences in the nucleus (NUMTs): insights into our African origins and the mechanism of foreign DNA integration. Hum Mutat. 2004;23:125–133. doi: 10.1002/humu.10304. [DOI] [PubMed] [Google Scholar]
- Moriyama EN, Powell JR. Gene length and codon usage bias in Drosophila melanogaster, Saccharomyces cerevisiae and Escherichia coli. Nucl Acids Res. 1998;26:3188–3193. doi: 10.1093/nar/26.13.3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nene V, Wortman JR, Lawson D, Haas B, Kodira C, Tu ZJ, Loftus B, Xi Z, Megy K, Grabherr M, Ren Q, Zdobnov EM, Lobo NF, Campbell KS, Brown SE, Bonaldo MF, Zhu J, Sinkins SP, Hogenkamp DG, Amedeo P, Arensburger P, Atkinson PW, Bidwell S, Biedler J, Birney E, Bruggner RV, Costas J, Coy MR, Crabtree J, Crawford M, Debruyn B, Decaprio D, Eiglmeier K, Eisenstadt E, El-Dorry H, Gelbart WM, Gomes SL, Hammond M, Hannick LI, Hogan JR, Holmes MH, Jaffe D, Johnston JS, Kennedy RC, Koo H, Kravitz S, Kriventseva EV, Kulp D, Labutti K, Lee E, Li S, Lovin DD, Mao C, Mauceli E, Menck CF, Miller JR, Montgomery P, Mori A, Nascimento AL, Naveira HF, Nusbaum C, O’leary S, Orvis J, Pertea M, Quesneville H, Reidenbach KR, Rogers YH, Roth CW, Schneider JR, Schatz M, Shumway M, Stanke M, Stinson EO, Tubio JM, Vanzee JP, Verjovski-Almeida S, Werner D, White O, Wyder S, Zeng Q, Zhao Q, Zhao Y, Hill CA, Raikhel AS, Soares MB, Knudson DL, Lee NH, Galagan J, Salzberg SL, Paulsen IT, Dimopoulos G, Collins FH, Birren B, Fraser-Liggett CM, Severson DW. Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007;316:1718–1723. doi: 10.1126/science.1138878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamilo P, Viljakainen L, Vihavainen A. Exceptionally high density of NUMTs in the honeybee genome. Mol Biol Evol. 2007;24:1340–1346. doi: 10.1093/molbev/msm055. [DOI] [PubMed] [Google Scholar]
- Podnar M, Haring E, Pinsker W, Mayer W. Unusual origin of a nuclear pseudogene in the Italian wall lizard: intergenomic and interspecific transfer of a large section of the mitochondrial genome in the genus Podarcis (Lacertidae) J Mol Evol. 2007;64:308–320. doi: 10.1007/s00239-005-0259-0. [DOI] [PubMed] [Google Scholar]
- Pons J, Vogler AP. Complex pattern of coalescence and fast evolution of a mitochondrial rRNA pseudogene in a recent radiation of tiger beetles. Mol Biol Evol. 2005;22:991–1000. doi: 10.1093/molbev/msi085. [DOI] [PubMed] [Google Scholar]
- Ricchetti M, Tekaia F, Dujon B. Continued colonization of the human genome by mitochondrial DNA. PLoS Biol. 2004;2:E273. doi: 10.1371/journal.pbio.0020273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly E, Leister D. NUMTs in sequenced eukaryotic genomes. Mol Biol Evol. 2004;21:1081–1084. doi: 10.1093/molbev/msh110. [DOI] [PubMed] [Google Scholar]
- Severson DW, Meece JK, Lovin DD, Saha G, Morlais I. Linkage map organization of expressed sequence tags and sequence tagged sites in the mosquito, Aedes aegypti. Insect Mol Biol. 2002;11:371–378. doi: 10.1046/j.1365-2583.2002.00347.x. [DOI] [PubMed] [Google Scholar]
- Stewart JB, Beckenbach AT. Insect mitochondrial genomics 2: The complete mitochondrial genome sequence of a giant stonefly, Pteronarcys princeps, asymmetric directional mutation bias, and conserved plecopteran A+T-region elements. Genome. 2006;49:815–824. doi: 10.1139/g06-037. [DOI] [PubMed] [Google Scholar]
- Sunnucks P, Hales DF. Numerous transposed sequences of mitochondrial cytochrome oxidase I–II in aphids of the genus Sitobion (Hemiptera: Aphididae) Mol Biol Evol. 1996;13:510–524. doi: 10.1093/oxfordjournals.molbev.a025612. [DOI] [PubMed] [Google Scholar]
- Triant DA, DeWoody JA. Extensive mitochondrial DNA transfer in a rapidly evolving rodent has been mediated by independent insertion events and by duplications. Gene. 2007;401:61–70. doi: 10.1016/j.gene.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Viljakainen L, Oliveira DC, Werren JH, Behura SK. Transfers of mitochondrial DNA to the nuclear genome in the wasp Nasonia vitripennis. Insect Mol Biol. 2010;19:27–36. doi: 10.1111/j.1365-2583.2009.00932.x. [DOI] [PubMed] [Google Scholar]
- Wilfert L, Gadau J, Schmid-Hempel P. Variation in genomic recombination rates among animal taxa and the case of social insects. Heredity. 2007;98:189–197. doi: 10.1038/sj.hdy.6800950. [DOI] [PubMed] [Google Scholar]
- Yamauchi A. Rate of gene transfer from mitochondria to nucleus: effects of cytoplasmic inheritance system and intensity of intracellular competition. Genetics. 2005;171:1387–1396. doi: 10.1534/genetics.104.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zischler H, Geisert H, von Haeseler A, Pääbo S. A nuclear ‘fossil’ of the mitochondrial D-loop and the origin of modern humans. Nature. 1995;378:489–492. doi: 10.1038/378489a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.