

Abstract
Many neurotoxic organophosphates (OPs) inhibit acetylcholinesterase (AChE) and as a result can cause a life threatening cholinergic crisis. Current medical countermeasures, which typically include atropine and oximes target the cholinergic crisis and are effective in decreasing mortality but do not sufficiently protect against delayed neurological deficits. There is, therefore, a need to develop neuroprotective drugs to prevent long-term neurological deficits. We used acute hippocampal slices to test the hypothesis that 4R,6R-cembratrienediol (4R) protects against functional damage caused by the OP paraoxon (POX). To assess hippocampal function, we measured synaptically evoked population spikes (PSs). Application of 4R reversed POX inhibition of PSs and the EC50 of this effect was 0.8 µM. Atropine alone did not protect against POX neurotoxicity, but it did enhance protection by 4R. Pralidoxime partially regenerated AChE activity and protected against POX inhibition of PSs. 4R did not regenerate AChE suggesting that under our experimental conditions, the deleterious effect of POX on hippocampal function is not directly related to AChE inhibition. In conclusion, 4R is a promising neuroprotective compound against OP neurotoxins.
INTRODUCTION
Organophosphates (OPs) are a diverse family of chemicals used in industry, agriculture, medicine and warfare. Many neurotoxic OPs inhibit acetylcholinesterase (AChE) and the resultant accumulation of acetylcholine (ACh) causes a muscarinic and, to a lesser degree, a nicotinic crisis that is often fatal (Newmark 2004). Cholinergic overstimulation disturbs glutamatergic and GABAergic transmission and causes glutamate mediated excitotoxicity. OP toxicity is not limited to the acute cholinergic phase. Lingering debilitating effects are reported even when medical help is provided relatively early after exposure. Many survivors of the Tokyo sarin attack were afflicted with delayed neurological complications 7 years after the incident (Miyaki et al. 2005). Current medical countermeasures primarily address the acute effects of OP and focus on increasing survival of acutely intoxicated individuals. There is a need for neuroprotective compounds that arrest the excitotoxic and delayed apoptotic neuronal damage. (1S,2E,4R,6R,7E,11E)-cembra-2,7,11-triene-4,6-diol (4R) is a cyclic diterpenoid from tobacco (Ferchmin et al. 2009), a noncompetitive inhibitor of the α7 nicotinic receptor (Castro et al. 2009) and a novel neuroprotective compound that acts through a nicotinic antiapoptotic mechanism to protect against NMDA-induced excitotoxicity in hippocampal slices (Ferchmin et al. 2005). In this study, we test the potential for 4R to protect slices against acute paraoxon (POX) neurotoxicity.
MATERIAL AND METHODS
Unless otherwise specified, chemicals were from Sigma-Aldrich (St. Louis, MO). Paraoxon (O,O-Diethyl-O-4-nitro-phenylthiophosphate) was from Supelco (Bellefonte, PA, USA). The cembranoid (1S,2E,4R,6R,7E,11E)-cembra-2,7,11-triene-4,6-diol (4R) was from American Analytical (State College, Pennsylvania) and from Dr. K. El Sayed (School of Pharmacy, University of Louisiana, Monroe, LA). 4R stock solution was prepared in 100% dimethylsulfoxide (DMSO) and diluted in buffer the day of the experiment.
Slice Preparation and Electrophysiological Recordings
Acute hippocampal slices were prepared from male Sprague-Dawley rats (120–200 g) from our colony. All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Universidad C. del Caribe, School of Medicine). A standard artificial cerebrospinal fluid (ACSF), containing (in mM) 125 NaCl, 3.3 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 25 NaHCO3, and 10 glucose, was used for dissection and incubation. Hippocampi were dissected at ice temperature. Transverse slices were cut 400 µm in thickness with a manual slicer and immediately transferred to the recording chamber. Recording of extracellular field potentials or population spikes (PSs) was done as described (Ferchmin et al. 2000). Briefly, the chamber contained three lanes with independent perfusion lines exposed to the same gaseous phase (Fig. 1). The lower part of the chamber was filled with H2O kept at 37.4 ± 1°C and continuously bubbled with 95% O2, 5% CO2. The slices were kept at the gas-liquid interface, on an acrylic plate covered with nylon mesh (Hanes) located above the H2O superfused with ACSF and kept at 34 ± 1°C. Before entering the chamber, the ACSF was continuously bubbled with 95% O2, 5% CO2 and warmed by flowing through a stainless steel capillary immersed in the lower part of the chamber. The exterior of the chamber was kept at 30±1°C. The temperature at the three levels (outside, nylon mesh, and water bath) was strictly controlled to minimize variability. The electrophysiological activity of the slices stabilizes one hour after dissection. At that time, PSs were determined in each slice. A concentric bipolar electrode placed in the stratum radiatum of the CA1 area was used to stimulate the Schaffer collateral–commissural fibers with a constant current for 0.2 ms. The population spikes (PSs) were recorded in the stratum pyramidale using a glass micropipette filled with 2 M NaCl with an impedance ranging from 1 to 5 MΩ.
Fig. 1.

The recording chamber with the top removed showing the 3 lanes with hippocampal slices in place, the 3 separate inlet tubes and the common outlet.
Procedure for Testing Neurotoxicity
The procedure used to test neurotoxicity was as described (Schurr et al. 1995; Schurr et al. 1995) and modified by us (Ferchmin et al. 2000). Ten to 30 slices were distributed among the three lanes; when slices from more than one animal were used, they were equally distributed among the lanes. Testing of slices started 1 h after dissection. Each slice was stimulated with a stimulus strength twice that required for eliciting a threshold PS. This initial PS was recorded and compared with the response elicited by the same stimulus, recorded from the same position, after the completion of the experimental treatment. The effect of neurotoxic insult and of neuroprotection was expressed as percent of the initial PS remaining in the final PS.
The cembranoid was dissolved in DMSO and vehicle controls were exposed to DMSO added at the same final concentration (<0.1% v/v). At this concentration, DMSO had no effect on the PSs. All other compounds used were tested for effects on the size and shape of the PSs and those that affected the field potentials in control conditions were not used.
Data Analysis
The areas of the PS (millivolts per millisecond) were acquired and analyzed with the Labman program (gift from Dr. T. J. Teyler WWAMI Medical Education Program, University of Idaho, Moscow, ID). The data were statistically analyzed using SigmaStat version 2.03 (SPSS Science, Chicago, IL). Analysis of variance was used whenever the data were distributed normally; otherwise, Kruskal-Wallis one-way analysis of variance on ranks was used followed in each case by the appropriate post hoc test. When two groups were compared, the t test was used. Curve fitting was done with the least square minimization with the Marquardt-Levenburg method using PSI-Plot software (Poly Software International, Version 7, Pearl River, NY).
Determination of AChE activity
AChE activity was measured using the Elman assay (Ellman et al. 1961). Each assay was done using either the entire hippocampus or six 400 µm thick slices superfused with 0.5 ml/min of ACSF for 3.5 hours ± 15 min at 37°C. The samples were weighed, frozen on dry ice and homogenized in buffer (sodium phosphate buffer 0.1M, pH 8.0 +1% Triton X-100) at a concentration of 100 mg wet weight per ml of buffer. The homogenates were centrifuged at 12000 g for 1 min, the supernatant was collected and tetra isopropyl pyrophosphoramide (100 µM) was included to inhibit butyrylcholinesterase. AChE activity was measured in triplicate wells. The color changes were read in spectrophotometer at 405 nm with 16 kinetic cycles using a minimal kinetic interval. Enzyme activity was normalized to protein concentration, which was determined using the Bradford reagent (Bradford 1976). Data are expressed as µmol substrate transformed/min/mg of protein using the following formula: Activity (DOD/min sample − (DOD/min blank)*0.2 (total volume, ml)/0.014 (extinction coefficient) * 0.01 (sample volume, ml).
RESULTS
Effects of POX on the PS area in the CA1 region of the acute hippocampal slice
The neurotoxic effect of POX was assessed by determining PSs in slices prior to superfusion with POX and then again after POX was washed with ACSF. Comparison of the initial and final PSs showed that PS area was decreased by POX application and this inhibition was not reversed by prolonged washing with ACSF. The percent difference between the initial and the final PS for each slice was used to quantify the neurotoxic effect. Fig. 2 shows PSs recorded in control slices kept in ACSF, treated with POX and treated with POX and 30 min later rescued by treatment with 4R. The difference between the initial and final PSs in slices exposed for 4 hours to ACSF did not show a significant decrease caused by rundown. However, a 10 min application of 200 µM POX significantly reduced the final PS area. This POX-induced decrease of PSs was prevented when 30 min after POX the slices were exposed during 1 hour to 10 µM 4R and 1 µM atropine. The onset of the toxic effect of POX was fast but incomplete: with 100 µM POX, a maximum inhibition of approximately 80% was reached in 10 min (Fig 3). POX inhibition of the PSs was concentration-dependent up to 100 µM; higher concentrations did not significantly augment the toxicity of POX. The concentration-inhibition curve displayed an IC50 of 39.8 µM and a maximum inhibition of about 40% (Fig 4). These experiments showed that acute exposure to POX produces an irreversible decrease of the PSs leaving from 20% to 40% of the PS area unaffected.
Fig. 2.
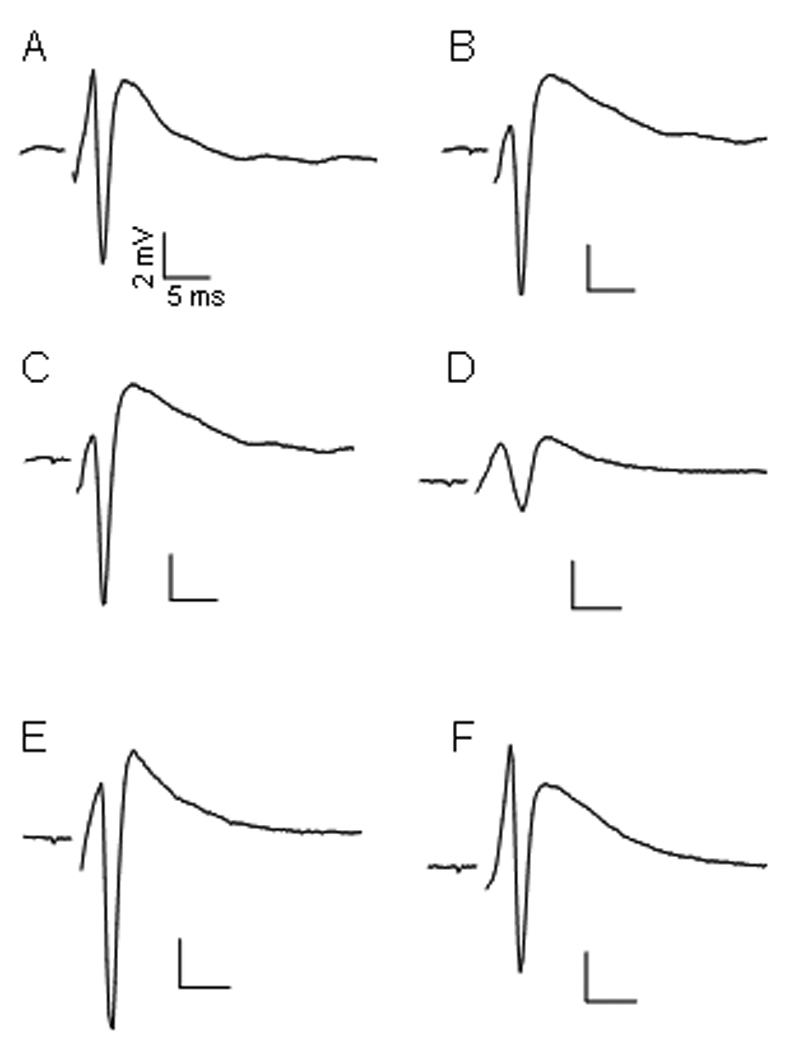
Representative PSs of 3 experimental conditions. A, C and E were the initial PS recorded 1 hour after dissection from representative slices from lanes 1 to 3, respectively (see Fig 1). The areas of these PSs were used as 100% to calculate the remaining percent PSs after treatment. B is a PS recorded from lane 1 after 2 additional hours of superfusion with ACSF. D was recorded from lane 2 after 10 min of 200 µM POX followed by 2 hours of ASCF washing. F was recorded from lane 3 after 10 min of 200 µM POX, washed for 30 min with ASCF and finally exposed for 1 hour to 10 µM 4R plus 1 µM atropine.
Fig. 3.
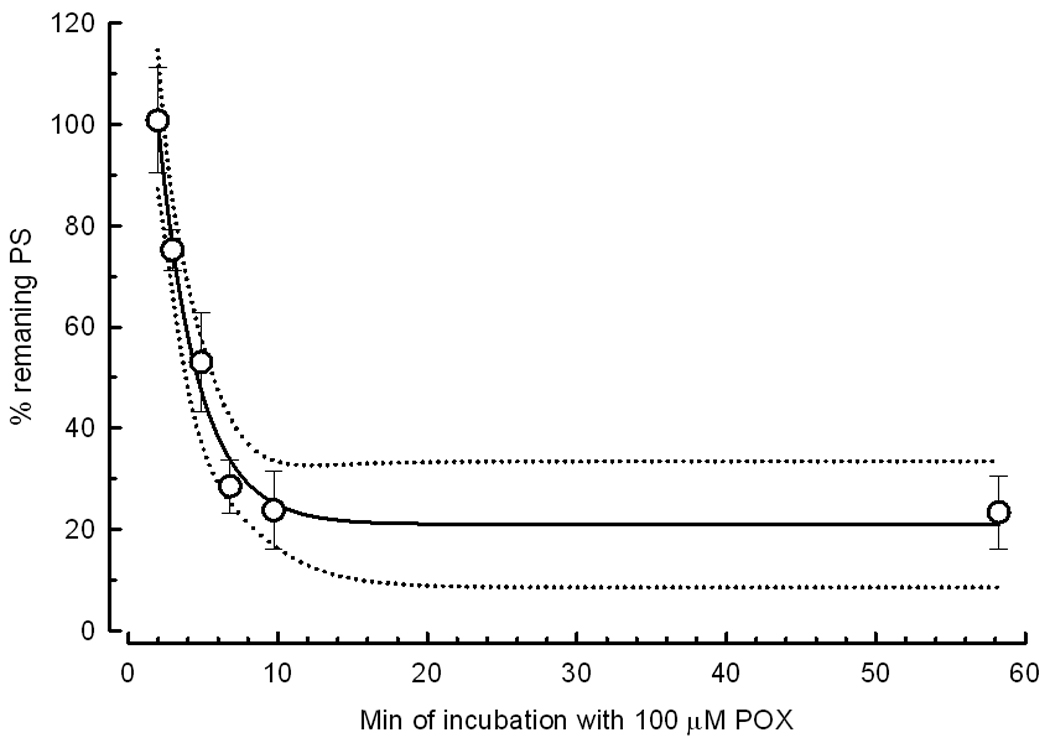
POX decreases the population spike area in a time-dependent manner. After the determination of the initial PS, slices were exposed to 100 µM POX for times varying from 2 to 60 min; the final PS was recorded after washing with ACSF for 1 hour. The symbols represent the mean ± SEM of PS areas from 14 slices. The dotted line is the 95% confidence interval. The solid line represents the equation: Y=((100−20.9)*EXP(−(X−2)/2.76))+20.9 where the values of the plateau (20.9%) and the time constant (2.76 min) represent the best fit to the data.
Fig. 4.
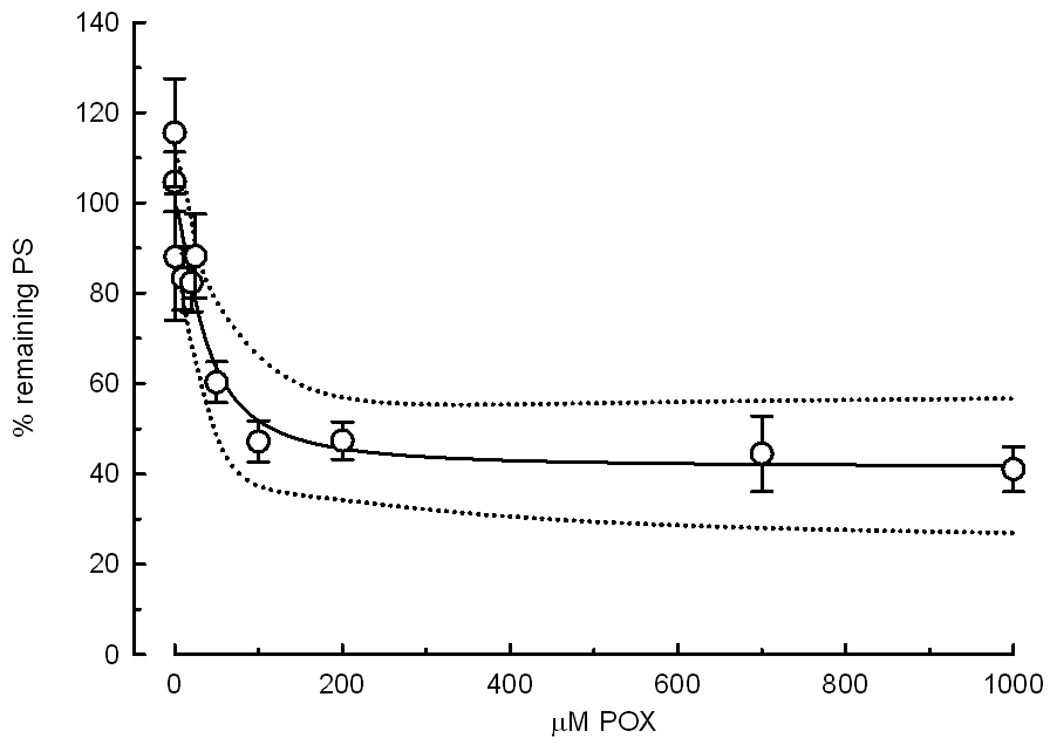
Curve of POX concentration versus the remaining PSs. After determination of initial PSs, slices were exposed to various concentrations of POX for 10 min. Final PSs were recorded after washing with ACSF for more than 1 hour. The remaining PS area is expressed as percent of the initial PS in the same slice vs. the concentration of POX. The symbols represent the mean ± SEM of PS areas from 14 or 21 slices. The dotted line is the 95% confidence interval. The solid line represents the equation: Y= ((100−37.6)/ (1+X/39.8))+37.6, where the values of the plateau (37.6 %) and the IC50 (39.8 µM) represent the best fit to the data.
4R protects the population spike from paraoxon toxicity
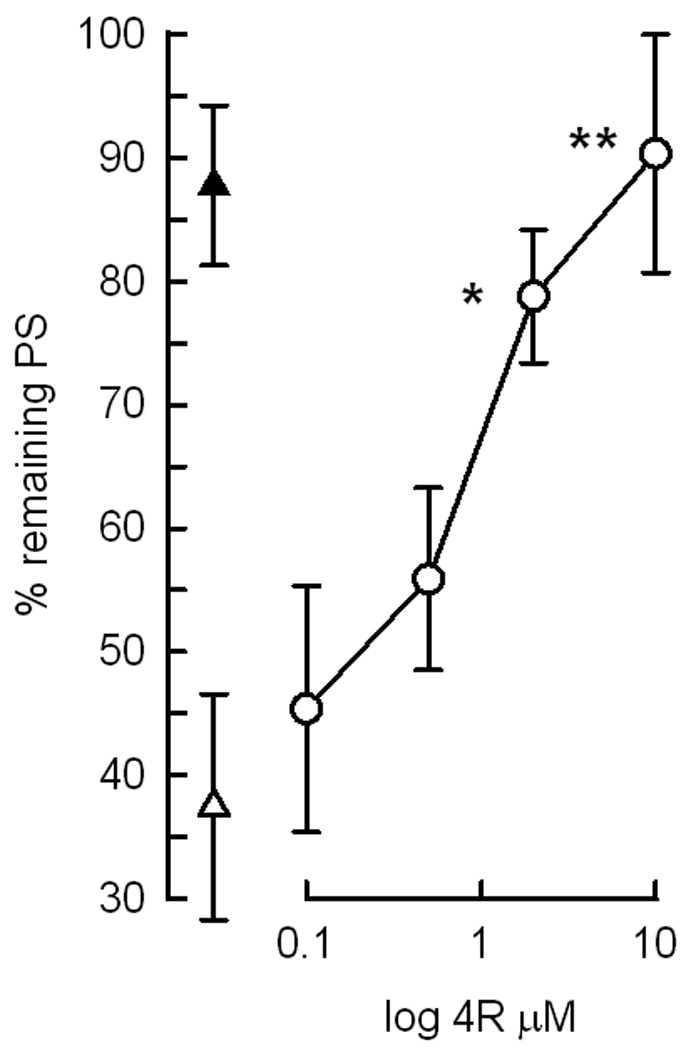
Since OPs are known to produce neuronal excitotoxicity and apoptosis (Carlson et al. 2000), we hypothesize that POX inhibition of PSs reflected a decreased the number of functional neurons as a consequence of POX-mediated neuronal apoptosis. 4R is a neuroprotective compound that ameliorates the effects of NMDA-induced excitotoxicity in hippocampal slices (Ferchmin et al. 2005). 4R acts by triggering an antiapoptotic mechanism and it is effective whether applied immediately before NMDA or 1h after NMDA. The experiments designed to test the effect of 4R on POX toxicity are summarized in Fig. 5. 4R was applied either before or after POX but was never present during POX application. 2 µM 4R was neuroprotective against 1 or 200 µM POX when applied either before or after POX (Fig. 5A) suggesting that 4R protects against POX toxicity indirectly, by activating a neuroprotective antiapoptotic cell signaling mechanism. The effect of 4R was concentration-dependent and 2 and 10 µM 4R significantly protected PSs against POX with an EC50 of 0.8 µM (Fig 5B).
Fig. 5.
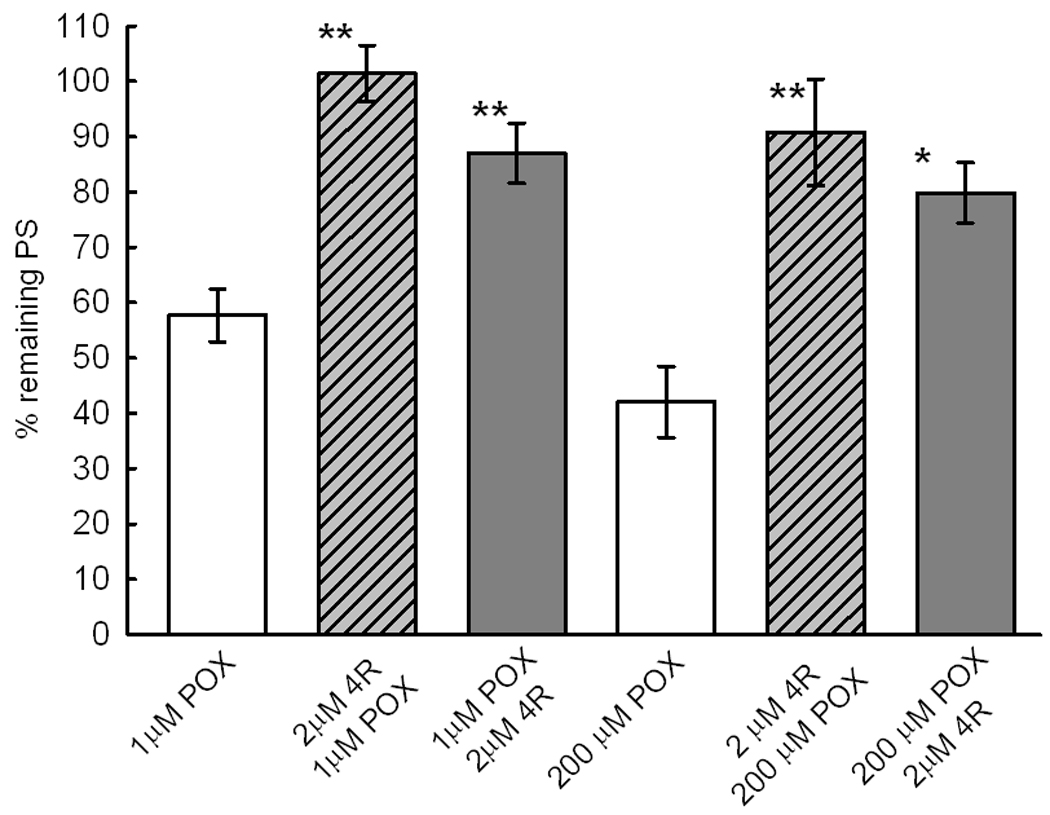
5A 4R applied either before or after 1 or 200 µM POX protects the PSs. The bars show the % of the initial PS remaining after the corresponding treatment ± S.E.M. The first white bar shows the effect 10 min exposure to 1 µM POX followed by 2 hours of washout with ACSF. The next (slashed bar) shows the % remaining PSs in slices superfused with 2 µM 4R for 1 hour before application of 1 µM POX followed by 1 hour of ACSF. The third, gray bar, shows the % remaining PS in slices exposed to 1 µM POX, washed for 30 min with ACSF and then exposed to 2µM 4R during 1 hour. There were 21 slices per experimental condition. The above experiment was replicated with 200 µM POX and 2 µM 4R. There were 14 slices per experimental condition in slices treated with 200 µM POX. The ** indicates p<0.001 and * p<0.002 significant difference with the corresponding POX control slice.
5B The dose response curve for 4R neuroprotection. One hour after dissection, the initial PSs were recorded. With the exception of ACSF controls (▲) which were superfused only with ACSF, all the other slices were exposed for 10 min to 200 µM POX, washed with ACSF for 30 min and superfused for 1 hour with ACSF (Δ) or with 0.1, 0.5, 2 or 10 µM 4R (○). After 15 min washing with ACSF to remove the 4R, the final PSs were recorded. The calculated EC50 for the neuroprotection by 4R was 0.8 µM. Significant difference between POX treatment and POX followed by 4R is indicated (**, p< 0.001 and *, p= 0.012). The number of slices per experimental group was 7,7,7,14,21 and 21. The solid line represents the equation: f1 = min + (max-min)/(1 + (x/EC50)^(−Hillslope)), where ‘min’ is the PS in absence of 4R, ‘max’ is the PS at saturation with 4R, EC50 is 4R concentration producing half-maximal effect and Hillslope is the slope coefficient; the best fitted parameters were (mean±SEM) : ‘min’ = 35.8±3.9, ‘max’ = 97.4118±10.0164, EC50 = 0.9±0.5, and ‘Hillslope’ = 0.9±0.3.
Effect of classical antidotes on paraoxon toxicity and 4R protection
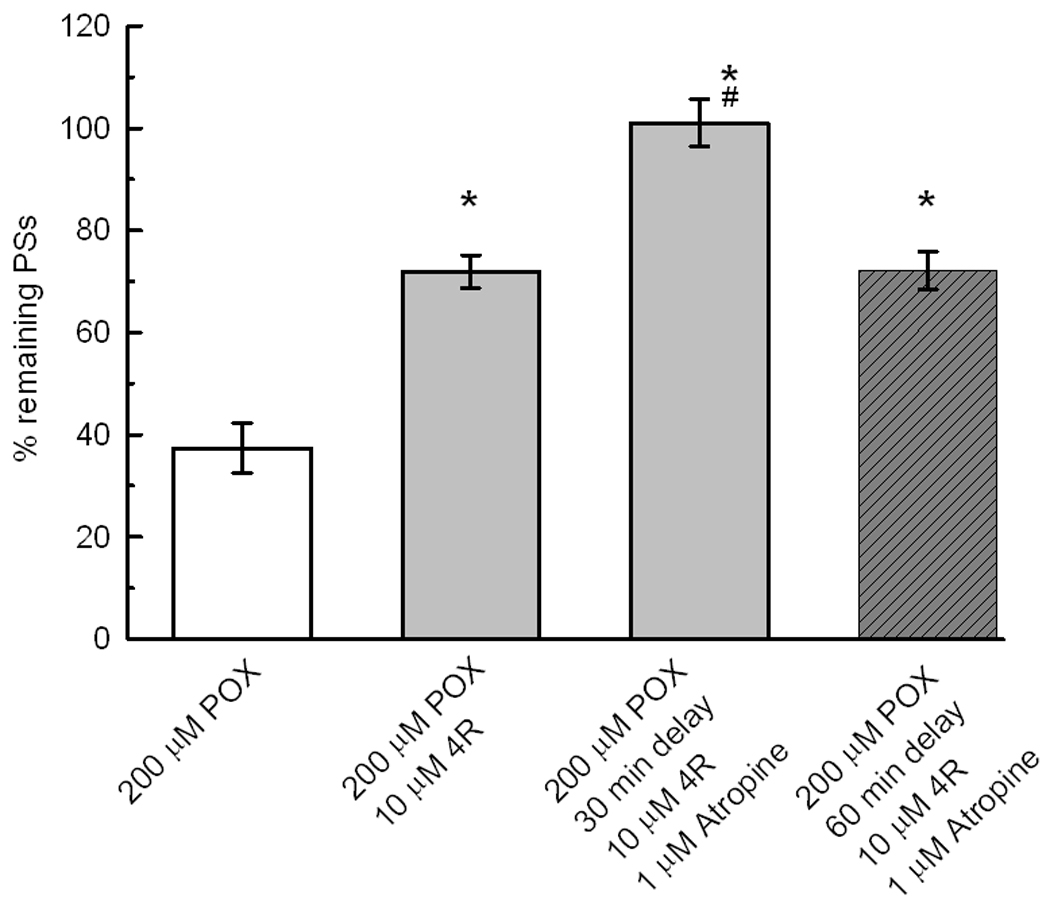
Atropine at 1 µM did not affect the PS area nor did it significantly protect PSs against POX toxicity (Fig 6A). Atropine per se did not show intrinsic toxicity. Exposure of slices for 1 hour to 1 or 50 µM atropine did not affect the size of PSs. Atropine applied at 1 or 50 µM for 90 min after POX had no significant effect on POX neurotoxicity (Fig 6A). However, Fig 6B shows that 1 µM atropine significantly increased the neuroprotection by 10 µM 4R applied after POX. The recovery of PSs by 10 µM 4R plus 1 µM atropine was nearly 100% and 72% when applied 30 min and 60 min after POX, respectively.
Fig. 6.
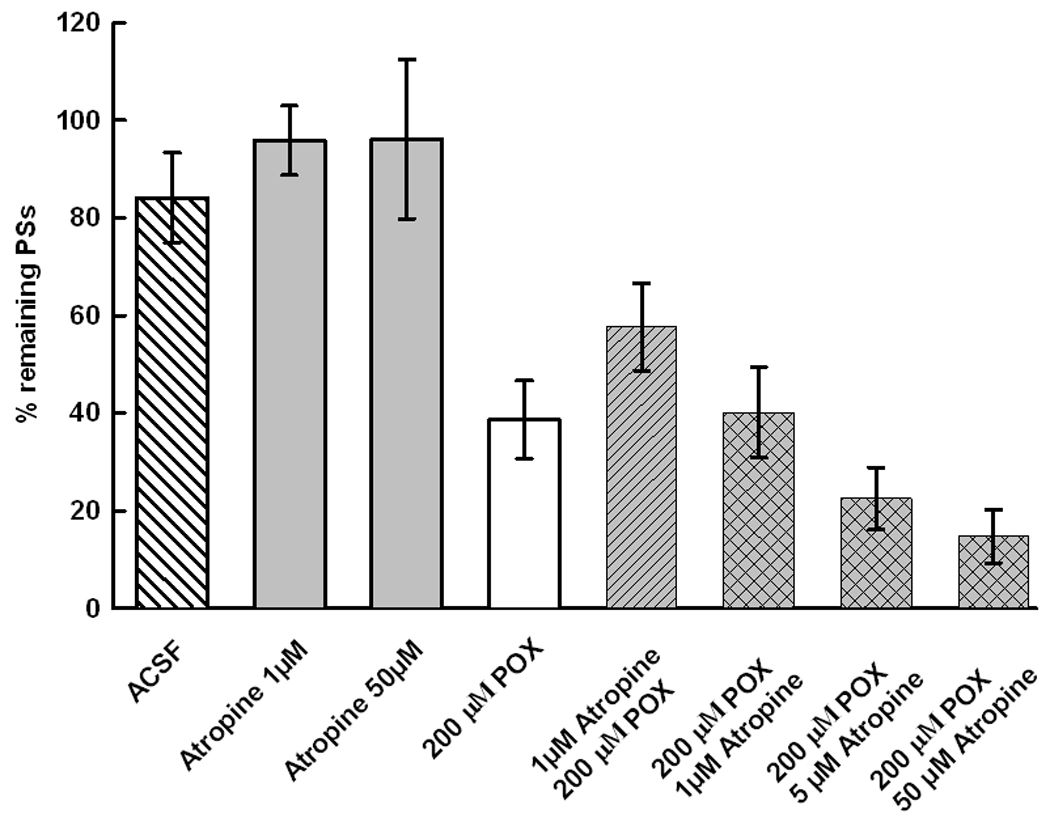
6A Atropine alone does not affect the PSs in the presence or absence of POX. The first 3 bars show that atropine did not affect the PSs in the absence of POX. Slices superfused for 3 hours with ACSF did no differ from slices exposed for 1 hour to 1 or 50 µM atropine followed by ACSF. 200 µM POX decreased the PSs in comparison the ACSF controls but 1 hour administration of 1 µM atropine before or after POX did not significantly protect the slices. Higher atropine concentrations applied after POX suggests an augmentation of POX toxicity but the effect was not significant. The bars represent the mean ± SEM of 7, 21, 7 for the first 3 bars and 14 slices for each of the remaining bars.
6B 1 µM atropine enhances 4R neuroprotection. After the determination of the initial PS, all slices were perfused with 200 µM POX for 10 min followed by ACSF. The POX control, white bar, shows the effect of 10 min of POX followed by ACSF for 2 hours. Application of 10 µM 4R for 1 hour with a delay of 30 min after POX produced a significant protection of the PSs (*, p<0.05). When 1 µM atropine was administered together with 10 µM 4R, the protection increased significantly over the protection with only 4R (#, p<0.05). 4R plus atropine applied with a delay of 60 min after POX protected significantly. The bars represent the mean ± SEM. The number of slices per bar was 35, 21, 21 and 14.
Pralidoxime applied 30 min after POX significantly protected the PSs. The preservation of the PSs by 2 to 100 µM pralidoxime was dose dependent. Under the same conditions, 2 and 10 µM 4R were equally or more neuroprotective than 100 µM pralidoxime (Fig 7).
Fig. 7.
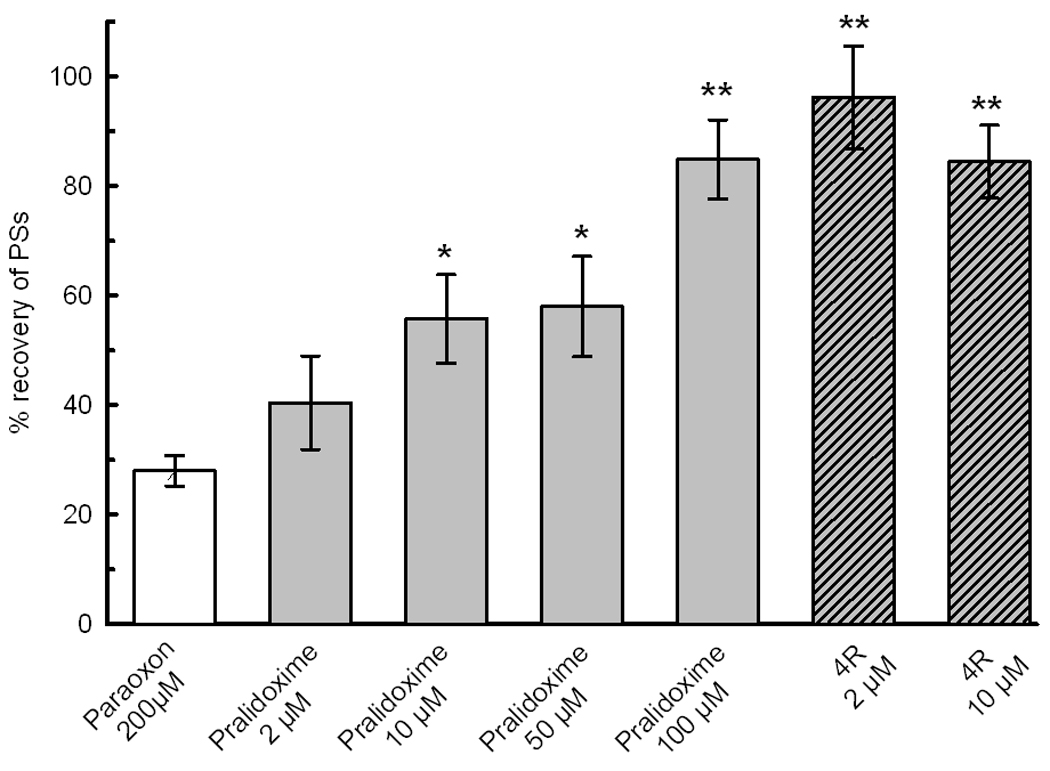
Pralidoxime protects the population spike from POX toxicity. Pralidoxime, 2, 10, 50 and 100 µM, applied 30 min after POX protected the slices against POX. The slashed bars show for the sake of comparison the effect of 2 and 10 µM 4R. Significant difference with POX controls is indicated as *, p<0.05; **, p<0.001. The number of slices per bar was 70, 14, 14, 14, 21, 14 and 21.
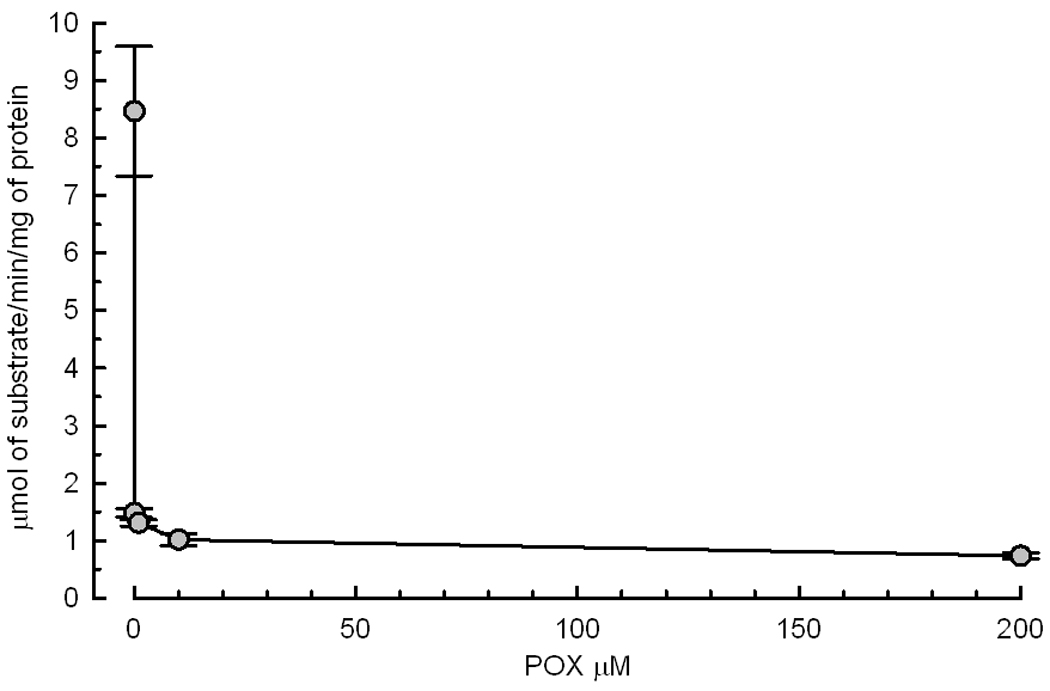
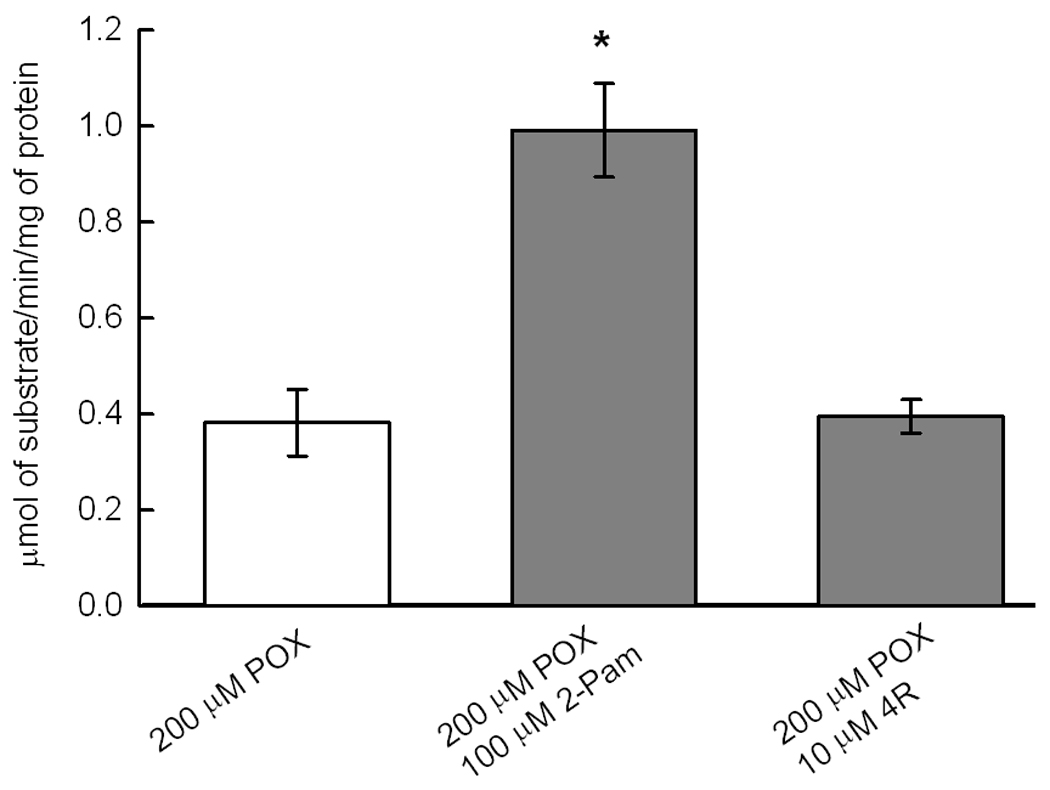
Since the primary molecular target of POX is AChE, we determined the effect of sample preparation and superfusion, POX and 4R on AChE activity. Surprisingly, AChE activity decayed by about 20% during the process of slicing the hippocampus and superfusion with ACSF for 1 hour caused an additional 90% decrease of AChE activity (Fig. 8A). The dose dependence of AChE inhibition by POX is shown in Fig 8B. Notably, a concentration of POX as low as 10 nM inhibited approximately 80% of AChE activity. The role of AChE inhibition in mediating the effect of POX on synaptically evoked PSs was investigated by determining AChE activity in control slices and in slices treated with either POX pralidoxime or 4R. Pralidoxime reactivated the AChE activity but 4R did not (Fig. 8C) yet both protected PSs (Fig 7).
Fig. 8.
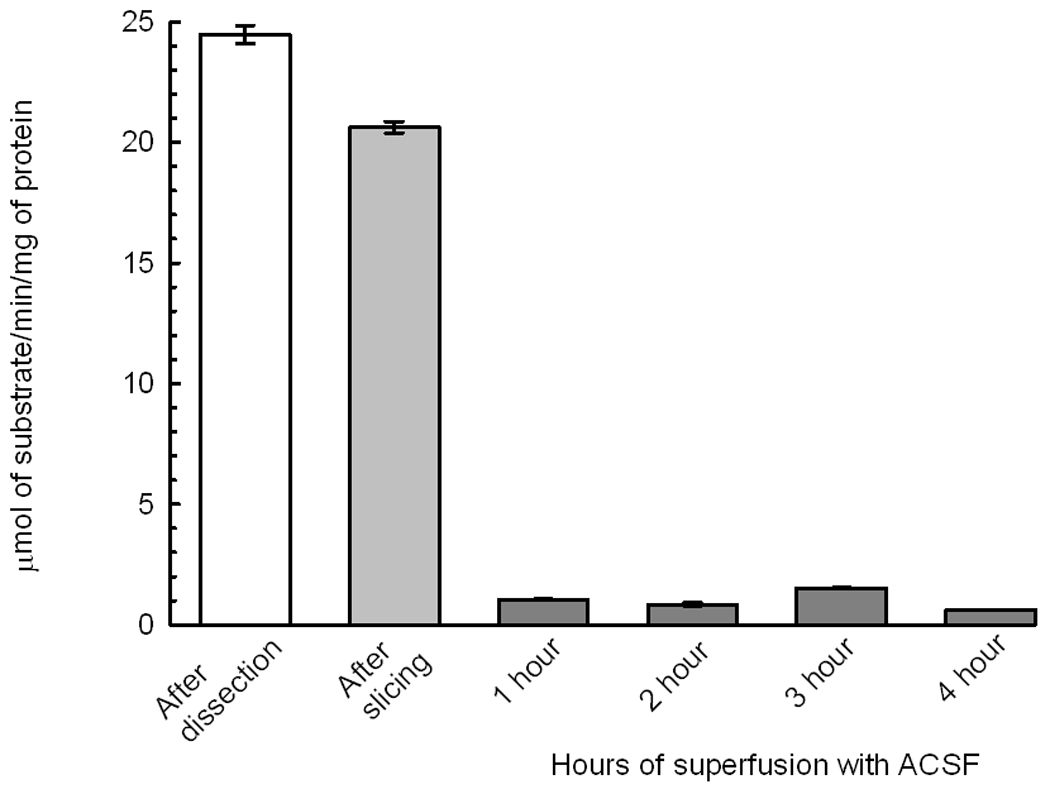
8A AChE activity decays drastically during the slicing process. The white bar shows the activity of AChE in rapidly dissected hippocampus. The process of slicing caused a loss of 20% of activity. Superfusion in standard conditions in the chamber for 1 hour caused a final dramatic loss of activity. The bars are the mean ± SEM of 8 to 9 determinations of AChE activity.
8B Effect of POX concentration on AChE activity. Slices were exposed for 10 min to the POX concentration as shown, washed with ACSF for 30 min before determination of AChE activity. The results are presented as AChE activity ± SEM and each point represents the average of 3 determination of a homogenate of 3 to 6 slices.
8C Pralidoxime reactivates AChE activity and 4R does not but both protect the PS against POX. After dissection, slices were superfused with ACSF for 1 hour, treated with 200 µM POX for 10 min, washed for 30 min with ACSF and exposed to either 1 hour of ACSF, 100 µM pralidoxime or 10 µM 4R. The slices were frozen and later assayed for AChE activity as described in Methods. There were from 8 to 9 slices per experimental condition. AChE activity in the pralidoxime treated slices was significantly higher than in POX control and the 4R treated group (*, p<0.001).
DISCUSSION
Acute hippocampal slices have been used for more than two decades to study the effect of anoxia, oxygen and glucose deprivation, and excitotoxic amino acids (Fountain and Teyler 1987; Schurr and Rigor 1989; Schurr et al. 1995; Schurr et al. 1995; Zhang and Lipton 1999; Ferchmin et al. 2000; Ferchmin et al. 2005). In acute slices, most of the circuitry of the original tissue is preserved; the ratio of interneurons to pyramidal neurons is unchanged relative to in vivo models. Stimulation of afferents allows measurement of synaptically elicited population spikes (PSs) from about 30 to 60 pyramidal neurons. The size of the PS is directly proportional to the number of functionally active pyramidal neurons (Andersen et al. 1971), thus, quantification of PSs provides a measure of the extent of neuronal damage. This preparation is well suited to the study of early functional neuronal damage before the onset of cell death. Collectively, these observations strongly support acute hippocampal slices as a an excellent model system for studying early synaptic excitotoxic and neuroprotective events of OP neurotoxicity. It might be more important to understand these early changes when intervention is still possible rather than studying neuronal death when intervention is not useful any more. Furthermore, comparisons of early effects of experimental ischemia on electric activity in acute slices versus delayed neuronal cell death in cultured slices is consistent with the concept that the loss of electrophysiological activity in acute slices and neuronal cell death slice cultures represent the same event in a different time scale (Small et al. 1997). This is supported by the work of Schurr and colleagues (Schurr and Rigor 1995) showing that drugs that protect against the loss of PSs in acute slices also protect against excitotoxicity in neuronal cell culture, organotypic slices or in vivo models.
Under the experimental conditions employed in our studies, the waveforms recorded before and after POX, application did not show any consistent alterations other than a decrease of the PS area (Fig 2). No seizures were observed during or after POX superfusion as discussed in (Harrison et al. 2004). POX decreased the PSs area in a time- and dose-dependent manner; however, there was a fraction of the PSs that was resistant despite increased exposure time or concentration. POX concentrations higher than 100 µM (Fig 4) or exposures longer than 10 min (Fig 3) did not increase POX inhibition of PSs.
AChE inhibition accounts for more than 90% of the neurotoxic effect of OPs in vivo and only 10% could be explained by other mechanisms (Maxwell et al. 2006). In acute slices continuously superfused with ACSF, the effects of AChE inhibition appear to be less prominent because ACh cannot accumulate as in vivo. The role of AChE in POX neurotoxicity in acute slices is discussed below. In addition to inhibiting AChE, OPs induce neuronal excitotoxicity and apoptosis (Carlson et al. 2000; Li et al. 2010), thus silencing a fraction of neurons and decreasing the area of the PS. Since 4R protects slices against NMDA excitotoxicity by a nicotinic anti-apoptotic mechanism (Ferchmin et al. 2005; Ferchmin et al. 2009) we tested here its effect against POX.
The neuroprotective effect of 4R applied 30 min after POX was dose dependent with an ED50 of 0.8 µM (Fig 5B). 4R applied before POX was marginally more efficacious than when applied 30 min after POX (Fig 5D). 4R protects with similar efficacy when applied before or after POX exposure (Fig 5A) suggesting that it does not act as an antidote but rather by activation of a cell survival signaling pathway.
Atropine, a muscarinic antagonist, is a life saving antidote for patients poisoned with OPs because it inhibits the muscarinic overstimulation cause by AChE inhibition. However, in slices atropine was not efficacious. Fig 6A shows that 1 or 50 µM atropine in the absence of POX did not affect the PS area. When applied prior to application of POX, 1 µM atropine did not significantly prevent the decrease of PSs by POX. The toxic effect of POX was not ameliorated by 1, 5 or 50 µM atropine applied 30 min after POX. Interestingly, 1 µM atropine seems to act synergistically with 10 µM 4R to protect against POX neurotoxicity when applied 30 min after POX (Fig 6B). The protection of PSs by 4R plus atropine was 100% when added 30 min after POX exposure, and still highly significant when added 1 hour after POX exposure. It is not possible to rule out the possibility that atropine acts in part by a nicotinic mechanism (Zwart and Vijverberg 1997, 1998).
Pralidoxime is another classic antidote used clinically on victims of OPs poisoning because of its ability to regenerate AChE activity (Petroianu et al. 2007). Interestingly, pralidoxime promoted the recovery of the PSs from POX poisoning albeit with an efficacy approximately 10 fold lower than 4R (Fig 7). The neuroprotective effects of pralidoxime on PSs were coincident with significant protection of AChE activity, suggesting a functional link between AChE activity and PS size. However, in contrast to pralidoxime, 4R did not reactivate AChE (Fig 8C) but yet protected the PSs. These data suggest there is no causal relationship between AChE activity and preservation of PSs. Further support for this conclusion are the findings that: 1) During dissection, slicing and incubation for 1 hour in the recording chamber, 90% of the AChE activity is lost (Fig 8A) while the electrophysiological activity remains stable for hours. 2) the discrepancy between the concentration-effect relationships for POX inhibition of AChE and POX effects on PSs. With respect to the latter, 10 nM and 10 µM POX inhibited 82% and 88% of AChE activity (Fig 8B) but 100 µM POX was needed to reach the plateau of PS inhibition (Fig 4). This conclusion, however, must be made with caution because the interaction of POX with different esterases is dynamic and complex (Estevez et al. 2011) and it is conceivable that a small pool of an esterase reactivated by pralidoxime reactivates the electrophysiological activity of slices. The lack of involvement of AChE is further supported by the finding that during dissection, slicing and incubation for 1 hour in the recording chamber 90% of the AChE activity is lost (Fig 8A) while the electrophysiological activity remains stable for hours with only minor deviations from in vivo models. Although we do not know the exact mechanism of AChE loss of activity in slices, there are reports, which suggest possible mechanisms. In the mammalian brain, AChE is present in various forms and in different compartments. Most of AChE is anchored in cell membranes by a transmembrane protein PRiMA (proline-rich membrane anchor) (Perrier et al. 2002; Xie et al. 2010). Not only PRiMA anchored AChE can detach but there are soluble forms that are amenable to be released. In addition, active secretion of AChE from neurons and PC12 cells was described (Llinas and Greenfield 1987; Schweitzer 1993).
Collectively, these data suggest that AChE inhibition is not a predominant mechanism of POX neurotoxicity in acute hippocampal slices probably because there is not a significant accumulation of ACh. Therefore, the acute slice preparation seems to be a uniquely suited model to study early events of OP neurotoxicity in the absence of a massive accumulation of ACh.
In conclusion, 4R protected the function of CA1 neurons against the neurotoxic effects of POX. Although the mechanism of neuroprotection in this model system was not elucidated, we hypothesize that 4R protects against POX by a mechanism similar to the one involved in protection against NMDA excitotoxicity (Ferchmin et al. 2005).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen P, Bliss TV, Skrede KK. Unit analysis of hippocampal polulation spikes. Exp Brain Res. 1971;13:208–221. doi: 10.1007/BF00234086. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Carlson K, Jortner BS, Ehrich M. Organophosphorus compound-induced apoptosis in SH-SY5Y human neuroblastoma cells. Toxicol Appl Pharmacol. 2000;168:102–113. doi: 10.1006/taap.2000.8997. [DOI] [PubMed] [Google Scholar]
- Castro W, Hann RM, Eterovic VA. Effects of 4R, 6R-cembratriene diol on human α7 nicotinic acetylcholine receptor Society for Neuroscience. Abstract Poster # 227.18/C14. 2009 [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Estevez J, Garcia-Perez A, Barril J, Vilanova E. Inhibition with Spontaneous Reactivation of Carboxyl Esterases by Organophosphorus Compounds: Paraoxon as a Model. Chem Res Toxicol. 2011;24:135–143. doi: 10.1021/tx100346c. [DOI] [PubMed] [Google Scholar]
- Ferchmin PA, Hao J, Perez D, Penzo M, Maldonado HM, Gonzalez MT, Rodriguez AD, De Vellis J. Tobacco cembranoids protect the function of acute hippocampal slices against NMDA by a mechanism mediated by alpha4beta2 nicotinic receptors. J Neurosci Res. 2005;82:631–641. doi: 10.1002/jnr.20666. [DOI] [PubMed] [Google Scholar]
- Ferchmin PA, Pagan OR, Ulrich H, Szeto AC, Hann RM, Eterovic VA. Actions of octocoral and tobacco cembranoids on nicotinic receptors. Toxicon. 2009;54:1174–1182. doi: 10.1016/j.toxicon.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferchmin PA, Perez D, Biello M. Spermine is neuroprotective against anoxia and N-methyl-D-aspartate in hippocampal slices. Brain Res. 2000;859:273–279. doi: 10.1016/s0006-8993(00)01973-9. [DOI] [PubMed] [Google Scholar]
- Fountain SB, Teyler TJ. Characterizing neurotoxicity using the in vitro hippocampal brain slice preparation: heavy metals. Prog Clin Biol Res. 1987;253:19–31. [PubMed] [Google Scholar]
- Harrison PK, Sheridan RD, Green AC, Scott IR, Tattersall JEH. A Guinea Pig Hippocampal Slice Model of Organophosphate-Induced Seizure Activity. Journal of Pharmacology and Experimental Therapeutics. 2004;310:678–686. doi: 10.1124/jpet.104.065433. [DOI] [PubMed] [Google Scholar]
- Li L, Cao Z, Jia P, Wang Z. Calcium signals and caspase-12 participated in paraoxon-induced apoptosis in EL4 cells. Toxicol In Vitro. 2010;24:728–736. doi: 10.1016/j.tiv.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Llinas RR, Greenfield SA. On-line visualization of dendritic release of acetylcholinesterase from mammalian substantia nigra neurons. Proc Natl Acad Sci U S A. 1987;84:3047–3050. doi: 10.1073/pnas.84.9.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell DM, Brecht KM, Koplovitz I, Sweeney RE. Acetylcholinesterase inhibition: does it explain the toxicity of organophosphorus compounds? Arch Toxicol. 2006;80:756–760. doi: 10.1007/s00204-006-0120-2. [DOI] [PubMed] [Google Scholar]
- Miyaki K, Nishiwaki Y, Maekawa K, Ogawa Y, Asukai N, Yoshimura K, Etoh N, Matsumoto Y, Kikuchi Y, Kumagai N, Omae K. Effects of sarin on the nervous system of subway workers seven years after the Tokyo subway sarin attack. J Occup Health. 2005;47:299–304. doi: 10.1539/joh.47.299. [DOI] [PubMed] [Google Scholar]
- Newmark J. Therapy for nerve agent poisoning. Arch Neurol. 2004;61:649–652. doi: 10.1001/archneur.61.5.649. [DOI] [PubMed] [Google Scholar]
- Perrier AL, Massoulie J, Krejci E. PRiMA: the membrane anchor of acetylcholinesterase in the brain. Neuron. 2002;33:275–285. doi: 10.1016/s0896-6273(01)00584-0. [DOI] [PubMed] [Google Scholar]
- Petroianu GA, Nurulain SM, Nagelkerke N, Shafiullah M, Kassa J, Kuca K. Five oximes (K-27, K-48, obidoxime, HI-6 and trimedoxime) in comparison with pralidoxime: survival in rats exposed to methyl-paraoxon. J Appl Toxicol. 2007;27:453–457. doi: 10.1002/jat.1224. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Heine MF, Rigor BM. Hypoxia, excitotoxicity, and neuroprotection in the hippocampal slice preparation. J. Neurosci. Methods. 1995;59:129–138. doi: 10.1016/0165-0270(94)00203-s. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Rigor BM. Protection by MK-801 against hypoxia-, excitotoxin-, and depolarization-induced neuronal damage in vitro. Neurochem. Int. 1995;26:519–525. doi: 10.1016/0197-0186(94)00148-n. [DOI] [PubMed] [Google Scholar]
- Schurr A, Rigor BM. Cerebral ischemia revisited: new insights as revealed using in vitro brain slice preparations. Experientia. 1989;45:684–695. doi: 10.1007/BF01974560. [DOI] [PubMed] [Google Scholar]
- Schurr A, Rigor BM. Brain slices in basic and clinical research. Boca Raton: CRC Press; 1995. [Google Scholar]
- Schweitzer ES. Regulated and constitutive secretion of distinct molecular forms of acetylcholinesterase from PC12 cells. J Cell Sci. 1993;106(Pt 3):731–740. doi: 10.1242/jcs.106.3.731. [DOI] [PubMed] [Google Scholar]
- Small DL, Monette R, Buchan AM, Morley P. Identification of calcium channels involved in neuronal injury in rat hippocampal slices subjected to oxygen and glucose deprivation. Brain Res. 1997;753:209–218. doi: 10.1016/s0006-8993(96)01385-6. [DOI] [PubMed] [Google Scholar]
- Xie HQ, Liang D, Leung KW, Chen VP, Zhu KY, Chan WK, Choi RC, Massoulie J, Tsim KW. Targeting acetylcholinesterase to membrane rafts: a function mediated by the proline-rich membrane anchor (PRiMA) in neurons. J Biol Chem. 2010;285:11537–11546. doi: 10.1074/jbc.M109.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lipton P. Cytosolic Ca2+ changes during in vitro ischemia in rat hippocampal slices: major roles for glutamate and Na+-dependent Ca2+ release from mitochondria. J. Neurosci. 1999;19:3307–3315. doi: 10.1523/JNEUROSCI.19-09-03307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, Vijverberg HP. Potentiation and inhibition of neuronal nicotinic receptors by atropine: competitive and noncompetitive effects. Mol Pharmacol. 1997;52:886–895. doi: 10.1124/mol.52.5.886. [DOI] [PubMed] [Google Scholar]
- Zwart R, Vijverberg HP. Four pharmacologically distinct subtypes of alpha4beta2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol Pharmacol. 1998;54:1124–1131. [PubMed] [Google Scholar]