

Summary
Background
Associations of C-reactive protein (CRP) concentration with risk of major diseases can best be assessed by long-term prospective follow-up of large numbers of people. We assessed the associations of CRP concentration with risk of vascular and non-vascular outcomes under different circumstances.
Methods
We meta-analysed individual records of 160 309 people without a history of vascular disease (ie, 1·31 million person-years at risk, 27 769 fatal or non-fatal disease outcomes) from 54 long-term prospective studies. Within-study regression analyses were adjusted for within-person variation in risk factor levels.
Results
Loge CRP concentration was linearly associated with several conventional risk factors and inflammatory markers, and nearly log-linearly with the risk of ischaemic vascular disease and non-vascular mortality. Risk ratios (RRs) for coronary heart disease per 1-SD higher loge CRP concentration (three-fold higher) were 1·63 (95% CI 1·51–1·76) when initially adjusted for age and sex only, and 1·37 (1·27–1·48) when adjusted further for conventional risk factors; 1·44 (1·32–1·57) and 1·27 (1·15–1·40) for ischaemic stroke; 1·71 (1·53–1·91) and 1·55 (1·37–1·76) for vascular mortality; and 1·55 (1·41–1·69) and 1·54 (1·40–1·68) for non-vascular mortality. RRs were largely unchanged after exclusion of smokers or initial follow-up. After further adjustment for fibrinogen, the corresponding RRs were 1·23 (1·07–1·42) for coronary heart disease; 1·32 (1·18–1·49) for ischaemic stroke; 1·34 (1·18–1·52) for vascular mortality; and 1·34 (1·20–1·50) for non-vascular mortality.
Interpretation
CRP concentration has continuous associations with the risk of coronary heart disease, ischaemic stroke, vascular mortality, and death from several cancers and lung disease that are each of broadly similar size. The relevance of CRP to such a range of disorders is unclear. Associations with ischaemic vascular disease depend considerably on conventional risk factors and other markers of inflammation.
Funding
British Heart Foundation, UK Medical Research Council, BUPA Foundation, and GlaxoSmithKline.
Introduction
C-reactive protein (CRP), a plasma protein synthesised by the liver, is a sensitive and dynamic systemic marker of inflammation.1 Its concentration in the circulation can increase by up to 10 000-fold during acute responses to serious infection or major tissue damage.2 In the absence of such spikes, however, the year-to-year within person variations in CRP concentration are similar to those in total cholesterol concentration and systolic blood pressure.3 The stability of this protein during long-term frozen blood storage and availability of standardised assays have assisted studies of CRP.4–8 Aside from whether measurement of CRP is useful in assessment of vascular risk,4,9 studies are needed to help find out if CRP is a mediator of vascular disease.10,11 CRP binds to LDL12,13 and is present in atherosclerotic plaques,14 so it has been proposed that CRP may have a causal role in coronary heart disease. In a literature-based meta-analysis7 of 22 prospective studies, the relative risk for coronary heart disease was 1·6 (95% CI 1·5–1·7) in a comparison of people in the top third (mean 2·4 mg/L) and bottom third (1·0 mg/L) of the CRP distribution.
To help judge the likelihood of causality, information is needed about the extent to which disease associations with CRP concentrations are independent of conventional risk factors.7 Previous studies were not adequately powered to assess whether CRP concentration is associated with different stroke subtypes,15–17 and whether concentration is associated exclusively with vascular disease or also with non-vascular diseases.18 The shape of the dose-response association between CRP concentration and the risk of vascular and non-vascular diseases has not been well characterised. Furthermore, powerful analyses are needed to find out whether the strength of association of CRP concentration and risk of disease varies by age, sex, or other clinically relevant subgroups.18 We therefore assessed the independence, specificity, magnitude, and shape of associations of CRP concentration with vascular and non-vascular outcomes under different circumstances.
Methods
Study design
Details of study selection, and data collation and harmonisation in the Emerging Risk Factors Collaboration have been described previously.19 Investigators from 116 prospective studies of cardiovascular risk factors, with a total of 1·2 million participants, shared individual records. In these studies, participants were not selected on the basis of having previously had cardiovascular disease; vascular morbidity and cause-specific mortality were recorded on the basis of clearly defined criteria; and accrued follow-up was more than 1 year. Analyses were restricted to participants without any known history of cardiovascular disease (ie, myocardial infarction, angina, or stroke defined according to study criteria). Information about CRP concentration, age, sex, and other conventional vascular risk factors was available in 54 studies, with a total of 160 309 participants with 27 769 first-ever non-fatal or fatal vascular and non-vascular disease outcomes (webappendix p 2 and p 15). Baseline information was not available for non-vascular diseases. Complete information was available for CRP concentration, age, sex, systolic blood pressure, smoking habits, history of diabetes mellitus (type 1 or 2, though usually not specified), body-mass index, and concentrations of triglycerides and total cholesterol in 109 742 participants from 37 studies. High-sensitivity CRP assays were used in 52 of 54 studies. The CRP standards used were identified in 43 studies (webappendix p 4). International Classification of Diseases codings (to at least three numbers) were used to register fatal diseases, which were ascertained from information provided on death certificates (webappendix p 6). Medical records, autopsy findings, and other supplementary sources were used to help classify deaths in 41 of 54 studies. Standard definitions of myocardial infarction based on Monitoring Trends and Determinants in Cardiovascular Disease (MONICA) or WHO criteria were reported in 49 studies.20 Diagnosis of stroke on the basis of typical clinical features and characteristic changes on brain imaging was reported in 40 studies, enabling attribution of stroke types (eg, cerebral ischaemia, cerebral haemorrhage, or subarachnoid haemorrhage).
The study was approved by an ethics review committee in Cambridgeshire, UK.
Statistical analyses
Details of the statistical methods have been previously provided.21 Normal distributions were obtained with natural logarithm (loge) transformation of positively skewed variables, including CRP concentration. The pooled SD for baseline loge CRP concentration was 1·11, which corresponds to a three-fold difference (ie, e1·11) in mg/L on the original scale of CRP measurement. Sex-specific associations of CRP with various risk factors were assessed, using a linear mixed model adjusted for age.22 The primary outcome was coronary heart disease (ie, first-ever myocardial infarction or fatal coronary heart disease), with subsidiary analyses of stroke by subtype, death from vascular disease, and aggregate of non-vascular death (with subdivisions modelled on the approach of a previous study23). Only first non-fatal vascular outcomes or deaths recorded at age 40 years or older were included in the primary analysis (ie, deaths preceded by non-fatal coronary heart disease or stroke were not included in the analyses). We did a subsidiary analysis for fatal outcomes without censoring for previous non-fatal outcomes. Analyses involved a two-stage approach with estimates of association calculated separately within each study before data from different studies were pooled by use of random-effects meta-analysis. We used fixed-effect models for parallel analyses. For the 42 studies analysed as prospective cohorts, hazard ratios were calculated with Cox proportional hazards regression stratified by sex (and, when appropriate, by trial group). The assumptions for the proportionality of hazards were met. For the 12 case-control studies nested within prospective cohorts, odds ratios were calculated with conditional or unconditional logistic regression models, as appropriate. Hazard ratios and odds ratios were assumed to represent nearly the same relative risk and are collectively described as risk ratios (RRs). As directly measured concentrations of LDL-cholesterol were available for 48 475 participants, the concentration of non-HDL cholesterol was used as the main marker of cholesterol content in proatherogenic lipoproteins rather than that of LDL-cholesterol estimated with the Friedewald formula.24,25
Study-specific RRs were calculated within overall quantiles of baseline concentrations of CRP, and were combined on the log scale by use of multivariate random-effects meta-analysis, and plotted against pooled mean usual concentrations within each quantile. 95% CIs were estimated from variances attributed to the groups to reflect the amount of information within each group (including the reference group). When associations were nearly log-linear, regression coefficients were calculated to estimate the RR associated with a 1-SD higher loge CRP (1·11 loge mg/L, corresponding to a three-fold higher concentration). RRs were adjusted progressively for age, sex, and several other conventional risk factors, with the evidence of association indicated by the Wald χ2 statistic.23 The sequence of introduction of variables into the model followed the example of a previous report.23 To help distinguish whether CRP concentration or inflammatory processes in general might be relevant to disease risk, we also adjusted for other circulating markers of inflammation (such as fibrinogen). I2 statistic was used as a measure of consistency across studies—ie, the percentage of variance in estimated loge RRs that was attributable to between study variation as opposed to sampling variation. Values of I2 close to 0 correspond to lack of heterogeneity. We investigated diversity at the study level by grouping studies according to recorded characteristics and meta-regression.
We corrected concurrently for regression dilution in CRP concentration and in potential confounding factors.26,27 Regression dilution ratios for each characteristic were calculated with regression of serial measurements on baseline concentrations, adjusted for conventional risk factors plus baseline loge CRP concentration and duration of follow-up.26,27 Correction for within-person variation in CRP concentration and potential confounders was achieved by use of conditional expectations of long-term average (usual) concentrations of CRP and confounders predicted from regression calibration models,27 and used in assessments of associations with disease risk. For analyses we used Stata (version 10.0).
Role of the funding source
SK and JD had full access to all data in the study and had final responsibility to submit the report for publication. The study was conducted and analysed independently from its funders.
Results
Mean age of participants at entry into the study was 60 years (SD 8); 76 171 (48%) were women; and 78 619 (49%) were European and 75 919 (47%) were North American. During 1·31 million person-years at risk (median 5·8 years to first outcome, IQR 2·5–9·5), there were 10 451 coronary heart disease outcomes (7381 non-fatal myocardial infarctions and 3070 deaths from coronary diseases), 2846 ischaemic strokes, 469 haemorrhagic strokes, 1180 unclassified strokes, and 1659 deaths from other vascular diseases, 10 236 from non-vascular diseases, and 860 from unknown causes. Mean loge CRP concentration varied between studies and increased with age (figure 1), but SDs were generally similar between studies (webappendix p 2). The overall median for baseline CRP concentration was 1·72 mg/L (5th percentile 0·25, 95th percentile 12·4).
Figure 1.
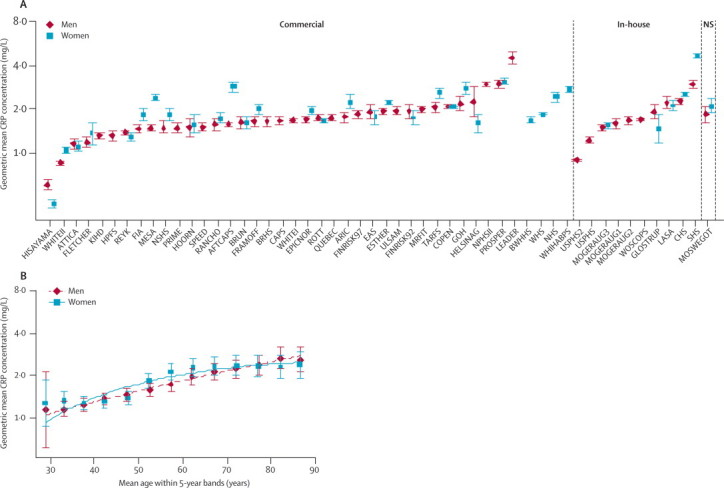
Geometric mean C-reactive protein (CRP) concentration in men and women according to cohort and assay source (A) and within 5-year bands adjusted for cohort (B)
Data from the third National Health and Nutrition Examination Survey (NHANESIII) and Population Study of Women in Göteborg (GOTOW) are not represented in the graph because they did not use high sensitivity CRP assays.19 NS=not stated. Error bars represent the 95% CIs.
CRP concentration was positively correlated with concentrations of non-HDL cholesterol, loge triglycerides, fibrinogen, and interleukin 6, systolic blood pressure, body-mass index, and loge leucocyte count; and inversely with concentrations of HDL cholesterol and albumin (figure 2). CRP concentration was higher in women than in men, in smokers than in non-smokers, and in people with diabetes than in those without, and was lower in alcohol drinkers than in non-drinkers and in physically active people than in those not physically active (figure 2; webappendix p 8). More than one CRP measurement was available in 22 124 participants with 24 222 serial measurements (mean interval 5·1 years, SD 2·4; webappendix p 16). The regression-dilution ratio of loge CRP concentration, adjusted for age and sex, was 0·58 (95% CI 0·52–0·63), indicating a year-to-year consistency that was generally similar to that for systolic blood pressure (0·54, 0·51–0·58), and concentrations of total cholesterol (0·59, 0·54–0·64), HDL cholesterol (0·74, 0·70–0·78), and fibrinogen (0·51, 0·46–0·56) in the same individuals.
Figure 2.
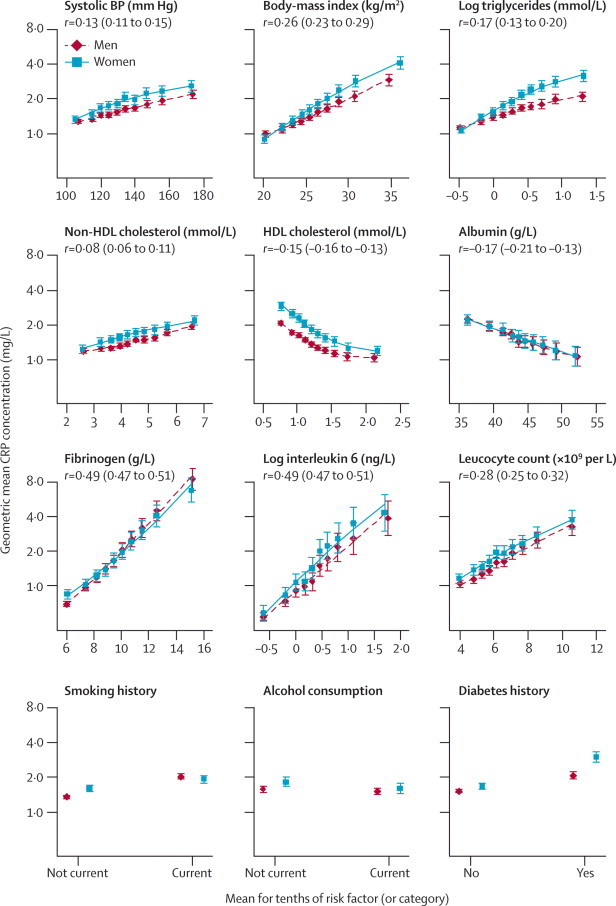
Cross-sectional associations between geometric mean C-reactive protein (CRP) concentration and some conventional risk factors and other characteristics
Mean CRP concentration was adjusted to age 50 years. Error bars represent the 95% CIs. BP=blood pressure. r=Pearson's correlation coefficient (95% CI) for association between the risk marker and loge CRP concentration in men and women combined.
In analyses adjusted for age and sex only, associations of loge CRP concentration with the risk of coronary heart disease and ischaemic stroke were nearly log-linear, with no obvious risk thresholds (figure 3A). The corresponding associations of CRP concentration with all vascular mortality and with all non-vascular mortality seemed curvilinear. RR for coronary heart disease, adjusted for age and sex only, per 1-SD higher usual loge CRP concentration (ie, three-fold higher CRP concentration) was 1·68 (95% CI 1·59–1·78), with the RR possibly higher for fatal (1·84, 1·59–2·14) than for non-fatal myocardial infarction (1·59, 1·45–1·75; p=0·04; webappendix p 9). For all strokes combined, the corresponding RR was 1·39 (1·29–1·51), with RRs of 1·46 (1·32–1·61) for ischaemic stroke, 1·07 (0·86–1·32) for haemorrhagic stroke, and 1·41 (1·22–1·63) for unclassified stroke. Figure 4 shows the RRs for all deaths from vascular and non-vascular causes. CRP concentration was significantly associated with a range of different conditions, including several cancers, lung diseases, and even external causes. There were generally too few cases of site-specific cancer mortality to enable reliable analyses. RRs were generally similar in analyses that excluded current smokers or were restricted to participants with first outcomes recorded after the first 5 years of follow-up (webappendix p 18).
Figure 3.
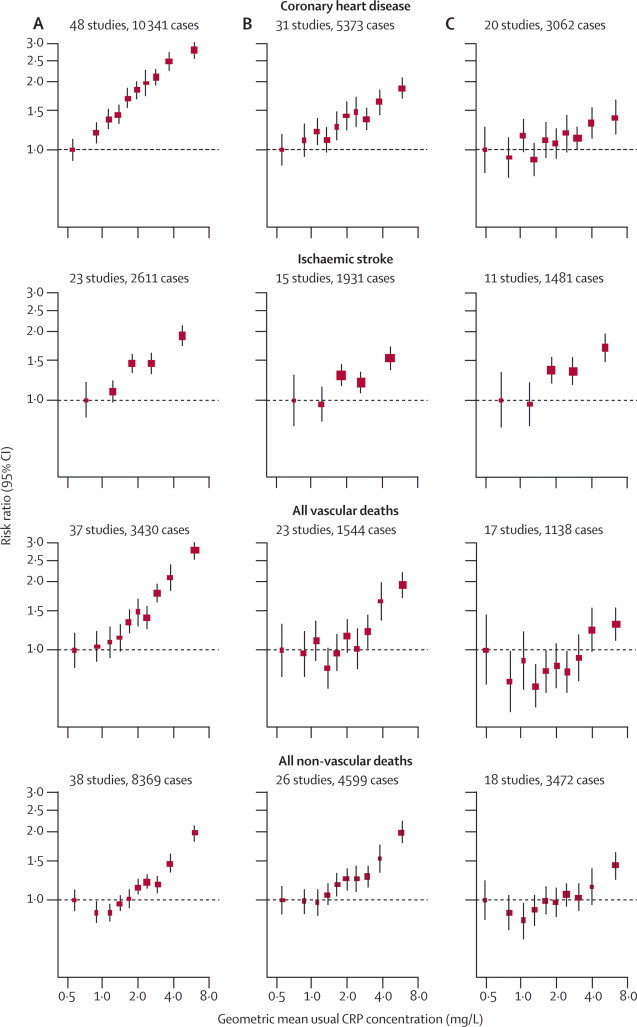
Risk ratios for major vascular and non-vascular outcomes by quantiles of C-reactive protein (CRP) concentration, with different degree of adjustment for potential confounders
Adjusted study-specific loge risk ratios were combined by use of multivariate random-effects meta-analysis. The adjustments were age, sex, and study only (A); age, sex, study, systolic blood pressure, smoking, history of diabetes, body-mass index, concentrations of loge triglycerides, non-HDL cholesterol, and HDL cholesterol, and alcohol consumption (B); and (A) plus (B) plus fibrinogen (C). Studies with fewer than ten cases of any outcome were excluded from the analysis of that outcome. Error bars represent the 95% CIs, calculated using floating absolute risk technique. The sizes of the boxes are proportional to the inverse of the variance of the risk ratios.
Figure 4.
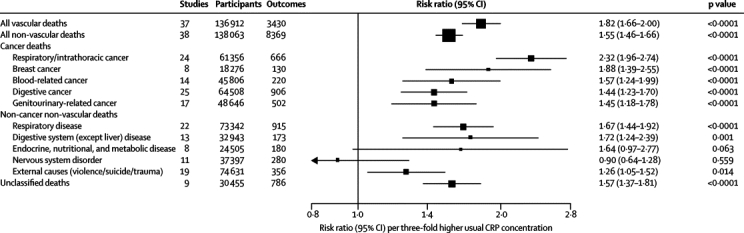
Age-adjusted and sex-adjusted risk ratios for mortality from vascular and non-vascular diseases per three-fold higher usual C-reactive protein (CRP) concentration
Data are numbers, unless otherwise indicated. Risk ratios (boxes) were adjusted only for age, and stratified (when appropriate), by sex and trial group. Studies with fewer than ten cases of any outcome were excluded from the analysis of that outcome. The risk ratios are presented per 1·11 higher loge CRP (ie, 1-SD), corresponding to a three-fold higher CRP concentration. Horizontal lines represent the 95% CIs. The sizes of the boxes are proportional to the inverse of the variance of the risk ratios.
In analyses involving adjustment for additional risk factors, RR for coronary heart disease was reduced after adjustment for age, sex, systolic blood pressure, smoking, history of diabetes, body-mass index, and concentrations of triglycerides and total cholesterol (table 1). Further adjustment for concentrations of non-HDL cholesterol and HDL cholesterol, and alcohol consumption, resulted in a further reduction in RR and in the corresponding Wald χ2 statistic (table 1; figure 3B). Adjusted RRs for coronary heart disease per 1-SD higher levels of each risk factor were 1·37 (95% CI 1·27–1·48) with CRP concentration, 1·28 (1·16–1·40) with non-HDL-cholesterol concentration, and 1·35 (1·25–1·45) with systolic blood pressure (webappendix p 19). The RRs with CRP were 1·55 (1·37–1·76) for vascular mortality and 1·54 (1·40–1·68) for non-vascular mortality after adjustment of conventional risk factors (webappendix p 10).
Table 1.
Risk ratios (RRs) for coronary heart disease and ischaemic stroke per three-fold higher usual C-reactive protein (CRP) concentration with progressive adjustment for usual levels of potential confounders
|
Basic adjustment* |
Further adjustment* |
|||||
|---|---|---|---|---|---|---|
| RR† (95% CI) usual loge CRP concentration | Wald χ21 | I2 (95% CI) | RR† (95% CI) usual loge CRP concentration | Wald χ21 | I2 (95% CI) | |
| Coronary heart disease‡ | ||||||
| Adjusted for age, sex, and study | 1·64 (1·54–1·75) | 231 | 44% (17–62) | 1·63 (1·51–1·76) | 149 | 51% (26–68) |
| Plus systolic blood pressure | 1·57 (1·48–1·67) | 201 | 37% (7–58) | 1·55 (1·44–1·67) | 134 | 42% (12–62) |
| Plus smoking | 1·50 (1·41–1·59) | 181 | 26% (0–51) | 1·48 (1·38–1·59) | 123 | 30% (0–55) |
| Plus history of diabetes | 1·45 (1·36–1·55) | 121 | 38% (7–58) | 1·43 (1·32–1·55) | 78 | 42% (10–62) |
| Plus body-mass index | 1·46 (1·37–1·55) | 137 | 22% (0–48) | 1·45 (1·34–1·56) | 85 | 31% (0–56) |
| Plus loge triglycerides concentration | 1·41 (1·32–1·51) | 103 | 26% (0–51) | 1·40 (1·29–1·52) | 64 | 35% (0–58) |
| Plus total cholesterol concentration | 1·42 (1·33–1·52) | 110 | 24% (0–50) | 1·41 (1·30–1·53) | 69 | 33% (0–57) |
| Plus non-HDL cholesterol concentration§ | .. | .. | .. | 1·40 (1·30–1·51) | 74 | 26% (0–53) |
| Plus HDL cholesterol concentration§ | .. | .. | .. | 1·39 (1·28–1·49) | 71 | 25% (0–52) |
| Plus alcohol consumption§ | .. | .. | .. | 1·37 (1·27–1·48) | 65 | 26% (0–53) |
| Ischaemic stroke¶ | ||||||
| Adjusted for age, sex, and study | 1·43 (1·32–1·55) | 80 | 0 (0–51) | 1·44 (1·32–1·57) | 68 | 7% (0–57) |
| Plus systolic blood pressure | 1·34 (1·23–1·45) | 50 | 0 (0–51) | 1·33 (1·23–1·45) | 46 | 0 (0–54) |
| Plus smoking | 1·31 (1·20–1·42) | 40 | 0 (0–51) | 1·31 (1·20–1·42) | 38 | 0 (0–54) |
| Plus history of diabetes | 1·25 (1·14–1·35) | 26 | 0 (0–51) | 1·24 (1·14–1·36) | 25 | 0 (0–54) |
| Plus body-mass index | 1·29 (1·18–1·41) | 29 | 0 (0–51) | 1·29 (1·18–1·42) | 28 | 0 (0–54) |
| Plus loge triglycerides concentration | 1·27 (1·16–1·40) | 26 | 0 (0–51) | 1·27 (1·15–1·40) | 24 | 0 (0–54) |
| Plus total cholesterol concentration | 1·28 (1·17–1·41) | 27 | 0 (0–51) | 1·28 (1·16–1·40) | 25 | 0 (0–54) |
| Plus non-HDL cholesterol concentration§ | .. | .. | .. | 1·28 (1·16–1·40) | 25 | 0 (0–54) |
| Plus HDL cholesterol concentration§ | .. | .. | .. | 1·27 (1·16–1·40) | 25 | 0 (0–54) |
| Plus alcohol consumption§ | .. | .. | .. | 1·27 (1·15–1·40) | 24 | 0 (0–54) |
Analyses were restricted to participants with complete information available for sex, trial group, and all confounding variables; studies with fewer than ten cases were excluded from the analysis of each outcome.
Per 1·11 higher loge CRP (ie, 1-SD), corresponding to a three-fold higher CRP concentration, progressively adjusted and stratified when appropriate by sex and trial group.
Basic adjustment: 37 studies, 109 742 participants, and 8056 events; further adjustment: 31 studies, 91 990 participants, and 5373 events.
Non-HDL cholesterol has been substituted for total cholesterol in these adjusted models.
Basic adjustment: 17 studies, 65 825 participants, and 2006 events; further adjustment: 15 studies, 60 763 participants, and 1931 events.
Figure 3C and table 2 show RRs for coronary heart disease and ischaemic stroke after further adjustment for fibrinogen (or other markers of inflammation). In participants with complete information available for CRP concentration, conventional risk factors, and fibrinogen concentration, RR for coronary heart disease associated with CRP after adjustment for conventional risk factors was reduced from 1·36 (95% CI 1·22–1·52) to 1·23 (1·07–1·42) after further adjustment for fibrinogen (Wald χ2 was reduced from 29 to 8; table 2). By comparison, the corresponding adjusted RR for coronary heart disease was 1·21 (1·08–1·35) per 1-SD higher usual fibrinogen after adjustment for several conventional risk factors and CRP concentration. RR for ischaemic stroke associated with CRP after adjustment for conventional risk factors did not differ after further adjustment for fibrinogen (table 2). RR for non-vascular death was 1·52 (1·37–1·69) with CRP concentration after adjustment for conventional risk factors, and 1·34 (1·20–1·50; webappendix p 11) after further adjustment for fibrinogen. When fibrinogen was replaced with another marker of inflammation (eg, leucocyte count) in the model, reduction in the RRs with CRP was slightly smaller (albeit based on fewer data). Since only about 10% of individuals had concomitant data for CRP concentration, conventional risk factors, fibrinogen concentration, and at least one other inflammatory marker, more detailed adjustment for inflammation was not possible.
Table 2.
Risk ratios (RRs) for coronary heart disease and ischaemic stroke per three-fold higher usual C-reactive protein (CRP) concentration with adjustment for usual levels of conventional risk factors plus different inflammatory markers
| Studies | Outcomes | RR*(95% CI) usual logeCRP concentration | Wald χ21 | I2 (95% CI) | ||
|---|---|---|---|---|---|---|
| Coronary heart disease | ||||||
| Adjusted for age, sex, and study (n=66 117) | 20 | 3062 | 1·65 (1·48–1·84) | 80 | 57% (30–74) | |
| Plus conventional risk factors† | 20 | 3062 | 1·36 (1·22–1·52) | 29 | 36% (0–63) | |
| Plus fibrinogen | 20 | 3062 | 1·23 (1·07–1·42) | 8 | 57% (30–74) | |
| Adjusted for age, sex, and study (n=32 958) | 12 | 2689 | 1·66 (1·48–1·86) | 78 | 52% (7–75) | |
| Plus conventional risk factors† | 12 | 2689 | 1·44 (1·29–1·62) | 40 | 35% (0–67) | |
| Plus albumin | 12 | 2689 | 1·38 (1·26–1·51) | 47 | 15% (0–54) | |
| Adjusted for age, sex, and study (n=21 917) | 11 | 2688 | 1·68 (1·48–1·91) | 65 | 60% (22–79) | |
| Plus conventional risk factors† | 11 | 2688 | 1·42 (1·26–1·60) | 32 | 37% (0–69) | |
| Plus loge leucocyte count | 11 | 2688 | 1·30 (1·16–1·45) | 20 | 36% (0–68) | |
| Adjusted for age, sex, and study (n=19 016) | 7 | 1547 | 1·77 (1·37–2·28) | 19 | 83% (67–91) | |
| Plus conventional risk factors† | 7 | 1547 | 1·42 (1·15–1·74) | 11 | 59% (5–82) | |
| Plus loge interleukin 6 | 7 | 1547 | 1·19 (1·01–1·41) | 4 | 31% (0–70) | |
| Ischaemic stroke | ||||||
| Adjusted for age, sex, and study (n=47 353) | 11 | 1481 | 1·49 (1·34–1·67) | 49 | 16% (0–57) | |
| Plus conventional risk factors† | 11 | 1481 | 1·32 (1·18–1·47) | 24 | 0 (0–60) | |
| Plus fibrinogen | 11 | 1481 | 1·32 (1·18–1·49) | 22 | 0 (0–60) | |
| Adjusted for age, sex, and study (n=19 382) | 6 | 890 | 1·37 (1·23–1·52) | 35 | 0 (0–75) | |
| Plus conventional risk factors† | 6 | 890 | 1·28 (1·14–1·44) | 16 | 0 (0–75) | |
| Plus albumin | 6 | 890 | 1·21 (1·07–1·35) | 10 | 0 (0–75) | |
| Adjusted for age, sex, and study (n=14 076) | 7 | 1252 | 1·40 (1·26–1·55) | 42 | 0 (0– 71) | |
| Plus conventional risk factors† | 7 | 1252 | 1·25 (1·11–1·41) | 13 | 0 (0–71) | |
| Plus loge leucocyte count | 7 | 1252 | 1·15 (1·02–1·29) | 5 | 0 (0–71) | |
| Adjusted for age, sex, and study (n=9918) | 3 | 480 | 1·63 (1·15–2·29) | 8 | 63% (0–89) | |
| Plus conventional risk factors† | 3 | 480 | 1·47 (1·09–1·98) | 6 | 42% (0–83) | |
| Plus loge interleukin 6 | 3 | 480 | 1·18 (0·98–1·41) | 3 | 0 (0–90) | |
Analyses were restricted to participants with complete information about sex, trial group, and all the conventional risk factors plus each inflammatory marker. Studies with fewer than ten cases were excluded from the analysis of each outcome. Data are number, unless otherwise indicated.
Per 1·11 higher loge CRP (ie, 1-SD), corresponding to a three-fold higher CRP concentration, progressively adjusted and stratified (when appropriate) by sex and trial group.
Age, sex, systolic blood pressure, smoking, history of diabetes, body-mass index, concentrations of loge triglycerides, non-HDL cholesterol, and HDL cholesterol, and alcohol consumption.
There may have been heterogeneity between studies in RRs for coronary heart disease (I2=26%, 95% CI 0–53). Apart from possibly higher RRs in men than in women (p=0·015), RRs for coronary heart disease did not vary in clinically relevant subgroups—such as people with diabetes, increased concentrations of lipids, and obesity, or in smokers—or with other participant or study characteristics (webappendix pp 20–21), or in exploratory analyses of individuals with morbid obesity or very high concentrations of lipids (webappendix p 20). Likewise, evidence of heterogeneity between studies in RRs for ischaemic stroke was not clear (I2=0, 0–54). Qualitatively similar results to those reported here were noted in analyses that did not correct for regression dilution; adjusted for smoking amount (webappendix p 12); used fixed-effect models (webappendix p 13); included fatal outcomes without censoring previous non-fatal outcomes (webappendix p 14); excluded the two studies in which high-sensitivity CRP assays were not used; compared larger versus smaller studies (webappendix p 24); and excluded the 10 188 participants with CRP values greater than 10 mg/L.
Discussion
The current study has shown that CRP concentration is as consistent within individuals during several years as are total cholesterol concentration and systolic blood pressure. In individuals without initial vascular disease, associations of CRP concentration with subsequent risk of coronary heart disease, ischaemic stroke, and deaths from vascular and non-vascular diseases (including several cancers and respiratory diseases) were generally log-linear in shape. These associations persisted in analyses in which smokers and the initial years of follow-up were excluded.
Associations of CRP with vascular and non-vascular outcomes were each of broadly similar size. These findings resemble results previously reported with other downstream markers of inflammation, notably fibrinogen concentration,23 leucocyte count,28 albumin concentration,28 or erythrocyte sedimentation rate.29 Like CRP, each of these factors is modulated by mediators of inflammatory cascades (eg, interleukin 6), and our data confirm that such inflammatory markers are associated with CRP. Although persistent low-grade inflammation is proposed to be relevant to vascular and malignant diseases,30 the mechanisms are not known. The associations that we noted between CRP concentration and death from external causes (eg, injury) might be attributed, at least in part, to confounding by comorbidity at baseline.
Adjustment for several conventional risk factors and plasma fibrinogen (also an acute phase reactant protein synthesised in the liver and modulated by interleukin 623,31,32) resulted in considerable weakening of associations of CRP concentration with risk of coronary heart disease. Such adjustment also attenuated associations of CRP concentration with ischaemic stroke and deaths from non-vascular diseases. Hence, although our results support the idea that some process related to persistent inflammation is associated with vascular disease and other chronic disorders, most of the association with ischaemic vascular disease depends on conventional risk factors and fibrinogen concentration.29,31–34 If fibrinogen mediates the association between CRP concentration and risk of coronary heart disease, then correction for fibrinogen could be an overadjustment.
In studies in which CRP-related genotypes are proxies for life-long CRP concentration (ie, Mendelian randomisation analyses), such difficulties in interpretation should be avoided, provided the genotypes are not correlated with fibrinogen. In previous analyses, including a study of 28 000 patients with coronary heart disease and 100 000 controls,35 null associations were reported between CRP-related genotypes and fibrinogen concentration,36 conventional risk factors,37 and risk of coronary heart disease.36,38,39 Hence, our findings—taken with available genetic data—reduce the likelihood of causality for CRP in coronary heart disease. Results from randomised trials have shown that statins reduce concentrations of CRP in healthy individuals and in those with stable vascular disease.40 Rosuvastatin reduced the risk of first-ever vascular disease in individuals who had lower-than-average LDL-cholesterol concentration and higher-than-average CRP concentration.41 But, since statins potently affect LDL-cholesterol concentration (an established causative risk factor in coronary disease), studies of these drugs cannot provide specific causal inferences about CRP concentration. Immunosuppressant or anti-inflammatory drugs, or vaccination, have not yet been adequately studied as prophylactic interventions to test the inflammation hypothesis.32
This meta-analysis differs from previous reports in several important ways. First, it was larger and more comprehensive. Second, use of individual participant data enabled a consistent approach to adjustment for potential confounders and exploration of subgroups. Third, associations were corrected for measurement error and within-person variation (ie, regression dilution26,27,42), using information from 22 124 participants with serial measurements. Fourth, individuals with a history of coronary heart disease or stroke were excluded, reducing any effects of clinically evident disease on CRP concentration. Although there was heterogeneity in the findings in relation to coronary heart disease (no greater in extent to that seen with non-HDL-cholesterol concentration in the same studies25), little of it could be attributed to the recorded characteristics. Since most disease outcomes were recorded among white participants, the findings do not necessarily apply to non-white individuals. Our results confirm that CRP concentration can differ substantially in different ethnic groups (eg, concentrations were 26% higher in black individuals and 16% lower in east Asian individuals than in white people).43
Large studies are needed that concurrently assess several proximal (eg, interleukin 6) and distal (eg, CRP, fibrinogen) markers of inflammation, and markers of rupture-prone plaque (eg, lipoprotein-associated phospholipase A244), as well as their genetic and lifestyle determinants. Further work is also needed to assess whether evidence of low-grade inflammation mainly indicates external triggers (eg, socioeconomic position or infection), insulin resistance, hereditary predisposition, or some combination of such factors.45–47
In subsequent analyses from this collaboration, we will assess CRP concentration for the prediction of vascular disease—a separate issue requiring analytical approaches that are different from those used in this meta-analysis.48–50
Correspondence to: Emerging Risk Factors Collaboration Coordinating Centre, Department of Public Health and Primary Care, University of Cambridge, Strangeways Research Laboratory, Cambridge CB1 8RN, UK
Acknowledgments
Acknowledgments
The ERFC Coordinating Centre is underpinned by a programme grant from the British Heart Foundation and supported by specific grants from the UK Medical Research Council and the BUPA Foundation, and an unrestricted educational grant from GlaxoSmithKline. A variety of sources have supported recruitment, follow-up, and laboratory measurements in the 116 cohorts contributing to the ERFC. Investigators of several of these studies have contributed to a list of some of these funding sources. We thank Juan-Pablo Casas, Aroon Hingorani, and Liam Smeeth for helpful comments. The writing committee accepts full responsibility for the content of this report.
Contributors
SK did the statistical analysis and drafted the report with JD. All members of the writing committee provided critical revisions. All investigators shared individual data and had an opportunity to contribute to the interpretation of the results and to the redrafting of the report. The data management team collated and standardised the data. All members of the coordinating centre contributed to the collection, standardisation, analysis, and interpretation of the data.
Emerging Risk Factors Collaboration (ERFC)
Writing Committee: Stephen Kaptoge (University of Cambridge), Emanuele Di Angelantonio (University of Cambridge), Gordon Lowe (University of Glasgow), Mark B Pepys (University College London Medical School), Simon G Thompson (Medical Research Council [MRC] Biostatistics Unit), Rory Collins (University of Oxford), John Danesh (University of Cambridge).
ERFC Investigators: AFTCAPS R W Tipping; ALLHAT C E Ford, S L Pressel; AMORIS G Walldius, I Jungner; ARIC A R Folsom, L Chambless, C M Ballantyne; ATTICA D Panagiotakos, C Pitsavos, C Chrysohoou, C Stefanadis; BHS M W Knuiman; BIP U Goldbourt, M Benderly, D Tanne; BRHS P Whincup, S G Wannamethee, R W Morris; BRUN S Kiechl, J Willeit, A Mayr, G Schett; BUPA N Wald; BWHHS S Ebrahim, D Lawlor; CaPS J Yarnell, J Gallacher; CASTEL E Casiglia, V Tikhonoff; CHARL P J Nietert, S E Sutherland, D L Bachman, J E Keil; CHS M Cushman, B M Psaty, R Tracy; COPEN A Tybjærg-Hansen, B G Nordestgaard, J Zacho, R Frikke-Schmidt; CUORE S Giampaoli, L Palmieri, S Panico, D Vanuzzo, L Pilotto; DRECE A Gómez de la Cámara, J A Gómez Gerique; DUBBO L Simons, J McCallum, Y Friedlander; EAS F G R Fowkes, A Lee; EPESEBOS J Taylor, J M Guralnik, C L Phillips; EPESEIOW R B Wallace, J M Guralnik, C L Phillips; EPESENCA D G Blazer, J M Guralnik, C L Phillips; EPESENHA C L Phillips, J M Guralnik; EPICNOR K-T Khaw; ESTHER H Brenner, E Raum, H Müller, D Rothenbacher; FIA J-H Jansson, P Wennberg; FINE_FIN A Nissinen; FINE_IT C Donfrancesco, S Giampaoli; FINRISK92, FINRISK97 V Salomaa, K Harald, P Jousilahti, E Vartiainen; FLETCHER M Woodward; FRAMOFF R B D'Agostino, P A Wolf, R S Vasan, E J Benjamin; GLOSTRUP E-M Bladbjerg, T Jørgensen, L Møller, J Jespersen; GOH R Dankner, A Chetrit, F Lubin; GOTO33, GOTO43 A Rosengren, L Wilhelmsen, G Lappas, H Eriksson; GOTOW C Björkelund, L Lissner, C Bengtsson; GRIPS P Cremer, D Nagel; HELSINAG R S Tilvis, T E Strandberg; HISAYAMA Y Kiyohara, H Arima, Y Doi, T Ninomiya; HONOL B Rodriguez; HOORN J Dekker, G Nijpels, C D A Stehouwer; HPFS E Rimm, J K Pai; IKNS S Sato, H Iso, A Kitamura, H Noda; ISRAEL U Goldbourt; KIHD J T Salonen, K Nyyssönen, T-P Tuomainen, J A Laukkanen; LASA D J H Deeg, M A Bremmer; LEADER T W Meade, J A Cooper; MALMO B Hedblad, G Berglund, G Engström; MCVDRFP W M M Verschuren, A Blokstra; MESA M Cushman, A R Folsom, B M Psaty, S Shea; MOGERAUG1, MOGERAUG2, MOGERAUG3 A Döring, W Koenig, C Meisinger; MORGEN W M M Verschuren, A Blokstra, H B Bueno-de-Mesquita; MOSWEGOT L Wilhelmsen, A Rosengren, G Lappas; MRFIT L H Kuller, G Grandits; NCS R Selmer, A Tverdal, W Nystad; NHANES I, NHANES II, NHANES III R F Gillum, M Mussolino; NHS E Rimm, S Hankinson, J E Manson, J K Pai; NORTH KARELIA V Salomaa, K Harald, P Jousilahti, E Vartiainen; NPHS I T W Meade, J A Cooper, C Knottenbelt; NPHS II J A Cooper, K A Bauer; NSHS K Davidson, S Kirkland, J Shaffer, M R Korin; OSAKA S Sato, A Kitamura, Y Naito, H Iso; OSLO I Holme, R Selmer, A Tverdal, W Nystad; OYABE H Nakagawa, K Miura; PARIS1 P Ducimetiere, X Jouven, G Luc; PRHHP C J Crespo, M R Garcia-Palmieri; PRIME P Amouyel, D Arveiler, A Evans, J Ferrieres; PROCAM H Schulte, G Assmann; PROSPER C J Packard, N Sattar, R G Westendorp, B M Buckley; QUEBEC B Cantin, B Lamarche, J-P Després, G R Dagenais; RANCHO E Barrett-Connor, D L Wingard, R R Bettencourt; REYK V Gudnason, T Aspelund, G Sigurdsson, B Thorsson; RIFLE M Trevisan; ROTT J Witteman, I Kardys, M M B Breteler, A Hofman; SHHEC H Tunstall-Pedoe, R Tavendale, G Lowe, M Woodward; SHS B V Howard, Y Zhang, L Best, J Umans; SPEED Y Ben-Shlomo, G Davey-Smith TARFS A Onat; TPT T W Meade; TROMSØ I Njølstad, E B Mathiesen, M-L Løchen, T Wilsgaard; ULSAM E Ingelsson, S Basu, T Cederholm, L Byberg; USPHS J M Gaziano, M Stampfer, P M Ridker; USPHS2 J M Gaziano, P M Ridker; VHMPP H Ulmer, G Diem, H Concin; VITA A Tosetto, F Rodeghiero; WHI-HaBPS S Wassertheil-Smoller, J E Manson; WHITE I M Marmot, R Clarke, R Collins, A Fletcher; WHITE II E Brunner, M Shipley; WHS P M Ridker, J Buring; WOSCOPS J Shepherd, S Cobbe, I Ford, M Robertson; XIAN Y He; ZARAGOZA A Marin Ibañez; ZUTE E J M Feskens.
Data Management Team: M Walker, S Watson.
Coordinating Centre: R Collins, E Di Angelantonio, S Erqou, S Kaptoge, S Lewington, L Pennells, P L Perry, K K Ray, N Sarwar, M Alexander, A Thompson, S G Thompson, M Walker, S Watson, I R White, A M Wood, J Danesh (principal investigator).
Conflicts of interest
JD has received research funding from the British Heart Foundation, BUPA Foundation, diaDexus, European Union, Evelyn Trust, Fogarty International Centre, GlaxoSmithKline, MRC, Merck, National Heart Lung and Blood Institute, National Institute of Neurological Disorders and Stroke, Novartis, Roche, and the Wellcome Trust. RC is paid by the British Heart Foundation, National Health Service, and UK Biobank, and has received research funding and reimbursement of costs for attending scientific meetings (but no honoraria or consultancy payments) from AstraZeneca, Bayer, Bristol-Myers Squibb, British Heart Foundation, Cancer Research UK, European Union, Kadoorie Trust, MRC, Merck, Roche, Sanofi, Schering, Solvay, and UK Biobank. SGT has received research funding from MRC, National Institute for Health Research, BUPA, British Heart Foundation, and European Union. The other authors declare that they have no conflicts of interest.
Web Extra Material
References
- 1.Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest. 2003;111:1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 3.Emberson JR, Whincup PH, Morris RW, Walker M, Lowe GD, Rumley A. Extent of regression dilution for established and novel coronary risk factors: results from the British Regional Heart Study. Eur J Cardiovasc Prev Rehabil. 2004;11:125–134. doi: 10.1097/01.hjr.0000114967.39211.e5. [DOI] [PubMed] [Google Scholar]
- 4.Pearson TA, Mensah GA, Alexander RW. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. doi: 10.1161/01.cir.0000052939.59093.45. [DOI] [PubMed] [Google Scholar]
- 5.Kuller LH, Tracy RP, Shaten J, Meilahn EN. Relation of C-reactive protein and coronary heart disease in the MRFIT nested case-control study. Multiple Risk Factor Intervention Trial. Am J Epidemiol. 1996;144:537–547. doi: 10.1093/oxfordjournals.aje.a008963. [DOI] [PubMed] [Google Scholar]
- 6.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 7.Danesh J, Wheeler JG, Hirschfield GM. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. 2004;350:1387–1397. doi: 10.1056/NEJMoa032804. [DOI] [PubMed] [Google Scholar]
- 8.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 9.Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds risk score. JAMA. 2007;297:611–619. doi: 10.1001/jama.297.6.611. [DOI] [PubMed] [Google Scholar]
- 10.Scirica BM, Morrow DA. Is C-reactive protein an innocent bystander or proatherogenic culprit? The verdict is still out. Circulation. 2006;113:2128–2134. doi: 10.1161/CIRCULATIONAHA.105.611350. [DOI] [PubMed] [Google Scholar]
- 11.Verma S, Devaraj S, Jialal I. Is C-reactive protein an innocent bystander or proatherogenic culprit? C-reactive protein promotes atherothrombosis. Circulation. 2006;113:2135–2150. [PubMed] [Google Scholar]
- 12.de Beer FC, Soutar AK, Baltz ML, Trayner IM, Feinstein A, Pepys MB. Low density lipoprotein and very low density lipoprotein are selectively bound by aggregated C-reactive protein. J Exp Med. 1982;156:230–242. doi: 10.1084/jem.156.1.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pepys MB, Rowe IF, Baltz ML. C-reactive protein: binding to lipids and lipoproteins. Int Rev Exp Pathol. 1985;27:83–111. [PubMed] [Google Scholar]
- 14.Zhang YX, Cliff WJ, Schoefl GI, Higgins G. Coronary C-reactive protein distribution: its relation to development of atherosclerosis. Atherosclerosis. 1999;145:375–379. doi: 10.1016/s0021-9150(99)00105-7. [DOI] [PubMed] [Google Scholar]
- 15.Bos MJ, Schipper CM, Koudstaal PJ, Witteman JC, Hofman A, Breteler MM. High serum C-reactive protein level is not an independent predictor for stroke: the Rotterdam Study. Circulation. 2006;114:1591–1598. doi: 10.1161/CIRCULATIONAHA.106.619833. [DOI] [PubMed] [Google Scholar]
- 16.Kaplan RC, McGinn AP, Baird AE. Inflammation and hemostasis biomarkers for predicting stroke in postmenopausal women: the Women's Health Initiative Observational Study. J Stroke Cerebrovasc Dis. 2008;17:344–355. doi: 10.1016/j.jstrokecerebrovasdis.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersson J, Johansson L, Ladenvall P. C-reactive protein is a determinant of first-ever stroke: prospective nested case-referent study. Cerebrovasc Dis. 2009;27:544–551. doi: 10.1159/000214217. [DOI] [PubMed] [Google Scholar]
- 18.Casas JP, Shah T, Hingorani AD, Danesh J, Pepys MB. C-reactive protein and coronary heart disease: a critical review. J Intern Med. 2008;264:295–314. doi: 10.1111/j.1365-2796.2008.02015.x. [DOI] [PubMed] [Google Scholar]
- 19.Emerging Risk Factors Collaboration. Danesh J, Erqou S, Walker M. The Emerging Risk Factors Collaboration: analysis of individual data on lipid, inflammatory and other markers in over 1·1 million participants in 104 prospective studies of cardiovascular diseases. Eur J Epidemiol. 2007;22:839–869. doi: 10.1007/s10654-007-9165-7. [DOI] [PubMed] [Google Scholar]
- 20.Tunstall-Pedoe H, Kuulasmaa K, Amouyel P. Myocardial infarction and coronary deaths in the World Health Organization MONICA Project. Registration procedures, event rates, and case-fatality rates in 38 populations from 21 countries in four continents. Circulation. 1994;90:583–612. doi: 10.1161/01.cir.90.1.583. [DOI] [PubMed] [Google Scholar]
- 21.Emerging Risk Factors Collaboration. Erqou S, Kaptoge S, Perry PL. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fibrinogen Studies Collaboration. Kaptoge S, White IR, Thompson SG. Associations of plasma fibrinogen levels with established cardiovascular disease risk factors, inflammatory markers, and other characteristics: individual participant meta-analysis of 154 211 adults in 31 prospective studies: the fibrinogen studies collaboration. Am J Epidemiol. 2007;166:867–879. doi: 10.1093/aje/kwm191. [DOI] [PubMed] [Google Scholar]
- 23.Fibrinogen Studies Collaboration. Danesh J, Lewington S, Thompson SG. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005;294:1799–1809. doi: 10.1001/jama.294.14.1799. [DOI] [PubMed] [Google Scholar]
- 24.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 25.Emerging Risk Factors Collaboration. Di Angelantonio E, Sarwar N, Kaptoge S. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fibrinogen Studies Collaboration. Wood AM, White I, Thompson SG, Lewington S, Danesh J. Regression dilution methods for meta-analysis: assessing long-term variability in plasma fibrinogen among 27 247 adults in 15 prospective studies. Int J Epidemiol. 2006;35:1570–1578. doi: 10.1093/ije/dyl233. [DOI] [PubMed] [Google Scholar]
- 27.Fibrinogen Studies Collaboration. Wood AM, White I, Thompson SG. Correcting for multivariate measurement error by regression calibration in meta-analyses of epidemiological studies. Stat Med. 2009;28:1067–1092. doi: 10.1002/sim.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danesh J, Collins R, Appleby P, Peto R. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: meta-analyses of prospective studies. JAMA. 1998;279:1477–1482. doi: 10.1001/jama.279.18.1477. [DOI] [PubMed] [Google Scholar]
- 29.Andresdottir MB, Sigfusson N, Sigvaldason H, Gudnason V. Erythrocyte sedimentation rate, an independent predictor of coronary heart disease in men and women: the Reykjavik Study. Am J Epidemiol. 2003;158:844–851. doi: 10.1093/aje/kwg222. [DOI] [PubMed] [Google Scholar]
- 30.Kundu JK, Surh YJ. Inflammation: gearing the journey to cancer. Mutat Res. 2008;659:15–30. doi: 10.1016/j.mrrev.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Danesh J, Kaptoge S, Mann AG. Long-term interleukin-6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PLoS Med. 2008;5:e78. doi: 10.1371/journal.pmed.0050078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 33.Clarke R, Emberson JR, Breeze E. Biomarkers of inflammation predict both vascular and non-vascular mortality in older men. Eur Heart J. 2008;29:800–809. doi: 10.1093/eurheartj/ehn049. [DOI] [PubMed] [Google Scholar]
- 34.Heikkila K, Harris R, Lowe G. Associations of circulating C-reactive protein and interleukin-6 with cancer risk: findings from two prospective cohorts and a meta-analysis. Cancer Causes Control. 2009;20:15–26. doi: 10.1007/s10552-008-9212-z. [DOI] [PubMed] [Google Scholar]
- 35.Elliott P, Chambers JC, Zhang W. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48. doi: 10.1001/jama.2009.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casas JP, Shah T, Cooper J. Insight into the nature of the CRP-coronary event association using Mendelian randomization. Int J Epidemiol. 2006;35:922–931. doi: 10.1093/ije/dyl041. [DOI] [PubMed] [Google Scholar]
- 37.Timpson NJ, Lawlor DA, Harbord RM. C-reactive protein and its role in metabolic syndrome: mendelian randomisation study. Lancet. 2005;366:1954–2009. doi: 10.1016/S0140-6736(05)67786-0. [DOI] [PubMed] [Google Scholar]
- 38.Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. 2008;359:1897–1908. doi: 10.1056/NEJMoa0707402. [DOI] [PubMed] [Google Scholar]
- 39.Lange LA, Carlson CS, Hindorff LA. Association of polymorphisms in the CRP gene with circulating C-reactive protein levels and cardiovascular events. JAMA. 2006;296:2703–2711. doi: 10.1001/jama.296.22.2703. [DOI] [PubMed] [Google Scholar]
- 40.Kinlay S. Low-density lipoprotein-dependent and -independent effects of cholesterol-lowering therapies on C-reactive protein: a meta-analysis. J Am Coll Cardiol. 2007;49:2003–2009. doi: 10.1016/j.jacc.2007.01.083. [DOI] [PubMed] [Google Scholar]
- 41.Ridker PM, Danielson E, Fonseca FA. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 42.Clarke R, Shipley M, Lewington S. Underestimation of risk associations due to regression dilution in long-term follow-up of prospective studies. Am J Epidemiol. 1999;150:341–353. doi: 10.1093/oxfordjournals.aje.a010013. [DOI] [PubMed] [Google Scholar]
- 43.Kelley-Hedgepeth A, Lloyd-Jones DM, Colvin A. Ethnic differences in C-reactive protein concentrations. Clin Chem. 2008;54:1027–1037. doi: 10.1373/clinchem.2007.098996. [DOI] [PubMed] [Google Scholar]
- 44.Packard CJ. Lipoprotein-associated phospholipase A2 as a biomarker of coronary heart disease and a therapeutic target. Curr Opin Cardiol. 2009;24:358–363. doi: 10.1097/HCO.0b013e32832bcb22. [DOI] [PubMed] [Google Scholar]
- 45.Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW. C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol. 1999;19:972–978. doi: 10.1161/01.atv.19.4.972. [DOI] [PubMed] [Google Scholar]
- 46.Lawlor DA, Smith GD, Rumley A, Lowe GD, Ebrahim S. Associations of fibrinogen and C-reactive protein with prevalent and incident coronary heart disease are attenuated by adjustment for confounding factors. British Women's Heart and Health Study. Thromb Haemost. 2005;93:955–963. doi: 10.1160/TH04-12-0805. [DOI] [PubMed] [Google Scholar]
- 47.Danesh J, Saracci R, Berglund G. EPIC-Heart: the cardiovascular component of a prospective study of nutritional, lifestyle and biological factors in 520 000 middle-aged participants from 10 European countries. Eur J Epidemiol. 2007;22:129–141. doi: 10.1007/s10654-006-9096-8. [DOI] [PubMed] [Google Scholar]
- 48.Fibrinogen Studies Collaboration. Pennells L, White IR, Wood AM, Kaptoge S, Sarwar N. Measures to assess the prognostic ability of the stratified Cox proportional hazards model. Stat Med. 2009;28:389–411. doi: 10.1002/sim.3378. [DOI] [PubMed] [Google Scholar]
- 49.Pencina MJ, D'Agostino RB, Sr, D'Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172. doi: 10.1002/sim.2929. [DOI] [PubMed] [Google Scholar]
- 50.Shah T, Casas JP, Cooper JA. Critical appraisal of CRP measurement for the prediction of coronary heart disease events: new data and systematic review of 31 prospective cohorts. Int J Epidemiol. 2009;38:217–231. doi: 10.1093/ije/dyn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.