

Abstract
Largazole is a macrocyclic depsipeptide originally isolated from the marine cyanobacterium Symploca sp., which is indigenous to the warm, blue-green waters of Key Largo, Florida (whence largazole derives its name). Largazole contains an unusual thiazoline-thiazole ring system that rigidifies its macrocyclic skeleton, and it also contains a lipophilic thioester side chain. Hydrolysis of the thioester in vivo yields largazole thiol, which exhibits remarkable antiproliferative effects and is believed to be the most potent inhibitor of the metal-dependent histone deacetylases (HDACs). Here, the 2.14 Å-resolution crystal structure of the HDAC8-largazole thiol complex is the first of an HDAC complexed with a macrocyclic inhibitor and reveals that ideal thiolate-zinc coordination geometry is the key chemical feature responsible for its exceptional affinity and biological activity. Notably, the core structure of largazole is conserved in romidepsin, a depsipeptide natural product formulated as the drug Istodax® recently approved for cancer chemotherapy. Accordingly, the structure of the HDAC8-largazole thiol complex is the first to illustrate the mode of action of a new class of therapeutically important HDAC inhibitors.
Histone deacetylases (HDACs) catalyze the hydrolysis of acetylated lysine side chains in histone and non-histone proteins, and these enzymes are implicated in a number of biological processes such as cell differentiation, proliferation, senescence, and apoptosis1-3. The metal-dependent enzymes are classified by amino acid sequence relationships as class I HDACs (1, 2, 3, and 8), class IIa HDACs (4, 5, 7, and 9), class IIb HDACs (6 and 10), and the class IV enzyme, HDAC114. These HDACs adopt the α/β fold first observed in arginase, a metalloenzyme that utilizes a Mn2+2 cluster to catalyze L-arginine hydrolysis5. However, the metal-dependent HDACs utilize only a single metal ion, either Zn2+ or Fe2+ in vivo, for catalytic function6. Aberrant HDAC activity is found in various diseases, most notably cancer, making these enzymes critical targets for therapeutic intervention7-9.
HDAC inhibitors block the proliferation of tumor cells by inducing cell differentiation, cell cycle arrest, and/or apoptosis, and these compounds comprise some of the leading therapies approved or in clinical trials for cancer chemotherapy7-11. The primary affinity determinant of an HDAC inhibitor is a functional group that coordinates to the active site Zn2+ ion, such as a hydroxamic acid. A hydroxamic acid will ionize to form an exceptionally stable 5-membered ring chelate with the active site Zn2+ ion, as first demonstrated in a thermolysin-hydroxamate complex12. Perhaps the best known hydroxamic acid inhibitor of the HDACs is suberoy-lanilide hydroxamic acid (Zolinza®), which was the first HDAC inhibitor approved for cancer chemotherapy13.
The Zn2+-binding moiety of an HDAC inhibitor is tethered to a “capping group” that interacts with the mouth of the active site cleft. The most structurally complex capping groups are found in macrocyclic peptide and depsipeptide inhibitors (a depsipeptide contains both amide and ester linkages)7. For example, romidepsin (Istodax®, Fig. 1) is a macrocyclic depsipeptide that was recently approved for the treatment of cutaneous T-cell lymphoma14,15. Romidepsin itself is actually a prodrug; upon disulfide bond reduction in vivo, one of the romidepsin thiol side chains is proposed to coordinate to the active site Zn2+ ion15. However, no crystal structure is available to confirm this proposal.
Figure 1.

The disulfide bond of romidepsin is reduced while the thioester linkage of largazole is hydrolyzed to form potent depsipeptide thiol inhibitors of HDACs. Structurally identical portions of each inhibitor are highlighted in red.
The 16-membered macrocyclic ring of romidepsin is comparable to that of the recently-identified marine natural product largazole (Fig. 1), a cyclic depsipeptide originally isolated from the cyanobacterium Symploca sp. indigenous to Key Largo, Florida16. In contrast with romidepsin, largazole contains nonpeptidic thiazole and 4-methylthiazoline groups that rigidify the macrocyclic ring. Like romidepsin, largazole is a prodrug; hydrolysis of its thioester side chain in vivo yields a free thiol group capable of coordinating to the catalytic Zn2+ ion of HDAC enzymes. Indeed, largazole thiol is believed to be the most potent inhibitor known of HDAC enzymes17, exhibiting low nanomolar inhibitory activity against several HDAC enzymes17,18 and remarkable antiproliferative effects16. Largazole was recently hailed in Newsweek as the latest victory in bioprospecting the vast gold mine of marine natural products for new disease therapies.19
We now report the X-ray crystal structure of HDAC8 complexed with largazole thiol at 2.14 Å resolution (Fig. 2); structure determination statistics are recorded in Table S1. This is the first structure of an HDAC complex with a macrocyclic depsipeptide inhibitor and the first structure of an HDAC complex in which thiolate-Zn2+ coordination is observed. Largazole thiol binds to each monomer in the asymmetric unit of the crystal with full occupancy and thermal B factors comparable to those of surrounding residues. The electron density map in Fig. 3a shows that the macrocyclic skeleton of the depsipeptide caps the mouth of the active site. The macrocyclic skeleton undergoes minimal conformational changes upon binding to HDAC8, since its backbone conformation is very similar to that of the uncomplexed macrocycle20. Thus, the thiazoline-thiazole moiety rigidifies the macrocyclic ring with a pre-formed conformation that is ideal for binding to HDAC8.
Figure 2.
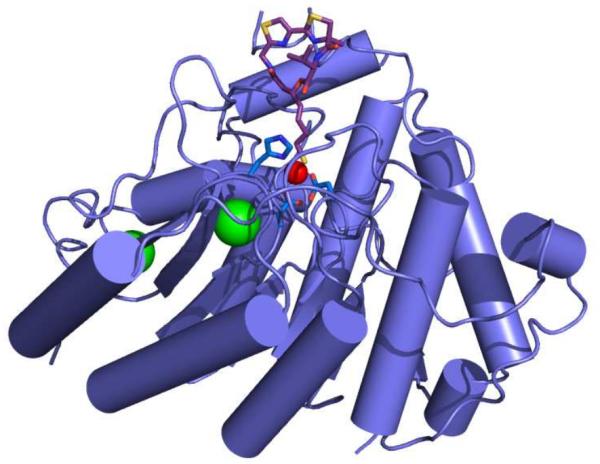
HDAC8-largazole thiol complex. The catalytic Zn2+ ion (red sphere) is coordinated by D178, H180, and D267 (blue sticks). Largazole thiol is shown as a stick figure (C = magenta, N = blue, O = red, and S = yellow). Structural K+ ions appear as green spheres.
Figure 3.
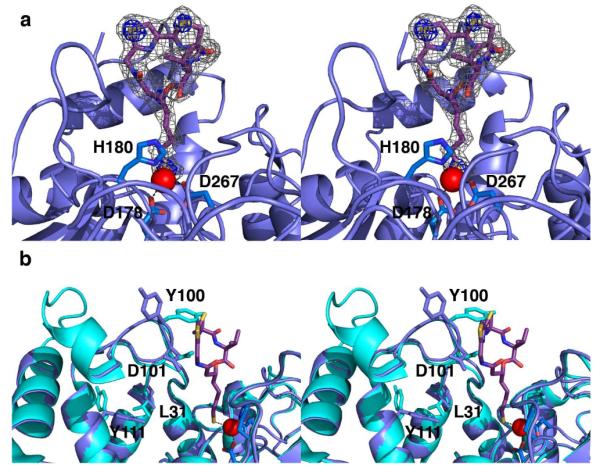
(a) Simulated annealing omit map contoured at 3.0 σ (gray mesh) showing largazole thiol bound in the active site of monomer A; contours at 8.0 σ (blue mesh) confirm the positions of electron-rich sulfur atoms. Atoms are color-coded as in Fig. 2, and metal coordination interactions are indicated by black dotted lines. (b) Superposition of the HDAC8-largazole complex (blue; largazole is colored as in Fig. 2) and the HDAC8-substrate complex (cyan; PDB code 3EWF, less substrate atoms).
Although no conformational changes in largazole are required for enzyme-inhibitor complexation, considerable conformational changes are required by HDAC8 to accommodate the binding of the rigid and bulky inhibitor. Most prominent are conformational changes in the L2 loop, specifically L98-F109, and especially Y100 (Fig. 3b). The Cα of Y100 shifts ~2 Å from its position in the H143A HDAC8-substrate complex21, and the side chain rotates nearly 180°. This conformational change is the direct consequence of inhibitor binding and is not observed in HDAC8 complexes with smaller inhibitors. Additionally, D101, a highly conserved residue that functions in substrate binding21,22, also undergoes a conformational change to accommodate inhibitor binding. Previously unobserved conformational changes that accommodate the binding of the bulky depsipeptide may reflect those that accommodate the large protein substrates of HDAC8 in vivo.
Additional conformational changes are evident in the L1 and L2 loops (Fig. 3b). The protein backbone in the L1 loop region, specifically L31-K36, shifts ~1 Å as a result of the movement of D101 (the influence of D101 on the conformation of L31 has been described21). These structural changes also affect Y111, which rotates approximately 145°. Apart from the conformational change of Y100, the conformational changes of D101, L31, and Y111 are similar to those triggered by the binding of the hydroxamate inhibitor M344 to HDAC821. Inhibitor atoms make hydrogen bond and polar interactions with D101, Y306, and two water molecules, as well as van der Waals interactions with numerous active site residues. Selected enzyme-inhibitor interactions are listed in Table S2.
The thiol side chain of largazole extends deep into the active site cleft, where the thiol moiety is likely ionized as the negatively-charged thiolate anion as it coordinates to the catalytic Zn2+ ion. The overall metal coordination geometry is nearly perfectly tetrahedral, with ligand-Zn2+-ligand angles ranging on average 107.6°-111.8°. The thiolate moiety exhibits preferred thiolate-metal coordination geometry23 with a thiolate S-Zn2+ separation of 2.3 Å, a C-S-Zn2+ angle of 97.5°, and a C-C-S-Zn2+ dihedral angle of 92.4° (values averaged across monomers A and B in the asymmetric unit). Ideal metal coordination geometry presumably makes a substantial contribution to enzyme-inhibitor affinity.
The structure of the HDAC8-largazole thiol complex provides a foundation for understanding structure-affinity relationships in myriad largazole derivatives recently synthesized and studied in various laboratories. For example, altering the length of the thiol side chain, converting the side chain olefin from a trans to a cis configuration, or changing the stereochemistry of the macrocycle-side chain linkage from (S) to (R), results in significant affinity losses24,25. Each of these structural changes would compromise the ideal Zn2+ coordination geometry observed in the HDAC8-largazole complex. In contrast, substitution of the methyl group of the 4-methylthiazoline moiety with a hydrogen atom has essentially no effect on affinity17; the structure of the enzyme-inhibitor complex shows that this methyl group is oriented parallel to, but does not contact, the protein surface. Possibly, modification with longer and/or larger substituents at this position would allow for the capture of additional affinity interactions on the protein surface.
Other studies demonstrate that the L-valine moiety of largazole thiol can be substituted with L-tyrosine, L-alanine, or glycine without significant loss of affinity24-26. It is clear in Fig. 3 that the L-valine side chain is pointing directly out toward solvent and does not directly influence the enzyme-inhibitor interface, so different L-amino acids (or possibly D-amino acids, or α,α-disubstituted amino acids) might be tolerated at this position as long as they do not perturb the conformation of the macrocyclic ring. Intriguingly, substitution of a pyridine ring for the thiazole ring enhances affinity 3-4 fold17. The structure of the enzyme-inhibitor complex shows that the thiazole ring is oriented away from the protein structure toward solvent, so it is possible that this position, too, could tolerate additional substitution without compromising affinity. Finally, given that the rigid thiazoline-thiazole system triggers the conformational change of Y100 toward solvent, it is perhaps not surprising that substitution of an oxazoline-oxazole system likely does the same while preserving or enhancing affinity to different HDAC isozymes17.
In closing, we note that the structure of the HDAC8-largazole thiol complex also provides a framework for understanding the binding of the drug romidepsin. Given that romidepsin and largazole share the core depsipeptide thiol structural motif (Fig. 1), it is reasonable to conclude that HDAC inhibition by romidepsin is similarly characterized by ideal thiolate-Zn2+ coordination geometry as observed in the HDAC8-largazole complex. Accordingly, the HDAC8-romidepsin complex is readily modeled based on the crystal structure coordinates of largazole (Fig. 4). This model will be a useful guide for the design of second-generation romidepsin analogues. Given that the active site mouths of HDAC isozymes can differ from one another, the structure-based derivatization of macrocyclic depsipeptide scaffolds may lead to designer depsipeptides capable of isozyme-specific inhibition, a crucial goal of HDAC inhibitor design7. This possibility will be addressed in future studies.
Figure 4.
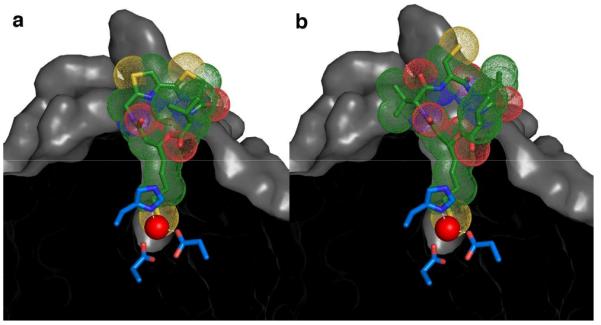
The experimentally determined structure of the HDAC8-largazole thiol complex (a) serves as a template for modeling the HDAC8-romidepsin thiol complex (b) simply by superimposing conserved depsipeptide features (i.e., the red portions of each inhibitor as illustrated in Fig. 1).
Supplementary Material
ACKNOWLEDGMENT
Crystallographic data for this study were measured at beam-line X29 of the National Synchrotron Light Source. Financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources of the National Institutes of Health grant number P41RR012408. K.E.C. and D.P.D. also thank Dr. Monica Ilies for assistance with largazole deprotection. This work was supported by the National Institutes of Health Grants GM49758 (D.W.C.) and CA110246 (A.J.P.).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Largazole preparation; structure determination, refinement, and crystallographic statistics (Table S1) for the HDAC8-largazole complex; and Table S2 of selected enzyme-inhibitor interactions. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The atomic coordinates and crystallographic structure factors of the HDAC8-largazole thiol complex have been deposited in the Protein Data Bank with accession code 3RQD.
REFERENCES
- (1).De Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Biochem. J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Haberland M, Montgomery RL, Olson EN. Nature Rev. Gen. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Emanuele S, Lauricella M, Tesoriere G. Int. J. Oncol. 2008;33:637–646. [PubMed] [Google Scholar]
- (4).Gregoretti IV, Lee YM, Goodson HV. J. Mol. Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- (5).Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Nature. 1996;383:554–557. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- (6).Gantt SL, Gattis SG, Fierke CA. Biochemistry. 2006;45:6170–6178. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- (7).Mwakwari SC, Patil V, Guerrant W, Oyelere AK. Curr. Topics Med. Chem. 2010;10:1423–1440. doi: 10.2174/156802610792232079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Marks PA, Wu WS. J. Cell. Biochem. 2009;107:600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lane AA, Chabner BA. J. Clin. Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- (10).Mei S, Ho AD, Mahlknecht U. Int. J. Oncol. 2004;25:1509–1519. [PubMed] [Google Scholar]
- (11).Bolden JE, Peart MJ, Johnstone RW. Nature Rev. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- (12).Holmes MA, Matthews BW. Biochemistry. 1981;20:6912–6920. doi: 10.1021/bi00527a026. [DOI] [PubMed] [Google Scholar]
- (13).Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. The Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- (14).Guan P, Fang H. Drug Disc. Ther. 2010;4:388–391. [PubMed] [Google Scholar]
- (15).Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- (16).Taori K, Paul VJ, Luesch H. J. Am. Chem. Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- (17).Bowers AA, West N, Newkirk TL, Troutman-Youngman AE, Schreiber SL, Wiest O, Bradner JE, Williams RM. Org. Lett. 2009;11:1301–1304. doi: 10.1021/ol900078k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ying Y, Taori K, Kim H, Hong J, Luesch H. J. Am. Chem. Soc. 2008;130:8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- (19).Behar M. Newsweek. 2010;156:40–41. [Google Scholar]
- (20).Seiser T, Kamena F, Cramer N. Angew. Chem. 2008;47:6483–6485. doi: 10.1002/anie.200802043. [DOI] [PubMed] [Google Scholar]
- (21).Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfí A, De Francesco R, Steinkühler C, Di Marco S. Embo Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chakrabarti P. Biochemistry. 1989;28:6081–6085. doi: 10.1021/bi00440a052. [DOI] [PubMed] [Google Scholar]
- (24).Zeng X, Yin B, Hu Z, Liao C, Liu J, Li S, Li Z, Nicklaus MC, Zhou G, Jiang S. Org. Lett. 2010;12:1368–1371. doi: 10.1021/ol100308a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ying Y, Liu Y, Byeon SR, Kim H, Luesch H, Hong J. Org. Lett. 2008;10:4021–4024. doi: 10.1021/ol801532s. [DOI] [PubMed] [Google Scholar]
- (26).Benelkebir H, Marie S, Hayden AL, Lyle J, Loadman PM, Crabb SJ, Packham G, Ganesan A. Bioorg. Med. Chem. 2011;15:3650–3658. doi: 10.1016/j.bmc.2011.02.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.