

Abstract
The estrogen receptor (ER) pathway and the epidermal growth factor receptor (EGFR) pathway play pivotal roles in breast cancer progression. Targeted therapies able to intercept ER or signaling downstream to EGFR and its kin, HER2, are routinely used to treat distinct groups of breast cancer patients. However, patient responses are limited by resistance to endocrine therapy, which may be due to compensatory HER2/EGFR signaling. This raises the possibility that simultaneous interception of HER2 and ER may enhance therapeutic efficacy. To address the question, we treated breast cancer cells with both fulvestrant (ICI 182780), an ER antagonist with no agonist effects, and lapatinib, an orally available tyrosine kinase inhibitor specific to EGFR and HER2. Our results indicate that the combination of drugs is especially effective when applied to HER2-overexpressing, ER-positive cancer cells. Interestingly, fulvestrant activated the mitogen-activated protein kinase (MAPK) pathway of these cells, but complete inhibition of MAPK signaling was observed on cotreatment with lapatinib. Taken together, our observations reinforce the possibility that the effectiveness of combining anti-ER and anti-HER2/EGFR drugs may be especially effective on a relatively small subtype of HER2-overexpressing, ER-positive tumors of the breast.
Introduction
Targeted therapies are in common clinical use for the treatment of breast cancer. Approximately 70% of breast cancers are estrogen receptor α (ERα)-positive [1,2], and 20% to 25% of mammary tumors present overexpression of HER2 (also called ErbB-2/neu), a receptor tyrosine kinase related to epidermal growth factor receptor (EGFR) [3]. Although most ERα-positive mammary tumors initially respond to therapy with antiestrogens such as tamoxifen, acquired patient resistance severely limits therapeutic efficacy [4,5]. Several mechanisms of endocrine resistance have been proposed [6]. They include deregulation of various components of the ER pathway itself, alterations in molecules responsible for cell cycle and cell survival, and the activation of escape pathways that can provide tumors with alternative proliferative and survival stimuli. Among these, increased expression or signaling of growth factor receptor pathways has been associated with both experimental and clinical resistance to endocrine therapy [7–9].
The ERBB family of receptor tyrosine kinases plays important roles in the development of resistance to endocrine therapy [10–14]. This family consists of four members, namely, EGFR, HER2/ERBB2, HER3/ERBB4, and HER4/ERBB4, which execute multiple functions such as cell growth, differentiation, motility, and regulation of apoptosis, through a complex interplay of homodimerization and heterodimerization of the four ERBB members [15]. HER2 is the main signal amplifier of this growth factor receptor family, and it was previously observed to regulate ERα expression and activity through neuregulins, HER3/HER4 ligands, which stimulate phosphoinositol 3-kinase signaling to protein kinase B [16]. In addition, both ErbB members and ERα use the mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) pathway as a major route of cellular activation [17].
Lapatinib (GW 2016) is a potent inhibitor of both the HER2 and the EGFR tyrosine kinase catalytic functions [18]. It has been shown that lapatinib cooperates with tamoxifen by inhibiting both cell proliferation and estrogen-dependent gene expression in breast cancer cells [19]. Moreover, when combined with lapatinib, letrozole, an aromatase inhibitor, significantly improved progression-free survival of patients with metastatic breast cancer that coexpresses hormone receptors and HER2 [20,21]. Fulvestrant (ICI 182780) is a pure antiestrogen, a steroidal 7-α-alkylsulphinyl analog of 17β-estradiol, which is structurally distinct from the nonsteroidal selective ER modulator tamoxifen [22]. Fulvestrant competitively inhibits binding of estradiol to the ER, thereby inducing a conformational change within the receptor, different from that of tamoxifen or estradiol [23]. Trastuzumab and mAb-431 are monoclonal antibodies against the HER2 receptor, of which trastuzumab is in common clinical use [24] and mAb-431 is a murine antibody specific to human HER2 [25]. As ERα and growth factor signaling pathways interact, combining fulvestrant and lapatinib/anti-HER2 mAbs might present a useful approach for targeting breast tumors coexpressing ERα and HER2. In this work, we tested whether the combination of lapatinib and fulvestrant is superior to the respective single treatments on ERα-positive mammary cell lines with variable levels of HER2, by analyzing effects on cell growth, cell cycle distribution, apoptosis, and protein expression levels. The results we present propose that the drug combination is especially effective when applied to HER2-overexpressing, ER-positive cancer cells, but it may also affect cancer cells expressing moderate levels of HER2.
Materials and Methods
Materials
Lapatinib was provided by GlaxoSmithKline (Brentford, UK). Fulvestrant (ICI 182,780) was supplied by Tocris Bioscience (Tocris Cookson Ltd, Bristol, UK). Trastuzumab was provided by Genentech, Inc (South San Francisco, CA). The previously described [25] monoclonal antibody to HER2, mAb-431, was produced by Adar Biotech (Rehovot, Israel). Antibodies against PDK1, p-PDK1, AKT-1, and p-AKT (Ser473) were purchased from Cell Signaling Technology, Inc (Boston, MA). Antibodies against ERα, ERK1, and p-ERK1/2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The secondary antibodies antirabbit-antibody HRP-linked immunoglobulin G was from Cell Signaling Technology, Inc, and the stabilized goat antimouse HRP-conjugated antibody was from Pierce (Rockford, IL).
Cell Cultures and Proliferation Assays
Cells were grown in Dulbecco modified Eagle medium (Sigma-Aldrich, Rehovot, Israel). Fetal bovine serum(10%) was added together with 1% penicillin/streptomycin. BT474, T47D, and MCF-7 cells were supplied by the European Collection of Cell Cultures (Salisbury, UK). Before the MTT tests, cells were grown for 24 hours in 96-well plates at a concentration of 3000 cells per well. In all experiments, we used phenol red-free medium, containing 1% charcoal-depleted fetal calf serum (PERBIO Science, Fischer, Germany) and penicillin/streptomycin at a final concentration of 1%. Estrogen was added to a final concentration of 10 nM. The Sigma M2128 Cell proliferation Kit I (MTT) was used according to the manufacturer's instructions. The XTT Cell Proliferation Assay Kit was obtained from Biological Industries (Beit HaEmek, Israel). The percentage of viable cells was calculated as follows: ([absorption sample - absorption blank] / [absorption control - absorption blank]) x 100.
Immunoblot Analyses
Total protein concentration of samples was evaluated by the BCA protein assay kit (Perbio Science Deutschland GmbH, Bonn, Germany). A total of 40 µg of protein was separated by 4% to 12% gradient SDS-PAGE and transferred to poly(vinylidene fluoride) membranes (BIO-RAD, Muenchen, Germany). Blocking was performed using PBS-Tween-20 (0.1%) with Roti-Block (Roth, Karlsruhe, Germany) at 4°C for 12 hours. Primary antibodies were incubated for 2 hours at room temperature. After thorough washing, membranes were incubated for 90 minutes with peroxidase-conjugated secondary antibodies. Signals were detected by chemiluminescence using the ECL detection system (Amersham Pharmacia Biotech, Freiburg, Germany).
Flow Cytometry Analyses
Cells were treated for 48 hours with dimethyl sulfoxide, lapatinib (0.1 µM), fulvestrant (0.1 µM), or their combination (0.1 µM each). After collection, cells were washed with saline and fixed with ethanol. Cells were again washed with saline and treated with solution 1, containing 100 µg/L RNase, 3.4 mM trisodium citrate, 0.1% Igepal, 1.5 mM sperm inetetrahydrochloride, and 0.5 mM tris(hydroxymethyl)aminomethane. Subsequently, cells were stained with solution 2 (208 mg/L propidium iodide, 3.4 mM trisodium citrate, 0.1% Igepal, 1.5 mM sperminetetrahydrochloride, and 0.5 mM tris-[hydroxymethyl]-aminomethane) and incubated for 15 minutes at room temperature. Around 20,000 cells were analyzed using a flow cytometer (FACS Calibur; Becton Dickinson, Heidelberg, Germany). Data were analyzed using ModFit software (Verity Software House, Topsham, ME) and confirmed in at least two independent experiments.
Morphogenesis Assays
Eight-well chamber slides (BD Biosciences) were coated using 35 µl of Matrigel per well. After regular growth in tissue culture dishes, cells were trypsinized and resuspended in phenol red-free assay medium supplemented with Matrigel (5%) and serum (5%).
Modeling and Quantification of Drug Synergy
Nonparametric modeling methods were applied to quantify synergism between two drugs. For each viability curve, the data set was interpolated by a linear piece-wise function. We then calculated (by means of a numerical integration) this interpolating curve and the horizontal line of 100% viability. These two steps were performed using Matlab “interp1” and “quad” functions. The resulting area, called viability area, can quantify the activity of the drug administered. The larger the calculated area is, the more effective the treatment applied will be. We assumed that if two drugs have a synergistic effect, the area in the case of drug combination should be closed by the mean of the single areas of the two drugs taken alone. In the case where the area resulting from the combination is larger than this mean, we considered that the effect was synergistic. The percentage of synergism between the two drugs was expressed as the rate of increase of the area of the combination with respect to the mean of the single areas of the two drugs when administered alone.
Results
In Vitro Growth-Inhibitory Effects of Lapatinib, Fulvestrant, and Their Combination on Mammary Tumor Cell Lines
To test the possible interactions between an anti-HER2 drug (lapatinib) and an anti-ER drug (fulvestrant) on the viability of ER-positive breast cancer, we used three cell lines that differ in their HER2 expression levels, namely MCF7 (low level of expression), T47D (medium level of expression), and BT474 cells, which overexpress HER2. Figure 1A presents an immunoblot showing the relative expression levels of HER2 and ER in the three cell lines, which reflect the general reciprocal relations between HER2 and ER levels. Cells were cultured for 24 hours and were thereafter treated for 72 hours with increasing concentrations of lapatinib and/or fulvestrant (Figure 1B). The combination of lapatinib and fulvestrant exerted a superior inhibitory effect on viability of BT474 cells at all drug concentrations we tested. In T47D cells, a superior effect was observed only with relatively high concentrations of the drug combination, and in MCF7 cells, we observed superiority of the combination at low drug concentrations, but at higher concentrations, fulvestrant showed a stronger effect than lapatinib. In conclusion, the drug combination exerted a superior combinatorial effect on ER-positive breast cancer cells according to the levels of HER2: the combination was stronger on BT474 cells than in the medium and low expressors T47D and MCF7 cells, respectively. The latter cells express relatively high levels of ER and, accordingly, displayed an enhanced sensitivity to high fulvestrant concentrations than to the combination of drugs. To address the question whether the improved combinatorial activity of lapatinib and fulvestrant could be due to the enhanced degradation of the ER, we probed cell extracts for ERα after 24 hours of treatment with the drugs. Our preliminary results (data not shown) indicated that fulvestrant, unlike lapatinib, caused partial degradation of ERα in T47D and MCF7 cells, but complete disappearance of ERα was noted after a combined treatment with the drugs. These observations therefore raise the possibility that the addition of an anti-HER2 targeting drug to fulvestrant may decrease ER levels. In addition, we compared the effects of fulvestrant and tamoxifen on the viability of BT474 and T47D cells but observed only minor differences (Figure 1C).
Figure 1.

Growth-inhibitory effects of lapatinib and fulvestrant on cultures of breast cancer cell lines. (A) BT474, T47D, and MCF7 cells were grown to semiconfluence. Afterward, cells were harvested and cleared lysates subjected to immunoblot analysis using the indicated antibodies. (B) BT474, T47D, and MCF7 cells were cultured for 24 hours; afterward, they were treated for 72 hours in the presence of increasing drug concentrations, as indicated. Thereafter, an MTT assay was performed as described in the Materials and Methods section, and cell viability was presented relative to control, untreated cells. Results are presented as mean and SD (bars) of six wells. The experiment was performed thrice. (C) BT474 and T47D cells were cultured for 24 hours; afterward, they were treated for 48 hours in the presence of increasing concentrations of the indicated drugs. Thereafter, an XTT assay was performed, and cell viability was presented relative to control, untreated cells. Results are presented as relative control, untreated cells. Mean and SD (bars) of six wells are shown. The experiment was repeated thrice.
To determine whether the observed superior effect of the combination of lapatinib and fulvestrant on BT474 cells was additive or synergistic, we quantified the cell viability results and used the nonparametric method as explained before. After a combinatorial treatment, a strong increase of the viability area could be observed in BT474 cells, and a smaller increase in comparison to single treatments could be observed in T47D cells. In MCF7 cells, the area with the highest viability was obtained by treating with fulvestrant (Figure 2A). To calculate whether the observed viability effects of the combinatorial drug treatments of BT474 and T47D cells were additive or synergistic, we set the estimated additive effect as a normalization reference (0%). The enhancement of inhibition of cell viability beyond the additive effect was 119% for BT474 cells, 21.8% for T47D cells, and 3.2% for MCF7 cells (Figure 2B). These results indicated a clear synergistic effect of the combination of lapatinib and fulvestrant when applied on BT474 cells, along with a mild synergistic effect on T47D cells.
Figure 2.

The combination of lapatinib and fulvestrant displays distinct effects on viability of ER-positive breast cancer cells. (A) BT474, T47D, and MCF7 cells were cultured for 24 hours and afterward treated with the indicated drugs for 72 hours. Thereafter, an MTT assay was performed, and cell viability was presented relative to control, untreated cells. The viability area was calculated from cell viability assays performed in the presence of increasing concentrations of fulvestrant, lapatinib, or their combination. (B) The relative increase of a combinatorial treatment of BT474, T47D, and MCF7 cells with lapatinib and fulvestrant (in comparison to additive effects) was calculated based on the assays presented in Figure 1.
Effects of Lapatinib, Fulvestrant, and Their Combination on the Cell Cycle Distribution of BT474, T47D, and MCF7 Cells
Ligand-occupied ER is known to promote cell cycle progression [26]. Hence, in the next step, we tested whether the observed drug-induced inhibition of cell viability was due to the effects on cell cycle distribution. BT474, T47D, and MCF7 cells were cultured for 48 hours with lapatinib (100 nM), fulvestrant (100 nM), or with a combination of both drugs, and the cells were subjected to cell sorting. An increase in the fraction of cells found within the G1 phase and a concomitant decrease of BT474 cells within the S phase was observed after treatment with fulvestrant or with the drug combination (Figure 3, upper panel). In contrast, the cell cycle distribution of T47D cells was less affected by the drug (Figure 3, middle panel), whereas in MCF7 cells, which express high levels of ER, fulvestrant and its combination with lapatinib exerted the most pronounced effects (Figure 3, bottom panel; a reduction in the fraction of cells in the G2 phase plus those from the S phase, from 19.4% of untreated cells to 5.2% of fulvestrant-treated cells). Interestingly, the effects of lapatinib on cell cycle distribution were much smaller than the effects of fulvestrant. In conclusion, the inhibition of ERα signaling in MCF7 cells, which are estrogen dependent and highly express ER, showed the most pronounced effect on cell cycle distribution. This is in accordance with early observations that antiestrogens, such as tamoxifen, can effectively block estrogen-mediated cell cycle progression [26,27].
Figure 3.
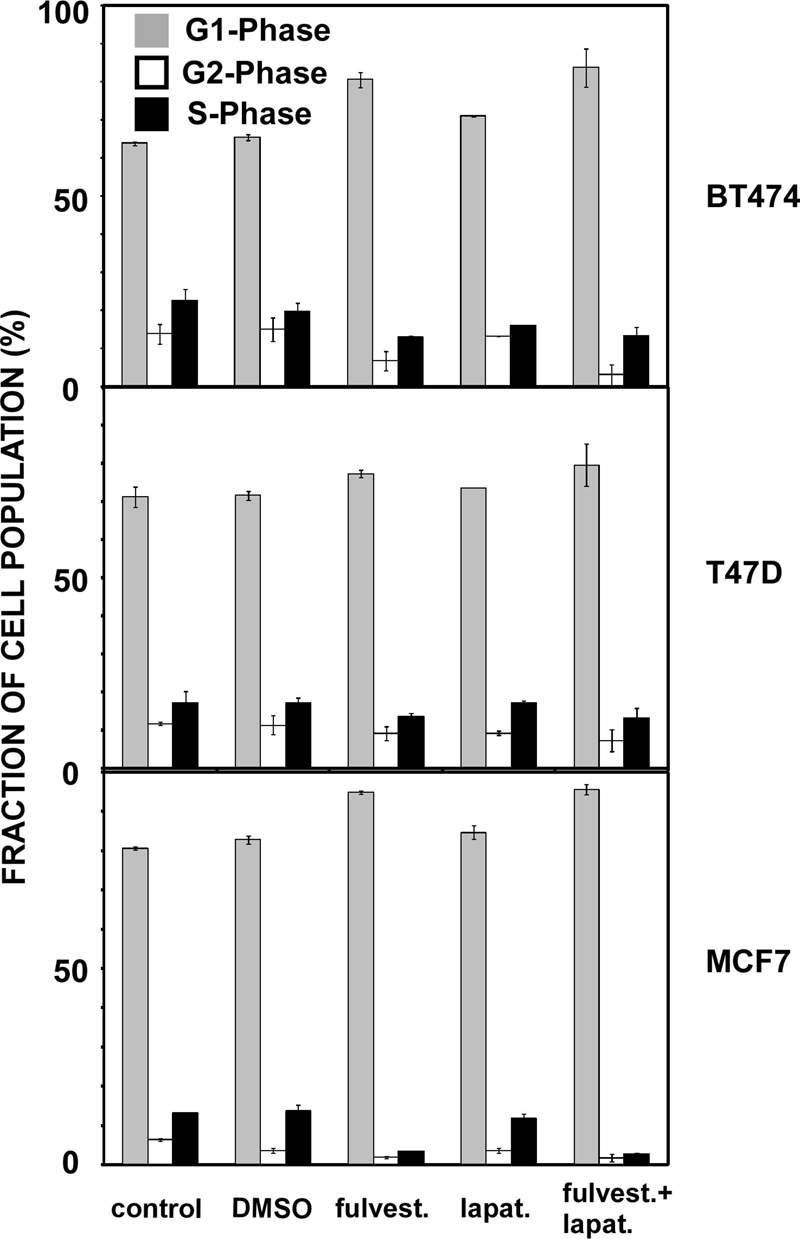
Effects of lapatinib, fulvestrant, and their combination on cell cycle distribution of ER-positive breast cancer cells. BT474, T47D, and MCF7 cells were cultured for 48 hours with lapatinib (100 nM), fulvestrant (100 nM), or their combination (100 nM each). Cells were harvested as described in the Materials and Methods section, and cell cycle assays were performed using 20,000 cells and a flow cytometer. Data analysis was performed using ModFit. Results are presented as mean and SD (bars) of three independent experiments.
Effects of Lapatinib, Fulvestrant, and Their Combination on Protein Kinase Signaling in Mammary Tumor Cells
Next, we examined the effects of lapatinib and fulvestrant on signaling along the main protein kinase pathways involved in HER2 signaling and endocrine resistance, namely, AKT and PDK1, as well as ERK, after single and combined treatments with lapatinib and fulvestrant. BT474 (Figure 4A), T47D (Figure 4A), and MCF7 (Figure 4B) cells were grown to semiconfluence and treated for 2 hours with relatively high concentrations of fulvestrant (1 µM), lapatinib (2 µM), or fulvestrant (1 µM) plus lapatinib (2 µM). Afterwards, cells were harvested and subjected to immunoblot analysis with the indicated antibodies.
Figure 4.

Effects of lapatinib, fulvestrant, and their combination on intracellular signaling of ER-positive breast cancer cells. Semiconfluent cultures of BT474 and T47D (A) and MCF7 (B) cells were treated for 2 hours with fulvestrant (1 µM), lapatinib (2 µM), or fulvestrant (1 µM) plus lapatinib (2 µM). Afterward, cells were harvested, and whole lysates were subjected to immunoblot analysis with antibodies specific to the phosphorylated forms of the indicated kinases (e.g., p-ERK) or to the respective general forms (e.g., g-ERK).
When tested on BT474 cells, lapatinib significantly reduced phosphorylation of ERK and AKT, but not PDK1. Nevertheless, fulvestrant slightly increased phosphorylation of ERK and AKT but weakly decreased PDK1 phosphorylation. Interestingly, the combination of lapatinib and fulvestrant almost erased phosphorylated ERK and AKT signals (Figure 4A), which may explain the growth-inhibitory effect of this combination on HER2-overexpressing cells. The levels of p-PDK1 were also reduced after treatment with the drug combination. In T47D cells (Figure 4A), lapatinib and the combination of fulvestrant and lapatinib reduced the levels of phosphorylated ERK to a similar extent, whereas fulvestrant and fulvestrant plus lapatinib reduced phosphorylation levels of PDK1 to a similar extent, suggesting distinct downstream effects of lapatinib and fulvestrant in T47D cells. In MCF7 cells (Figure 4B), the combination of drugs induced a stronger effect on the reduction of levels of phosphorylated ERK, whereas the levels of phosphorylated AKT and PDK1 were not affected. Interestingly, p-ERK levels increased under single fulvestrant treatment in BT474 cells (Figure 4A), pointing toward a compensatory ERK signaling when HER2 is highly expressed. This effect could be completely blocked by lapatinib. It is notable that BT474 cells exhibited higher sensitivity than the other two cell lines to the combination of lapatinib and fulvestrant, and these cells also showed the highest synergistic drug effect on cell viability (Figure 1A).
Compensatory activation of EGFR and concomitant ERK signaling were previously observed after treatment of mammary cells with tamoxifen [28]. Although fulvestrant is a complete ER antagonist, in contrast with the mixed activity of tamoxifen, the observed activation of ERK in fulvestrant-treated cells may reflect a shared compensatory mechanism, which is likely mediated by EGFR or HER2 because lapatinib completely blocked ERK activation in fulvestrant-treated cells.
Effects of Lapatinib, Fulvestrant, and Monoclonal Antibodies on the Proliferation of BT474 and T47D Cells
To extend the observations made using HER2-overexpressing BT474 cells and the moderate expressors, T47D, we tested whether the superior effect of the drug combination would be reflected by a mode of HER2 inhibition other than lapatinib. Hence, we tested proliferation of BT474 and T47D cells when treated with fulvestrant alone, or in combination with lapatinib, trastuzumab, or mAb-431, a murine anti-HER2 monoclonal antibody [25] (Figure 5). In consistency with our previous results, all combinations we tested were clearly superior to the respective single drug treatments when applied to BT474 cells. Likewise, T47D cells displayed much milder combinatorial effects, in line with their lower abundance of HER2: the combinations lapatinib-fulvestrant and trastuzumab-fulvestrant were more effective than the respective single treatments, whereas the impact of combining mAb-431 with fulvestrant was less effective. Interestingly, the fulvestrant-trastuzumab combination was more effective than the fulvestrant-lapatinib pair, probably due to the relatively low concentration of lapatinib (25 nM) used in this experiment. Notably, at this concentration, lapatinib was ineffective when singly applied, but it clearly synergized with fulvestrant when the combination was applied on BT474 cells. In conclusion, these results propose that blocking HER2 by either a kinase inhibitor or a monoclonal antibody can synergize with an ER antagonist, especially when the combinations are applied on HER2-overexpressing breast cancer cells.
Figure 5.

Effects of lapatinib, anti-HER2 antibodies, and fulvestrant, as well as their combinations, on proliferation of BT474 and T47D breast cancer cells. BT474 and T47D cells were cultured for 24 hours and afterward treated for 48 hours with the indicated drugs. The following concentrations were used, either alone or in combinations: fulvestrant (125 nM), lapatinib (25 nM), mAb-431 (2.5 µg/ml), and trastuzumab (2.5 µg/ml). An XTT assay was performed as described in the Materials and Methods section, and the results are presented as percentage of control untreated cells. Mean and SD (bars) of triplicates are shown. The experiment was repeated thrice.
Effects of Lapatinib, Fulvestrant, and Monoclonal Anti-HER2 Antibodies on BT474 Mammospheres
We previously demonstrated that three-dimensional spheroids of mammary cells grown in extracellular matrix (Matrigel) offer sensitive readouts when comparing effects of drugs on cell viability [29]. Hence, to extend the observations made with cellular monolayers, we cultured BT474 as spheroids embedded in cellular matrix (Figure 6). On day 4, lapatinib (50 nM), trastuzumab (5 µg/ml), mAb-431 (5 µg/ml), fulvestrant (250 nM), or their combinations were added to the culture medium. The images of 15-day-old spheroids (Figure 6A) were used to calculate cross-sectional areas (Figure 6B; on average, 95 spheroids were used per measurement). As we previously demonstrated, the cross-sectional area of a spheroid is a reliable parameter of cell viability and proliferation. Remarkably, all three drug combinations we used, namely, trastuzumab plus lapatinib, fulvestrant plus lapatinib, and mAb-431 plus lapatinib, exhibited a statistically superior ability to reduce spheroid size in comparison with the respective single treatments, which were quite effective on their own. In line with viability assays performed in cell monolayers, the combinations of fulvestrant with either lapatinib or trastuzumab yielded maximal inhibitory effects in this assay. In summary, our observations indicate that the combination of fulvestrant and lapatinib is able to strongly reduce viability of ERα-positive, HER2-overexpressing BT474 breast cancer cells grown either in monolayers or in spheroids. This inhibition is attributable to an inhibitory effect of the drug mixture on two kinase cascades, ERK and AKT. Like lapatinib, monoclonal antibodies specific to HER2 enhanced the effect of the ER antagonist, thus confirming the molecular mechanism and propose ways to translate these observations to clinical applications.
Figure 6.

Effects of lapatinib, trastuzumab, mAb-431, fulvestrant, and their combinations on BT474 spheroids grown in the extracellular matrix. (A) BT474 cells were seeded in the Matrigel to form spheroids. The following treatments were initiated 4 days later: lapatinib (50 nM), trastuzumab (5 µg/ml), mAb-431 (5 µg/ml), fulvestrant (250 nM), as well as the indicated drug combinations. The respective media were refreshed every 4 days. Spheroid images shown were captured on day 15. (B) Images of 15-day-old BT474 spheroids, which were obtained and treated as in A, were analyzed, and cross-sectional areas were quantified using ImageJ (National Institutes of Health, Bethesda, MD). On average, 95 spheroids were analyzed after each treatment, and two independent experiments were performed. Each treatment was compared with the untreated control group using the Mann-Whitney test and SPSS (Armonk, NY). Bonferroni corrections of calculated P values were used. Asterisks refer to statistically significant differences.
Discussion
Approximately two-thirds of newly diagnosed breast tumors are ER-positive [1,2], and almost one quarter of all breast cancers overexpress HER2 [3]. Close to half of the latter group presents ER positivity, leading to enhanced cell proliferation. Both anti-endocrine- and anti-HER2-targeted therapies proved their clinical efficacy in the treatment of ER-positive and HER2-positive breast cancer, respectively. Nevertheless, primary and evolving (secondary) resistance severely limit anti-endocrine therapy [10,30,31]. Because resistance commonly entails activation of compensatory signaling pathways, understanding the underlying routes of growth factor signaling, as well as their cross talk and interfaces with the steroid hormone axis of breast cancer, represents a prerequisite for the development of new combinatorial therapeutic strategies able to improve clinical efficacy [6,32].
A recent clinical study examined the question whether the combination of an antihormonal agent and trastuzumab (without chemotherapy) could prove useful as a treatment of HER2/ER-positive metastatic breast cancer in postmenopausal patients [21]. Trastuzumab plus anastrozole improved outcomes in the examined subgroup in the median progression-free survival (4.8 vs 2.4 months). Yet, another recently reported clinical trial [20] examined the question whether the addition of lapatinib to letrozole in the treatment of postmenopausal breast cancer patients with HER2- and ER-positive metastatic breast cancer could provide added efficacy. In this trial, combination therapy compared with letrozole alone significantly enhanced progression-free survival (8.2 vs 3.0 months). Thus, there is experimental evidence that combining anti-HER2 and endocrine treatments is clinically beneficial. The results of our in vitro studies are in line with these observations as the maximal combinatorial effect was observed in BT474, representing HER2-overexpressing, ERα-positive breast cancer cells. Nevertheless, some effects of the drug combination were also observed with the other cell lines, although their HER2 levels are lower. Therefore, we assume that a drug combination strategy may be extended to ER-positive breast lesions, which express moderate levels of HER2. Notably, a recent clinical trial reported that a higher dose of fulvestrant (500 vs 250 mg/month) significantly increased progression-free survival of postmenopausal women with ER-positive advanced breast cancer, whose conditions progressed under prior endocrine therapy [33]. In relation to the in vitro results, it is relevant that higher doses of fulvestrant might also be beneficial in a combination treatment regimen because maximal antiproliferative effects of the drug combination were observed at relatively high fulvestrant doses in all three cell lines we tested.
Acquired resistance to tamoxifen was previously shown to be correlated with markedly increased abundance of EGFR and HER2 in the MCF7 xenograft model [10]. This study concluded that the EGFR/HER2 compensatory route might mediate tamoxifen resistance in ER-positive breast cancer, despite continued suppression of ER genomic functions under endocrine therapy. The bidirectional cross talk between growth factor receptors and hormone receptors seems responsible for another type of resistance, to lapatinib. A model system based on the HER2-overexpressing BT474 cells, which became resistant to lapatinib after chronic treatment, showed derepression of the transcription factor FOXO3a and consequent activation of ER signaling in the resistant cells [34]. To directly address the possible influence of HER2 abundance, our study compared the effects of lapatinib and fulvestrant on ER-positive breast cancer cell lines, which express HER2 at different levels. The results we obtained indicate that the best inhibitory action was achieved in BT474 cells, which moderately express ERα and overexpress HER2. In additional experiments, we were able to extend this observation to an alternative HER2-blocking strategy, namely, the combination of fulvestrant with either trastuzumab or mAb-431 (an inhibitor of HER2 dimerization), in both two-dimensional and three-dimensional cell culture formats. Combining lapatinib and fulvestrant also proved some superior efficacy compared with the single-drug applications in T47D cells, which moderately express HER2, implying that HER2 can influence cell growth and survival even before achieving full overexpression. In line with this observation, our previous experiments showed that targeting HER2 in non-HER2-overexpressing cell lines led to reduced cell viability and proliferation in two-dimensional and three-dimensional culture formats [29]. A recent work defined the EGF-upregulated transcriptional program in MCF7 cells and found it to be correlated with the most highly expressed genes in HER2-expressing breast cancer [35]. Interestingly, HER2-induced ERK signaling was previously observed to be important for the cytoplasmic localization of ERα, which is involved in the onset of tamoxifen resistance of HER2-overexpressing cell lines [36]. Our experiments showed an increase in the phosphorylated levels of ERK in BT474 cells after treatment with fulvestrant. This effect could be completely blocked by the combination of lapatinib and fulvestrant, indicating once again the importance of pathway cross talk and inhibition of complementary mechanisms.
In summary, we propose a dual combination strategy for the treatment of ER-positive breast tumors that overexpress HER2 but also for ER-positive breast tumors that express intermediate levels of the oncoprotein. The strategy comprises mixing of fulvestrant and either lapatinib or a monoclonal antibody against HER2. Theoretical considerations predict that simultaneous blockade of two major hubs of signaling networks would be very effective, especially when the inhibited hubs generate partly redundant signals [37], because such networks have not been trained to overcome uncommon perturbations [38]. Our in vitro studies support these notions and also predict that the combined treatment would delay onset of resistance to steroid hormone blockers. Future studies in vitro and in animals may improve our understanding as well as determine the exact level of HER2 abundance needed for synergistic drug interactions, paving the way for additional clinical trials combining anti-ER and anti-HER2 drugs.
Footnotes
The authors declare no conflict of interest. This work was supported by research grants from the Moross Cancer Institute, the Estate of David Levinson, the Estate of John M. Lang, the Goldhirsh Foundation, the US National Cancer Institute (CA072981), the Seventh Framework Program (FP7) of the European Commission, the Israel Science Foundation, the German Research Foundation (DFG), the Dr Miriam and Sheldon G. Adelson Medical Research Foundation, the Marc Rich Foundation for Education, Culture and Welfare, and the MD Moross Institute for Cancer Research. A.E. was supported by a MINERVA Fellowship. G.M. was supported by a Sergio Lombroso Fellowship.
References
- 1.Clark GM, McGuire WL. Steroid receptors and other prognostic factors in primary breast cancer. Semin Oncol. 1988;15:20–25. [PubMed] [Google Scholar]
- 2.Allred DC, Brown P, Medina D. The origins of estrogen receptor α-positive and estrogen receptor α-negative human breast cancer. Breast Cancer Res. 2004;6:240–245. doi: 10.1186/bcr938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al. Studies of the HER-2/neu protooncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 4.Schiff R, Massarweh SA, Shou J, Bharwani L, Mohsin SK, Osborne CK. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res. 2004;10:331S–336S. doi: 10.1158/1078-0432.ccr-031212. [DOI] [PubMed] [Google Scholar]
- 5.Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998;339:1609–1618. doi: 10.1056/NEJM199811263392207. [DOI] [PubMed] [Google Scholar]
- 6.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–247. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipton A, Leitzel K, Ali SM, Demers L, Harvey HA, Chaudri-Ross HA, Evans D, Lang R, Hackl W, Hamer P, et al. Serum HER-2/neu conversion to positive at the time of disease progression in patients with breast carcinoma on hormone therapy. Cancer. 2005;104:257–263. doi: 10.1002/cncr.21202. [DOI] [PubMed] [Google Scholar]
- 8.Meng S, Tripathy D, Shete S, Ashfaq R, Haley B, Perkins S, Beitsch P, Khan A, Euhus D, Osborne C, et al. HER-2 gene amplification can be acquired as breast cancer progresses. Proc Natl Acad Sci USA. 2004;101:9393–9398. doi: 10.1073/pnas.0402993101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, Lee AV. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26:4078–4085. doi: 10.1200/JCO.2007.13.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68:826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 11.Dowsett M. Overexpression of HER-2 as a resistance mechanism to hormonal therapy for breast cancer. Endocr Relat Cancer. 2001;8:191–195. doi: 10.1677/erc.0.0080191. [DOI] [PubMed] [Google Scholar]
- 12.Massarweh S, Osborne CK, Jiang S, Wakeling AE, Rimawi M, Mohsin SK, Hilsenbeck S, Schiff R. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res. 2006;66:8266–8273. doi: 10.1158/0008-5472.CAN-05-4045. [DOI] [PubMed] [Google Scholar]
- 13.Arpino G, Green SJ, Allred DC, Lew D, Martino S, Osborne CK, Elledge RM. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a southwest oncology group study. Clin Cancer Res. 2004;10:5670–5676. doi: 10.1158/1078-0432.CCR-04-0110. [DOI] [PubMed] [Google Scholar]
- 14.Ellis MJ, Tao Y, Young O, White S, Proia AD, Murray J, Renshaw L, Faratian D, Thomas J, Dowsett M, et al. Estrogen-independent proliferation is present in estrogen-receptor HER2-positive primary breast cancer after neoadjuvant letrozole. J Clin Oncol. 2006;24:3019–3025. doi: 10.1200/JCO.2005.04.3034. [DOI] [PubMed] [Google Scholar]
- 15.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 16.Stoica GE, Franke TF, Wellstein A, Czubayko F, List HJ, Reiter R, Morgan E, Martin MB, Stoica A. Estradiol rapidly activates Akt via the ErbB2 signaling pathway. Mol Endocrinol. 2003;17:818–830. doi: 10.1210/me.2002-0330. [DOI] [PubMed] [Google Scholar]
- 17.Wong A, Lamothe B, Lee A, Schlessinger J, Lax I. FRS2α attenuates FGF receptor signaling by Grb2-mediated recruitment of the ubiquitin ligase Cbl. Proc Natl Acad Sci USA. 2002;99:6684–6689. doi: 10.1073/pnas.052138899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, Owens G, Alligood KJ, Spector NL. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. 2002;21:6255–6263. doi: 10.1038/sj.onc.1205794. [DOI] [PubMed] [Google Scholar]
- 19.Chu I, Blackwell K, Chen S, Slingerland J. The dual ErbB1/ErbB2 inhibitor, lapatinib (GW572016), cooperates with tamoxifen to inhibit both cell proliferation- and estrogen-dependent gene expression in antiestrogen-resistant breast cancer. Cancer Res. 2005;65:18–25. [PubMed] [Google Scholar]
- 20.Johnston S, Pippen J, Jr, Pivot X, Lichinitser M, Sadeghi S, Dieras V, Gomez HL, Romieu G, Manikhas A, Kennedy MJ, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmeno hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009;27:5538–5546. doi: 10.1200/JCO.2009.23.3734. [DOI] [PubMed] [Google Scholar]
- 21.Kaufman B, Mackey JR, Clemens MR, Bapsy PP, Vaid A, Wardley A, Tjulandin S, Jahn M, Lehle M, Feyereislova A, et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol. 2009;27:5529–5537. doi: 10.1200/JCO.2008.20.6847. [DOI] [PubMed] [Google Scholar]
- 22.Dauvois S, White R, Parker MG. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J Cell Sci. 1993;106(Pt 4):1377–1388. doi: 10.1242/jcs.106.4.1377. [DOI] [PubMed] [Google Scholar]
- 23.Dowsett M, Nicholson RI, Pietras RJ. Biological characteristics of the pure antiestrogen fulvestrant: overcoming endocrine resistance. Breast Cancer Res Treat. 2005;93(suppl 1):S11–S18. doi: 10.1007/s10549-005-9037-3. [DOI] [PubMed] [Google Scholar]
- 24.Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–2648. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 25.Klapper LN, Vaisman N, Hurwitz E, Pinkas-Kramarski R, Yarden Y, Sela M. A subclass of tumor-inhibitory monoclonal antibodies to ErbB-2/HER2 blocks crosstalk with growth factor receptors. Oncogene. 1997;14:2099–2109. doi: 10.1038/sj.onc.1201029. [DOI] [PubMed] [Google Scholar]
- 26.Sutherland RL, Reddel RR, Green MD. Effects of oestrogens on cell proliferation and cell cycle kinetics. A hypothesis on the cell cycle effects of antioestrogens. Eur J Cancer Clin Oncol. 1983;19:307–318. doi: 10.1016/0277-5379(83)90127-x. [DOI] [PubMed] [Google Scholar]
- 27.Sutherland RL, Green MD, Hall RE, Reddel RR, Taylor IW. Tamoxifen induces accumulation of MCF7 human mammary carcinoma cells in the G0/G1 phase of the cell cycle. Eur J Cancer Clin Oncol. 1983;19:615–621. doi: 10.1016/0277-5379(83)90177-3. [DOI] [PubMed] [Google Scholar]
- 28.Gee JM, Harper ME, Hutcheson IR, Madden TA, Barrow D, Knowlden JM, McClelland RA, Jordan N, Wakeling AE, Nicholson RI. The antiepidermal growth factor receptor agent gefitinib (ZD1839/Iressa) improves antihormone response and prevents development of resistance in breast cancer in vitro. Endocrinology. 2003;144:5105–5117. doi: 10.1210/en.2003-0705. [DOI] [PubMed] [Google Scholar]
- 29.Emde A, Pradeep CR, Ferraro DA, Ben-Chetrit N, Sela M, Ribba B, Kam Z, Yarden Y. Combining epitope-distinct antibodies to HER2: cooperative inhibitory effects on invasive growth. Oncogene. 2011;30:1631–1642. doi: 10.1038/onc.2010.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bender LM, Nahta R. Her2 cross talk and therapeutic resistance in breast cancer. Front Biosci. 2008;13:3906–3912. doi: 10.2741/2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Creighton CJ, Massarweh S, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, Shou J, Malorni L, Schiff R. Development of resistance to targeted therapies transforms the clinically associated molecular profile subtype of breast tumor xenografts. Cancer Res. 2008;68:7493–7501. doi: 10.1158/0008-5472.CAN-08-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen FL, Xia W, Spector NL. Acquired resistance to small molecule ErbB2 tyrosine kinase inhibitors. Clin Cancer Res. 2008;14:6730–6734. doi: 10.1158/1078-0432.CCR-08-0581. [DOI] [PubMed] [Google Scholar]
- 33.Di Leo A, Jerusalem G, Petruzelka L, Torres R, Bondarenko IN, Khasanov R, Verhoeven D, Pedrini JL, Smirnova I, Lichinitser MR, et al. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J Clin Oncol. 2010;28:4594–4600. doi: 10.1200/JCO.2010.28.8415. [DOI] [PubMed] [Google Scholar]
- 34.Xia W, Bacus S, Hegde P, Husain I, Strum J, Liu L, Paulazzo G, Lyass L, Trusk P, Hill J, et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci USA. 2006;103:7795–7800. doi: 10.1073/pnas.0602468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lupien M, Meyer CA, Bailey ST, Eeckhoute J, Cook J, Westerling T, Zhang X, Carroll JS, Rhodes DR, Liu XS, et al. Growth factor stimulation induces a distinct ER(α) cistrome underlying breast cancer endocrine resistance. Genes Dev. 2010;24:2219–2227. doi: 10.1101/gad.1944810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Z, Barnes CJ, Kumar R. Human epidermal growth factor receptor 2 status modulates subcellular localization of and interaction with estrogen receptor α in breast cancer cells. Clin Cancer Res. 2004;10:3621–3628. doi: 10.1158/1078-0432.CCR-0740-3. [DOI] [PubMed] [Google Scholar]
- 37.Amit I, Wides R, Yarden Y. Evolvable signaling networks of receptor tyrosine kinases: relevance of robustness to malignancy and to cancer therapy. Mol Syst Biol. 2007;3:151. doi: 10.1038/msb4100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stelling J, Sauer U, Szallasi Z, Doyle FJ, III, Doyle J. Robustness of cellular functions. Cell. 2004;118:675–685. doi: 10.1016/j.cell.2004.09.008. [DOI] [PubMed] [Google Scholar]