

Abstract
We have investigated four ~1.6-Mb microduplications and 55 smaller 350–680-kb microduplications at 15q13.2–q13.3 involving the CHRNA7 gene that were detected by clinical microarray analysis. Applying high-resolution array-CGH, we mapped all 118 chromosomal breakpoints of these microduplications. We also sequenced 26 small microduplication breakpoints that were clustering at hotspots of nonallelic homologous recombination (NAHR). All four large microduplications likely arose by NAHR between BP4 and BP5 LCRs, and 54 small microduplications arose by NAHR between two CHRNA7-LCR copies. We identified two classes of ~1.6-Mb microduplications and five classes of small microduplications differing in duplication size, and show that they duplicate the entire CHRNA7. We propose that size differences among small microduplications result from preexisting heterogeneity of the common BP4–BP5 inversion. Clinical data and family histories of 11 patients with small microduplications involving CHRNA7 suggest that these microduplications might be associated with developmental delay/mental retardation, muscular hypotonia, and a variety of neuropsychiatric disorders. However, we conclude that these microduplications and their associated potential for increased dosage of the CHRNA7-encoded α7 subunit of nicotinic acetylcholine receptors are of uncertain clinical significance at present. Nevertheless, if they prove to have a pathological effects, their high frequency could make them a common risk factor for many neurobehavioral disorders.
Keywords: microduplication, CHRNA7, NAHR, hypotonia, autism spectrum disorder
Introduction
Genomic regions flanked by low-copy repeats (LCRs, or segmental duplications) with DNA sequence identity greater than 95–97% are prone to recurrent microdeletions, microduplications, and inversions mediated by nonallelic homologous recombination (NAHR) [Lupski, 1998; Stankiewicz and Lupski, 2002]. The LCR-rich proximal region of chromosome 15 (15q11–q14) is one of the most unstable regions in the human genome [Knoll et al., 1993; Makoff and Flomen, 2007; Toth-Fejel et al., 1995]. DNA copy-number variations (CNVs) in this region include deletions, peri- and paracentric inversions, duplications, triplications, translocations, and supernumerary inv dup(15) chromosomes [Jauch et al., 1995; Rivera et al., 1990; Schinzel et al., 1994; Woodage et al., 1994]. Breakpoints of these rearrangements are located within complex sets of LCRs named BP1 to BP6 (Fig. 1). Deletions between BP1 or BP2 on the proximal side and BP3 on the distal side result in Prader-Willi or Angelman syndromes [Amos-Landgraf et al., 1999; Carrozzo et al., 1997; Christian et al., 1999; Robinson et al., 1998]. Duplications of the region flanked by BP1 or BP2 and BP3 are associated with learning disabilities, autism, and seizures [Cook et al., 1997; Dennis et al., 2006].
Figure 1.
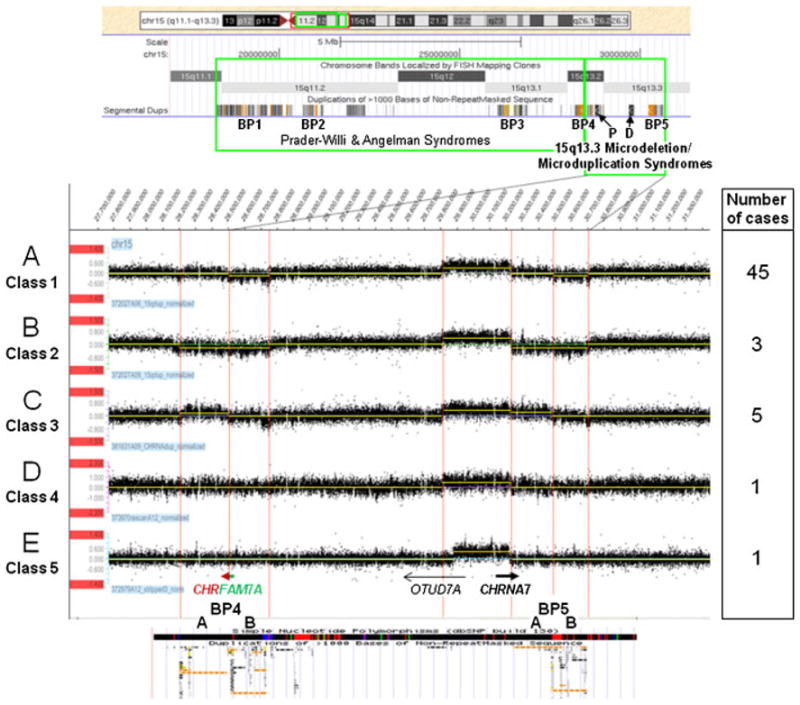
Results of aCGH analysis of 55 small microduplications in 15q13.3 using region-specific high-resolution 12 × 135K oligonucleotide microarrays (NimbleGen). BP1, BP2, and BP3 are LCRs flanking the common recurrent Prader-Willi and Angelman syndrome deletions; P and D are proximal and distal copies of the CHRNA7-LCR, respectively. A: One copy of the B segment of BP4 or BP5 is deleted. B: One copy of both A and B segments of BP4 or BP5 is deleted. C: One copy of the A segment of BP4 or BP5 is duplicated. D: Neither BP4 nor BP5 show a change in copy number compared to the control. E: A smaller sized CHRNA7 microduplication with proximal breakpoint mapping closer to the 5′ end of the OTUD7A gene. All gains and losses are relative to the control sample used.
An ~1.6 Mb recurrent microdeletion of a region between more distal LCRs, BP4, and BP5, and harboring six RefSeq genes: MTMR15, MTMR10, TRPM1 (MIM# 603561), KLF13 (MIM# 605328), OTUD7A (MIM# 612024), CHRNA7 (MIM# 118511), and one miRNA hsamir211 gene, has been found in patients with developmental delay/mental retardation, epilepsy, autism, schizophrenia, and other neurocognitive disorders [Ben-Shachar et al., 2009; Dibbens et al., 2009; International Schizophrenia Consortium, 2008; Helbig et al., 2009; Miller et al., 2009; Pagnamenta et al., 2009; Sharp et al., 2008; Stefansson et al., 2008; van Bon et al., 2009].
In many human populations, the BP4 LCR in this genomic region also harbors the chimeric CHRFAM7A gene. CHRFAM7A has not been found in nonhuman primates, and its occurrence in individuals of African descent is significantly lower than in Caucasian populations [Gault et al., 1998; Stassen et al., 2000]. Recently, Sinkus et al. [2009] suggested that CHRFAM7A has arisen during human evolution. CHRFAM7A is composed of a copy of the exon 5–10 portion of the CHRNA7 gene fused with a copy of the FAM7A gene. Its expression pattern and function are not well known.
Sharp et al. [2008] have shown that the BP4–BP5 region is inverted in 44% of individuals of varied ethnicities in the study population. This inversion likely results from NAHR between oppositely oriented BP4 and BP5 LCRs, and apparently predisposes to 15q13.3 microdeletions. Recently, we have described recurrent small 680-kb deletions and suggested that haploinsufficiency of CHRNA7 is causative for the majority of neurodevelopmental phenotypes observed in the 15q13.3 microdeletion syndrome [Shinawi et al., 2009]. CHRNA7 encodes the α7 subunit of the neuronal nicotinic acetylcholine receptor (nAChR), which forms pentameric ligand-gated cation channels.
Little is known, however, about the reciprocal 15q13.3 microduplications. Van Bon et al. [2009] reported four patients with cognitive impairment and psychiatric disorders coinciding with a BP4–BP5 microduplication. However, due to the limited number of individuals enrolled in those studies, it is difficult to conclude that this microduplication contributes to the etiology of neurodevelopmental phenotypes.
Here, we report the molecular characterization of four ~1.6 Mb BP4–BP5 and 55 small 350–680 kb microduplications involving CHRNA7, along with clinical phenotyping of 11 index patients with small microduplications and analysis of their extended pedigrees.
Subjects and Methods
Subjects
Fifty-five small 15q13.3 microduplications involving CHRNA7 [Shinawi et al., 2009] and four large BP4–BP5 microduplications were detected in 8,832 unrelated subjects referred for Chromosomal Microarray Analysis (CMA) at the Medical Genetics Laboratories (MGL) at Baylor College of Medicine (BCM). In this series, there were predominantly children with developmental delay/mental retardation, multiple congenital anomalies, dysmorphic features, autism or autistic spectrum, seizures, or others. CMA was performed using array-based comparative genomic hybridization (aCGH) with oligonucleotide-based version 6 (V6 OLIGO, 44K) (~4,000 patients), version 7 (V7 OLIGO, 105K) (~4,000 patients), and version 8 (V8 OLIGO, 180K) (~900 patients), designed by Baylor Medical Genetics Laboratories (http://www.bcm.edu/geneticlabs/) and manufactured by Agilent Technology (Santa Clara, CA) as previously described [El-Hattab et al., 2009; Lu et al., 2007; Ou et al., 2008]. Anonymized DNA samples were obtained from 55 individuals with small microduplications and four subjects with BP4–BP5 microduplications. Eleven probands with small microduplications were examined by C.P.S. at the outpatient Genetics Clinic at Texas Children’s Hospital and informed consent was obtained (approved by the Institutional Review Board for Human Subject Research at Baylor College of Medicine, H-12971 and H-24566). Five of these patients were of European, four of Hispanic and two of Asian descent. In 15 of 46 patients, additional CNVs were identified (Table 1, Supp. Table S1).
Table 1.
Phenotypic Features in 11 Unrelated Individuals with Small Microduplication of CHRNA7
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | M | M | M | M | M | M | M | F | M | M | M | |
| Age | 2 yo | 3 yo | 3 yo | 3 yo | 5 yo | 6 yo | 6 yo | 8 yo | 9 yo | 10 yo | 12 yo | |
| Class of CHRNA7 microduplication | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 3 | 1 | 2 | 1 | |
| Inheritance | mat | pat | pat | unknown (not mat) | mat | unknown | mat | mat | pat | unknown | mat | 4/8 mat 4/8 pat |
| Other CMA abnormalities | none | dup 5p15.2 (mat)a | none | dup Xq25 (mat)b | del 4q22.3 (mat)c | none | del 8q24.13q24.21 (de novo)d | none | none | none | dup 6q27 (not mat, father unavailable)e | 5/11 |
| DD/MR | Speech delay | GDD | GDD | GDD | GDD | Speech delay | GDD | Moderate MR | Mild MR | Moderate MR | Mild MR | 11/11 |
| Neuropsychiatric abnormalities | Pica (severe) | − | − | − | ASD | ASD, Anxiety disorder | − | − | ASD | ASD, Disruptive behavior disorder | ADHD, Bipolar disorder | 6/11 |
| Hypotonia | − | + | + | + | + | − | + | − | − | + | − | 6/11 |
| Seizures | − | − | + | − | − | − | − | − | − | − | − | 1/11 |
M, male; F, female; yo, years old; pat, paternal; mat, maternal; CMA, chromosome microarray; dup, duplication; del, deletion; DD, developmental delay; MR, mental retardation; GDD, global developmental delay; ASD, autism spectrum disorder; ADHD, attention-deficit/hyperactivity disorder.
arr 5p15.2(9021335-9243037) × 3.nuc ish 5p15.2(RP11-109L5 × 3)mat Gain in copy number in the 5p15.2 region of the short arm of chromosome 5, spanning a minimum of 0.222 Mb and a maximum of 0.261 Mb.
arr Xq25(123313165-123849841) × 2.nuc ish Xq25(RP11-107O17 × 2)mat Gain in copy number in the long arm of chromosome X, spanning a minimum of 0.537 Mb and a maximum of 0.571 Mb.
arr 4q22.3(RP11-369I16) × 1.ish del(4)(q22.3q22.3)(RP11-369I16dim)mat. This loss is approximately 140 kb in the proximal region covered by the clone. The nearest adjacent clone with no copy number change is 5.4 Mb proximal to the deleted segment.
arr 8q24.13q24.21(127185755-128672802) × 1.ish del(8)(q24.13q24.13)(RP11-248A1-)dn. Loss in copy number in the 8q24.13q24.21 region of the long arm of chromosome 8, spanning a minimum of 1.487 Mb and a maximum of 1.564 Mb.
arr 6q27(168458422-168622241) × 3.nuc ish 6q27(RP11-673P11 × 3) Gain in copy number in the 6q27 region of the long arm of chromosome 6, spanning a minimum of 0.164 Mb and a maximum of 0.932 Mb. FISH analysis with clone RP11-673P11 on chromosome 6 showed no evidence of the same gain in the mother.
DNA Isolation
DNA was prepared from peripheral blood using the Puregene DNA isolation kit (Gentra Systems, Minneapolis, MN).
aCGH
Recombination breakpoints were mapped using the 15q11.2–q14 region-specific high-resolution oligonucleotide aCGH. The 12 × 135K custom-designed microarrays (NimbleGen Systems, Madison, WI) were hybridized according to the manufacturer’s protocol. Briefly, 0.5 μg of patient genomic DNA and 0.5 μg of a normal male reference DNA sample were labeled by random priming with Cy3 and Cy5, respectively, using the NimbleGen Dual Color labeling kit. Microarrays were scanned on the GenePix 4000B scanner (Molecular Devices, Sunnyvale, CA). Scans were processed using NimbleScan v2.5, and analyzed with the SignalMap v1.9 (NimbleGen Systems). Array data have been deposited in GEO database under accession number GSE21268.
Verification of this analysis was performed using high-resolution 60K microarrays designed with the use of eArray (Agilent Technologies, Santa Clara, CA). The array consisted of 50,508 variable length probes with uniform Tm mapping between chr15:27,779,649 and 30,810,128 (hg18), or approximately one probe every 60 bp. Two μg of test sample and control DNA in 100 μl of H2O, was sheared to an average size of 300 bp using a Fisher Scientific Sonic Dismembrator Model 500 (three times for 15 sec at 14% power interrupted by 30-sec pause intervals). Test and control samples were labeled with Cy5 and Cy3, respectively, using the Bioprime Array CGH Genomic Labelling System (Invitrogen, Carlsbad, CA). From each sample, 900 ng were cohybridized for 20 hr. Slides were washed and scanned using an Agilent DNA Microarray Scanner. Data were normalized and plotted using Origin 8 software (OriginLab, Northampton, MA).
Multiplex Ligation-Dependent Probe Amplification (MLPA)
Probes for MLPA analysis were designed using the freely available H-MAPD Web server (http://genomics01.arcan.stony-brook.edu/mlpa/cgi-bin/mlpa.cgi) and mapped in exons for the following genes: APBA2, NDNL2, TJP1, ARHGAP11B, MTMR15, MTMR10, TRPM1, KLF13, and OTUD7A. For CHRNA7, probes were placed in exons 2 and 4. For CHRNA7/CHRFAM7A, probes were placed in exons 5 and 10 (Supp. Table S2). MLPA reactions were carried out using SALSA MLPA reagents and P300 reference probe mix as per instructions (MRC-Holland, Amsterdam). MLPA product (1.1 μl) and 0.25 μl of GS500 Liz Size Standard was added to 10 μl of formamide and loaded onto an ABI 3730xl capillary electrophoresis machine (Applied Biosystems, Foster City, CA). Data were analyzed using GeneMarker MLPA analysis software (SoftGenetics, State College, PA).
Long-Range PCR and DNA Sequencing
Long-range PCR primers were designed to harbor at least three to four nucleotides specific in one primer to proximal CHRNA7-LCR and in the other primer to distal CHRNA7-LCR, to allow preferential amplification of the predicted chimeric fragment containing the junction between parts of proximal and distal CHRNA7-LCRs, but not fragments of the original CHRNA7-LCRs [Shinawi et al., 2009]. The primers were design using Primer 3 software (http://www.frodo.wi.mit.edu/primer3). Amplification of 10–20 kb fragments was performed using Takara LA Taq Polymerase (TaKaRa Bio USA, Madison, WI) following the manufacturer’s protocol. Briefly, we used 25 μl reaction mixtures containing 100 ng genomic DNA, 0.4 mM dNTP (each), 0.2 μM primers (each), and 1.25 U of Taq polymerase. PCR conditions were: 94°C for 1 min, followed by 30 cycles at 94°C for 30 sec, and 68°C for 12 min, and 72°C for 10 min. In cases with residual amplification of the normal alleles from the normal control DNA, we adjusted the specificity of PCR conditions by increasing the annealing temperature to 70°C. The PCR products were treated with ExoSAPIT (USB, Cleveland, OH) to remove unconsumed dNTPs and primers, and directly sequenced by the dyeterminator method (Lone Star Labs, Houston, TX) using the primers used to amplify these DNA fragments and primers specific for both proximal and distal copies of the CHRNA7-LCR (Supp. Table S3).
DNA Sequence Analysis
The genomic sequences defined by coordinates identified in the aCGH experiments, were downloaded from the UCSC genome browser (NCBI build 36, March 2006, http://www.genome.ucsc.edu) and assembled using the Sequencher v4.8 software (Gene Codes, Ann Arbor, MI). Interspersed repeat sequences were identified using RepeatMasker (http://www.repeatmasker.org). DNA secondary structure was analyzed with Mfold (http://mfold.bioinfo.rpi.edu). The NAHR site sequences have been deposited in GenBank database with the accession numbers HM040991–HM041008.
Results
Structural Heterogeneity of Microduplications at 15q13.2–13.3
Microduplications were initially detected using Agilent clinical targeted arrays covering the BP4–BP5 region at 15q13.2–q13.3. Chromosomal breakpoints of the microduplications were more precisely mapped using two separate custom-designed high resolution microarray platforms: 135K NimbleGen and 60K Agilent oligonucleotide arrays. Additionally, the microduplications were further verified using MLPA. An example of the data for one class of microduplication is shown in Supp. Figure S1.
Fifty-four of the 55 small microduplications were ~430–680 kb in size and included the interval between the distal CHRNA7-LCR and BP5 (Fig. 1, Table 2). The majority of both the small and the large microduplications were accompanied by deletion or duplication of the A or B segment of BP4 or BP5 (Figs. 1–3).
Table 2.
Genomic Coordinates (hg 18) of the Randomly Selected Small and BP4–BP5 Microduplications in 15q13.3 Representing All Different Classes Derived from Region-Specific High-Resolution Array-CGH Analyses
| Microduplication | Class | Patient | Proximal breakpoint | Distal breakpoint |
|---|---|---|---|---|
| Small | 1 | 41 | 29,806,749 | 30,232,261 |
| 29,807,003 | 30,232,515 | |||
| 26 | 29,805,857 | 30,232,349 | ||
| 29,807,071 | 30,232,933 | |||
| 5 | 29,806,475 | 30,232,349 | ||
| 29,806,829 | 30,232,279 | |||
| 9 | 29,806,829 | 30,232,349 | ||
| 29,806,917 | 30,233,195 | |||
| 67 | 29,806,151 | 30,232,515 | ||
| 29,806,749 | 30,232,933 | |||
| 24 | 29,806,475 | 30,232,515 | ||
| 29,806,749 | 30,233,021 | |||
| 3 | 29,806,829 | 30,233,022 | ||
| 29,806,917 | 30,233,279 | |||
| 4 | 29,806,749 | 30,232,515 | ||
| 29,806,917 | 30,232,933 | |||
| 2 | 29,806,151 | 30,232,515 | ||
| 29,806,917 | 30,233,021 | |||
| 11 | 29,806,321 | 30,232,449 | ||
| 29,806,917 | 30,232,933 | |||
| 7 | 29,806,645 | 30,232,349 | ||
| 29,806,917 | 30,233,091 | |||
| 63 | 29,807,003 | 30,232,349 | ||
| 29,807,155 | 30,232,933 | |||
| 2 | 34 | 29,806,231 | 30,232,515 | |
| 29,806,645 | 30,233,021 | |||
| 13 | 29,806,829 | 30,232,515 | ||
| 29,806,917 | 30,232,933 | |||
| 10 | 29,806,577 | 30,232,349 | ||
| 29,806,917 | 30,233,021 | |||
| 3 | 15 | 29,806,645 | 30,470,910 | |
| 29,806,829 | 30,472,460 | |||
| 37 | 29,806,645 | 30,487,330 | ||
| 29,806,749 | 30,487,932 | |||
| 42 | 29,806,475 | 30,486,308 | ||
| 29,806,829 | 30,487,598 | |||
| 48 | 29,806,645 | 30,482,408 | ||
| 29,806,829 | 30,487,770 | |||
| 8 | 29,806,829 | 30,483,158 | ||
| 29,806,917 | 30,487,256 | |||
| 4 | 64 | 29,812,737 | 30,232,261 | |
| 29,813,858 | 30,233,279 | |||
| 5 | 35 | 29,876,190 | 30,218,795 | |
| 29,876,670 | 30,221,965 | |||
|
| ||||
| BP4-BP5 | 1 | 19 | 28,156,762 | 30,711,795 |
| 28,157,626 | 30,712,543 | |||
| 57 | 28,157,016 | 30,712,809 | ||
| 28,157,358 | 30,713,402 | |||
| 2 | 65 | 28,655,169 | 30,232,449 | |
| 28,655,539 | 30,233,021 | |||
Figure 3.

aCGH analysis of four BP4–BP5 microduplications using 12 × 135K oligonucleotide microarrays (NimbleGen). In one out of four cases, only the upstream portion of CHRNA7 is duplicated (B). Note that BP4 and BP5 segments are either (A) duplicated or (B) unchanged.
The most common was the association of the ~430 kb microduplication with the deletion of a copy of the B segment of BP4 or BP5. It was found in 45 of 55 microduplication cases (class 1, Fig. 1). The mechanisms of formation of these microduplications proposed in Figure 2A and B assume that BP4–BP5 region, both in the common and inverted orientation, carry a copy of CHRFAM7A gene. Matters are even more complicated as explained in the Discussion, because there is limited information as to the proportion of chromosomes with the common or inverted BP4–BP5 region that do or do not carry a copy of CHRFAM7A.
Figure 2.

Schematic representation of the proposed recombination events leading to the variety of small microduplications involving CHRNA7. CHRNA7-LCRs are depicted in red. The yellow rectangle represents the ~90-kb segment in BP4 that is duplicated in patients with small microdeletion and deleted in patients with reciprocal microduplication (class 1 or 3). A, B: The most common class 1/1* microduplication is shown. A: Class 1 microduplication with inversion breakpoints mapping within the A segment of BP4 and BP5 (brackets), but further downstream of CHRFAM7A and CHRNA7, respectively. NAHR between proximal and distal CHRNA7-LCRs leads to the loss of a copy of the BP4B repeat. B: Class 1* microduplication with inversion breakpoints (vertical dashed lines) mapping in intron 4 of CHRNA7 and its homologous part of CHRFAM7A within the A portion of BP5 and BP4, respectively. Note that the entire true copy of CHRNA7 is duplicated in A and C, and the fusion of the upstream portion of the true copy of CHRNA7 (exons 1–4) with the downstream portion of the CHRFAM7A gene (homologous to exons 5–10 of CHRNA7) is generated due to BP4–BP5 inversion. C: Class 3 of the small microduplications is depicted. NAHR between proximal and distal CHRNA7-LCRs leads to the duplication of the A repeat of BP5.
The class 1 microduplications could arise on the background of two slightly different inversions. One of the inversions would result in a novel fusion of at least first four exons of CHRNA7 with the remaining paralogous CHRNA7 exons that are part of the fusion CHRFAM7A gene (class 1*). We designate this novel fusion gene as CHRNA7*. In the second inversion, the entire true copy of CHRNA7 would be inverted (class 1). Depending on whether the breakpoint in the inversion chromosome occurred in intron 4 fusing all of the 5–10 exons of the CHRFAM7A to CHRNA7 exons 1–4, occurred downstream of exon 10 leaving the entire CHRNA7 gene intact, or occurred in an intermediate site, the inverted CHRNA7 can be comprised entirely of exons derived from exons 5–10 of either CHRNA7 or CHRFAM7A or of varying combinations of proximal exons from CHRNA7 and distal exons from CHRFAM7A. The microarray data presented in Figure 1 cannot distinguish between class 1 and class 1* microduplications. We do not know the relative proportion of class 1 versus class 1* microduplications among the 45 microduplications of the broadly defined class 1. Class 1 microduplications might be more likely to overexpress CHRNA7, while the CHRNA7* copy in class 1* microduplication might produce a protein of mutant or normal structure.
Deletion of the A and B segments of BP4 or BP5 was found in three small ~430 kb microduplications (class 2, Figs. 1 and 2A and B), and duplications of the A segment of BP5 were identified in five ~680-kb microduplication cases (class 3, Figs. 1 and 2C). The class 3 microduplications likely represent the reciprocal products of the small ~680-kb microdeletions that occurred through NAHR between the proximal and distal CHRNA7-LCRs on the normal and inverted chromosomes 15 [Shinawi et al., 2009]. Moreover, we found two classes of small microduplications not associated with deletions or duplications within BP4, BP5, or other regions within at least 1 Mb upstream and downstream of CHRNA7 (classes 4 and 5, Fig. 1). Interestingly, in one of these cases, the duplicated region was ~80 kb shorter (class 5) than that in the most common ~430 kb microduplications (Fig. 1).
In three out of four ~1.6 Mb BP4–BP5 microduplications, in which the entire CHRNA7 was duplicated, we observed also duplication of BP4 or BP5 LCRs (Fig. 3, Table 2).
The microduplication was inherited in all 21 small microduplication and two BP4–BP5 microduplication cases, in which both parents were studied (data not shown).
NAHR Hotspots in the Proximal and Distal CHRNA7-LCRs
In 11 microduplication cases, DNA sequencing of the long-range PCR products amplified from the patients’ DNA enabled us to narrow the recombination sites to 100–400 bp regions (flanked by two informative cis-morphic nucleotides) (Fig. 4). In two cases (patients 38 and 46), the NAHR region containing the recombination site could not be narrowed to less than 600 bp.
Figure 4.

Localization of NAHR hotspots within the proximal and distal CHRNA7-LCRs. NAHR site regions 1–6 are shown as black boxes. Genomic coordinates (hg18) of the proximal CHRNA7-LCR are shown in parenthesis.
In addition, we mapped and sequenced the NAHR site of the previously published small ~680-kb microdeletion (patient 4 in [Shinawi et al., 2009]) (Fig. 4). Consistent with the previous data for small ~680-kb microdeletions, the majority of NAHR sites of small microduplications cluster in the same two recombination hotspots, regions 1 and 6 (Fig. 4). In two cases, the patterns of alternating sequences of proximal and distal LCR-specific cis-morphisms in the chimeric LCRs (recombination site 2, 3, and 5) likely represent the results of earlier crossovers or gene conversion events (Fig. 4).
We analyzed the DNA structure of the two recombination hotspots for the presence of structural features that might have contributed to meiotic recombinogenic activity. NAHR hotspot 1 resides at the end of a retrotransposon LINE1 and hotspot 6 maps to a DNA segment harboring the LTR81AB of a Gypsy retrotransposon. We also found that the 7-mer CCTCCCT motif, often associated with meiotic recombination hotspots in humans [Myers et al., 2008], is located 125 bp downstream of the NAHR site 2. Further, we found several copies of this motif, carrying just a single nucleotide mismatch, located in the NAHR site regions 1, 5, and 6. Interestingly, two of the five imperfect copies of the recombination motif found in the hotspot region 6 match the 5′ half of a larger 13-mer, CCNCCNTNNCCNC, that has been suggested to be critical in recruiting crossover in 40% of human recombination hotspots [Myers et al., 2008]. However, CCTCCCT motifs and their derivatives are also scattered along the entire CHRNA7-LCRs, and their presence cannot, by themselves, serve as a predictor of NAHR hotspot location.
To investigate whether there is a common feature of the DNA secondary structures shared by the identified NAHR hotspots, we analyzed them in silico using the Mfold software. We found that the predicted thermodynamically most stable secondary structures of these recombination hotspot regions contain centrally localized palindromes that are more complex and longer than the neighboring palindromes (Supp. Fig. S2). Interestingly, the identified variants of CCTCCCT recombination motif reside consistently within the loop (head) regions of these palindromes.
Clinical Phenotyping and Pedigree Analysis
Eleven of 55 index patients (ages 3–12 years) with small microduplications involving CHRNA7 were examined, and extended pedigrees were obtained (Supp. Fig. S3). These 11 patients represent all cases for which consent could be obtained. All 11 patients displayed mild to moderate developmental delay or mental retardation (Table 1). Muscular hypotonia was present in six of 11 probands, with marked hypotonia in some of the younger patients and associated delays in motor development. No common dysmorphic facial features were noted in this patient population. Patient 1 had a history of congenital heart disease (hypoplastic left heart and coarctation of the aorta).
Interestingly, six of 11 index patients displayed neuropsychiatric abnormalities, including autism spectrum disorder in four patients, and bipolar disorder, anxiety disorder, disruptive behavior disorder, and severe pica in one patient each. Analysis of extended pedigrees showed that multiple family members carrying CHRNA7 small microduplication were affected with neuropsychiatric disorders, including major depressive disorder, bipolar disorder, anxiety, and alcoholism. The youngest individual with a microduplication involving CHRNA7 diagnosed with bipolar disorder is the 5-year-old brother of propositus 2; the 5-year-old brother manifested irritable mood, racing thoughts, distractibility, aggressive and hypersexual behaviors, pyromania, and suicidal ideation.
Pedigree analysis showed that there is significant variability in the expression of phenotypes in individuals with a microduplication involving CHRNA7. Penetrance was incomplete, as there are several family members, who have been identified to be carriers of CHRNA7 microduplications with normal intellectual performance and no history of neuropsychiatric abnormalities. Of note, five of 11 index patients carried at least one additional different CNV of potential clinical relevance, representing a complex genomic load [Lupski, 2007] that could be in line with the recently proposed “two-hit model” for severe developmental delay [Girirajan et al., 2010].
Whereas epilepsy has been strongly associated with microdeletions of CHRNA7 [Helbig et al., 2009; Shinawi et al., 2009], it is present only in one of 11 index patients with small microduplication involving CHRNA7 (Table 1). Patient 2, a 3-year-old male, has generalized myoclonic epilepsy and infantile spasms. Furthermore, out of the 10 family members to our index patients shown to carry a small microduplication involving CHRNA7, none has a history of seizures.
Both parents were available for microduplication testing in six of 11 families and the testing of the mother alone was possible in three families. The microduplication was inherited in six of six families where both parents were available for testing and in two families where only the mother was available (Table 1). The inheritance was maternal in five of eight cases and paternal in three of eight index cases. No de novo case of small microduplication of CHRNA7 has been identified to date. In five families (F2, F5, F8, F9, F11), the microduplication was inherited from a parent who had a history of neuropsychiatric problems themselves (four with major depressive disorder, one with a history of alcohol abuse). The mothers of patients 1 and 11 had learning disabilities by report. In two families (F3, F7), the microduplication was inherited from an apparently normal parent, and these parents were perceived by themselves, family members, and physicians as being normal based on education, employment, and rearing of a family. They did not have a history of cognitive impairment or neuropsychiatric disorder, although formal assessments of cognition and behavior were not performed.
Families of the carriers of BP4–BP5 microduplications were not available for clinical examination.
Discussion
Structures
The multiple large and highly homologous LCRs in chromosomal region 15q13.2–q13.3 are responsible for its extensive structural variability, including microdeletions, microduplications, and inversions. The opposite orientation of BP4 and BP5 LCRs is likely responsible for a recurrence of the paracentric ~1.6 Mb inversion between BP4 and BP5, found in 44% of randomly selected individuals [Sharp et al., 2008]; however, it does not explain the origin of the recurrent BP4–BP5 microdeletions and reciprocal microduplications. Recently, Makoff and Flomen [2009] postulated that a small inversion within the A portion of BP4 results in BP4A and BP5A segments being in direct orientation relative to each other; this in turn, could predispose to NAHR-mediated recurrent reciprocal BP4–BP5 microdeletions and microduplications.
Phenotypic and mechanistic analyses of BP4–BP5 and small microduplications at 15q13.2–q13.3 are complicated by the presence of the chimeric CHRFAM7A gene at the junction of the A and B segments in BP4. Recently, a 2-bp deletion in the CHRNA7 derived segment (exon 6) of CHRFAM7A, which results in a downstream premature stop codon, was found to be associated with schizophrenia [Sinkus et al., 2009], but no replication is yet available.
In classes 1/1*, and 3 of small microduplications, the NAHR recombination sites of the BP4–BP5 inversion are likely located within A segments for 1/1* or B segments for 3 of BP4 and BP5. This results in duplication of the entire original copy of CHRNA7 in class 1, and in the presence of one copy each for CHRNA7 and CHRNA7* in class 1* microduplications with no copy of CHRFAM7A in either class 1 or 1* microduplications (Fig. 2A and B). For class 3 microduplications, we hypothesize that the microduplication structure includes two copies of CHRNA7, one of CHRFAM7A, and none of CHRNA7* (Fig. 2C). The functional possibilities for microduplications with two copies of CHRNA7 and for those with one copy each of CHRNA7 and CHRNA7* are quite different. In some cases of class 1* microduplications, where the inversion breakpoints map within CHRNA7 (between intron 4 and exon 10) (Fig. 2B), an inactivating mutation within the CHRNA7 portion of CHRFAM7A (e.g., the previously described 2-bp deletion in exon 6 of CHRFAM7A) would alter the new CHRNA7* fusion transcript. Mutant forms of CHRNA7, derived either from CHRFAM7A or entirely from CHRNA7, could in turn, cause significant perturbation of neuronal homeostasis, for example, by a potential dominant negative effect on the oligomerization of nicotinic receptors in the brain. Further studies are needed to identify the pathophysiology of different classes of microduplications involving CHRNA7. Challenges include the relatively low expression of the α7 subunit of nAChR in peripheral tissues and the difficulty of working with antibodies to this receptor [Moser et al., 2007].
In addition to CHRNA7, small microduplications duplicate exon 1 of only one isoform of OTUD7A, whereas BP4–BP5 microduplications duplicate the entire OTUD7A gene. However, duplication of OTUD7A is unlikely to result in disease phenotype given OTUD7A is a putative deubiquitinase [Kayagaki et al., 2007], and changes in gene dosage for enzymes are rarely deleterious.
Benign or Pathological?
Phenotypic characterization of patients with microduplication of CHRNA7 revealed a similar spectrum of cognitive deficits and neurobehavioral abnormalities as seen in patients with microdeletion of CHRNA7, including developmental delay mental retardation, depression, bipolar disease, autism spectrum disease, attention-deficit/hyperactivity disorder (ADHD), and disruptive behavior disorder. However, this spectrum is not significantly different from the findings for all patients undergoing clinical array testing. In contrast to the reported incidences of epilepsy in patients with microdeletion of CHRNA7 [Dibbens et al., 2009; Helbig et al., 2009; Shinawi et al., 2009], seizures were only reported in one of 11 propositi with microduplication of CHRNA7, and zero of nine family members carrying the small microduplication, for which past medical history was obtained in this study.
The ability to determine whether particular CNVs are benign or pathological is often, as for microduplications studied here, limited by the availability of robust frequency data for carefully characterized controls. The lower the penetrance and the milder the phenotypes, the more difficult it is to determine pathological significance. Here, the limited data for frequency of the small microduplication in controls (one in 180 in [Helbig et al., 2009]) is not significantly different than that observed in patients. In addition, the small microduplication is inherited in all informative families to date, suggesting that genetic selection against the microduplication genotype is low. However, the frequency of mild neuropsychiatric phenotypes including major depression, ADHD, obsessive/compulsive disorder (OCD), and alcohol abuse in microduplication positive relatives of propostii leads us to be concerned that there may be phenotypic risks associated with the small microduplication. Control samples ordinarily are not screened for depression, ADHD, OCD, and alcohol abuse. If such phenotypic effects do exist, they would be important because of the relatively high frequency of the microduplications and because overexpression of CHRNA7 might provide pathophysiological and therapeutic insights. The situation for the small microduplications is additionally complicated by the fact that their different subtypes may have different functional properties, and no subtype information is yet available for controls. We believe that it will be necessary to study control populations screened for major depression, ADHD, OCD, and alcohol abuse, to determine whether each subclass of small microduplications is benign or pathological. Comparison of microduplication positive and microduplication negative first-degree relatives of microduplication cases may also help to clarify pathogenicity as distinct from assortative mating effects.
The pathogenic significance of the larger BP4–BP5 microduplication, involving CHRNA7, is reported as uncertain, although it may be associated with cognitive impairment and psychiatric disease [Ben-Shachar et al., 2009; van Bon et al., 2009]. Because the 350–680-kb small microduplications (1) were reported to be common in one control cohort, (2) are inherited in all cases to date, and (3) are often accompanied by other CNVs of uncertain significance, it is possible that some or all of the five classes of 350–680-kb microduplications are entirely benign and that the findings simply reflect bias of ascertainment. Alternatively, the presence of concomitant CNVs in five of 11 index patients in our study might suggest that the microduplication of CHRNA7 could be similar to a digenic effect (double heterozygotes) and cause an imbalance of neuronal homeostasis, predisposing to neurodevelopmental and neuropsychiatric phenotypes influenced by the presence or absence of other genetic modifiers. If the small microduplications are causing phenotypic effects in some cases, analysis of extended pedigrees would reveal what could be variable expressivity and incomplete penetrance of cognitive deficits and neuropsychiatric disorders in individuals with microduplication of CHRNA7. Phenomena such as variable expressivity and incomplete penetrance of the clinical phenotype, and assortative mating among individuals with psychatric disease (with or without detectable copy number variants) have been reported for other CNVs [McCarthy et al., 2009]. The clinical indications for CMA in 46 patients with small CHRNA7 microduplications referred to MGL are summarized in Supp. Table S1.
More studies are warranted to determine the frequency of CHRNA7 microduplications in healthy individuals, who have undergone extensive neuropsychiatric testing. Additional genetic modifiers probably influence the clinical phenotype of the individual patient. Those genetic modifiers could include (1) the sum of the expression level of all the copies of CHRNA7 and CHRNA7*; (2) copy number, expression level, and function, if any, of the CHRFAM7A fusion gene; (3) other monogenetic modifiers; or (4) the presence of concomitant copy number variants in the same patient, as seen in 15 of 46 patients enroled in this study (Supp. Table S1) and reported as a general phenomenon [Girirajan et al., 2010; Veltman and Brunner, 2010].
Mechanisms
We were able to sequence the NAHR sites of 13 small microduplications. In the majority of these cases (Fig. 4), we found that the microduplication NAHR sites overlap with those for the small ~680-kb microdeletions, further confirming the proposed double NAHR model of their formation [Shinawi et al., 2009]. By in-depth computational analyses of these regions, we found a positive correlation between the presence of NAHR hotspots and the relative increase of length and complexity of the predicted DNA hairpin structures. Moreover, we found imperfect copies of a common recombination-associated motif (CCTCCCT) within these hotspots, localizing to the head-part of the hairpins (Supp. Fig. S2). In addition to being associated with the CCTCCCT motif, the recombination hotspot 1 resides at the 3′ end of a non-LTR repetitive element LINE1, and a part of hotspot 6 maps within an LTR of a Gypsy retrotransposon. Interestingly, retrotransposons were already reported to be overrepresented in historical recombination hotspots [Cullen et al., 2002; Jeffreys et al., 1998; Lindsay et al., 2006], and the presence of the CCTCCCT motif within retrotransposon THE1A/B would dramatically increase probability of locating a historical hotspot within this mobile element [Myers et al., 2005]. However, distal sequences or epigenetic factors may contribute to hotspot activity equally well or better than local sequences [Arnheim et al., 2007; Bagshaw et al., 2006; Lupski, 2004; Ptak et al., 2005; Wu and Lichten, 1995].
Because the small microduplications most likely occur through NAHR between the CHRNA7-LCRs located within normal and inverted BP4–BP5 regions, we propose that the observed variability of the small microduplications and the associated variation in copy number of the A and B segments of BP4 and BP5 result from different locations of the inversion breakpoints within BP4 and BP5. We suggest that the most common variant of the small microduplications at 15q13.3 (class 1, Fig. 1), associated with the deletion of BP4B, arose on chromosome 15 with the inversion breakpoints located within the A segments of BP4 and BP5 (Fig. 2A and B). The BP5A breakpoint could be located anywhere within BP5A, including the 3′-terminal portion (exons 5–10) of the CHRNA7 gene, which overlaps with BP5A (Fig. 2B). The BP4A breakpoint might be located within CHRNA7-derived intron 4 of the CHRFAM7A gene, the CHRNA7 portion of CHRFAM7A, or further upstream within BP4A. The most plausible mechanism explaining the small microduplication associated with duplication of the A segment of BP5 assumes that the BP4–BP5 inversion breakpoints reside within the B segments of BP4 and BP5 (Fig. 2C). The mechanisms causing the small microduplication associated with the remaining (less common) CNVs within BP4 and BP5 seem more complex and likely involve different haplotypes of the BP4–BP5 inversions. We suggest that the BP4–BP5 inversion with breakpoints mapping within the CHRNA7 and CHRFAM7A genes may also lead to the functional inactivation of CHRNA7 through the transfer of the 2-bp polymorphism from the CHRFAM7A gene [Sinkus et al., 2009].
Future studies should include larger cohorts of individuals with microduplications involving CHRNA7 to better understand the complex genomotype–phenotype correlations in these families. In particular, identification of de novo cases will be very helpful to show whether parental BP4–BP5 inversions truly predispose to CHRNA7 microduplications in their offspring. Biological and biochemical studies are needed to determine whether partial duplications of CHRNA7 undergo nonsense mediated decay or are translated to truncated protein products and how this affects the oligomerization of the different nicotinic receptor subunits.
Supplementary Material
Acknowledgments
We thank P. Magoulas, K. Plunkett, P. Furman, P. Zimmerman, and S. Galvan for their tremendous help and assistance in this study. We thank J.R. Lupski, H.Y. Zoghbi, C.M.B. Carvalho, W. Gu, and C.G. Gonzaga-Jauregui for valuable discussions. P.S. was supported in part by grant R13-0005-04/2008 from the Polish Ministry of Science and Higher Education.
Footnotes
Communicated by Jacques S. Beckmann
Declaration of Competing Financial Interests
C.A.B., S.L., S.-H.K., A.P., S.W.C., A.L.B., and P.S. are based in the Department of Molecular and Human Genetics at BCM, which offers extensive genetic laboratory testing, including use of arrays for genomic copy number analysis, and derives revenue from this activity.
Additional Supporting Information may be found in the online version of this article.
References
- Amos-Landgraf JM, Ji Y, Gottlieb W, Depinet T, Wandstrat AE, Cassidy SB, Driscoll DJ, Rogan PK, Schwartz S, Nicholls RD. Chromosome breakage in the Prader–Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet. 1999;65:370–386. doi: 10.1086/302510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnheim N, Calabrese P, Tiemann-Boege I. Mammalian meiotic recombination hot spots. Annu Rev Genet. 2007;41:369–399. doi: 10.1146/annurev.genet.41.110306.130301. [DOI] [PubMed] [Google Scholar]
- Bagshaw AT, Pitt JP, Gemmell NJ. Association of poly-purine/poly-pyrimidine sequences with meiotic recombination hot spots. BMC Genomics. 2006;7:179. doi: 10.1186/1471-2164-7-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar S, Lanpher B, German JR, Qasaymeh M, Potocki L, Nagamani SC, Franco LM, Malphrus A, Bottenfield GW, Spence JE, Amato S, Rousseau JA, Moghaddam B, Skinner C, Skinner SA, Bernes S, Armstrong N, Shinawi M, Stankiewicz P, Patel A, Cheung SW, Lupski JR, Beaudet AL, Sahoo T. Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J Med Genet. 2009;46:382–388. doi: 10.1136/jmg.2008.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozzo R, Rossi E, Christian SL, Kittikamron K, Livieri C, Corrias A, Pucci L, Fois A, Simi P, Bosio L, Beccaria L, Zuffardi O, Ledbetter DH. Inter- and intrachromosomal rearrangements are both involved in the origin of 15q11–q13 deletions in Prader–Willi syndrome. Am J Hum Genet. 1997;61:228–231. doi: 10.1086/513907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH. Large genomic duplicons map to sites of instability in the Prader–Willi/Angelman syndrome chromosome region (15q11–q13) Hum Mol Genet. 1999;8:1025–1037. doi: 10.1093/hmg/8.6.1025. [DOI] [PubMed] [Google Scholar]
- Cook EH, Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C, Courchesne E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60:928–934. [PMC free article] [PubMed] [Google Scholar]
- Cullen M, Perfetto SP, Klitz W, Nelson G, Carrington M. High-resolution patterns of meiotic recombination across the human major histocompatibility complex. Am J Hum Genet. 2002;71:759–776. doi: 10.1086/342973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis NR, Veltman MW, Thompson R, Craig E, Bolton PF, Thomas NS. Clinical findings in 33 subjects with large supernumerary marker(15) chromosomes and 3 subjects with triplication of 15q11–q13. Am J Med Genet. 2006;140A:434–441. doi: 10.1002/ajmg.a.31091. [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Mullen S, Helbig I, Mefford HC, Bayly MA, Bellows S, Leu C, Trucks H, Obermeier T, Wittig M, Franke A, Caglayan H, Yapici Z, Sander T, Eichler EE, Scheffer IE, Mulley JC, Berkovic SF EPICURE Consortium. Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum Mol Genet. 2009;18:3626–3631. doi: 10.1093/hmg/ddp311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hattab AW, Smolarek TA, Walker ME, Schorry EK, Immken LL, Patel G, Abbott MA, Lanpher BC, Ou Z, Kang SH, Patel A, Scaglia F, Lupski JR, Cheung SW, Stankiewicz P. Redefined genomic architecture in 15q24 directed by patient deletion/duplication breakpoint mapping. Hum Genet. 2009;126:589–602. doi: 10.1007/s00439-009-0706-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault J, Robinson M, Berger R, Drebing C, Logel J, Hopkins J, Moore T, Jacobs S, Meriwether J, Choi MJ, Kim EJ, Walton K, Buiting K, Davis A, Breese C, Freedman R, Leonard S. Genomic organization and partial duplication of the human alpha7 neuronal nicotinic acetylcholine receptor gene (CHRNA7) Genomics. 1998;52:173–185. doi: 10.1006/geno.1998.5363. [DOI] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, Mefford HC, Kidd JM, Browning SR, Browning BL, Dickel DE, Levy DL, Ballif BC, Platky K, Farber DM, Gowans GC, Wetherbee JJ, Asamoah A, Weaver DD, Mark PR, Dickerson J, Garg BP, Ellingwood SA, Smith R, Banks VC, Smith W, McDonald MT, Hoo JJ, French BN, Hudson C, Johnson JP, Ozmore JR, Moeschler JB, Surti U, Escobar LF, El-Khechen D, Gorski JL, Kussmann J, Salbert B, Lacassie Y, Biser A, McDonald-McGinn DM, Zackai EH, Deardorff MA, Shaikh TH, Haan E, Friend KL, Fichera M, Romano C, Gécz J, DeLisi LE, Sebat J, King MC, Shaffer LG, Eichler EE. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, Kron KL, Steinich I, Kleefuss-Lie AA, Leu C, Gaus V, Schmitz B, Klein KM, Reif PS, Rosenow F, Weber Y, Lerche H, Zimprich F, Urak L, Fuchs K, Feucht M, Genton P, Thomas P, Visscher F, de Haan GJ, Møller RS, Hjalgrim H, Luciano D, Wittig M, Nothnagel M, Elger CE, Nürnberg P, Romano C, Malafosse A, Koeleman BP, Lindhout D, Stephani U, Schreiber S, Eichler EE, Sander T. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauch A, Robson L, Smith A. Investigations with fluorescence in situ hybridization (FISH) demonstrate loss of the telomeres on the reciprocal chromosome in three unbalanced translocations involving chromosome 15 in the Prader-Willi and Angelman syndromes. Hum Genet. 1995;96:345–349. doi: 10.1007/BF00210421. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Murray J, Neumann R. High-resolution mapping of crossovers in human sperm defines a minisatellite-associated recombination hotspot. Mol Cell. 1998;2:267–273. doi: 10.1016/s1097-2765(00)80138-0. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Phung Q, Chan S, Chaudhari R, Quan C, O’Rourke KM, Eby M, Pietras E, Cheng G, Bazan JF, Zhang Z, Arnott D, Dixit VM. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- Knoll JHM, Wagstaff J, Lalande M. Cytogenetic and molecular studies in the Prader-Willi and Angelman syndromes: an overview. Am J Med Genet. 1993;46:2–6. doi: 10.1002/ajmg.1320460103. [DOI] [PubMed] [Google Scholar]
- Lindsay SJ, Khajavi M, Lupski JR, Hurles ME. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am J Hum Genet. 2006;79:890–902. doi: 10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Shaw CA, Patel A, Li J, Cooper ML, Wells WR, Sullivan CM, Sahoo T, Yatsenko SA, Bacino CA, Stankiewicz P, Ou Z, Chinault AC, Beaudet AL, Lupski JR, Cheung SW, Ward PA. Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases. PLoS One. 2007;2:e327. doi: 10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Hotspots of homologous recombination in the human genome: not all homologous sequences are equal. Genome Biol. 2004;5:242. doi: 10.1186/gb-2004-5-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Structural variation in the human genome. N Engl J Med. 2007;356:1169–1171. doi: 10.1056/NEJMcibr067658. [DOI] [PubMed] [Google Scholar]
- Makoff AJ, Flomen RH. Detailed analysis of 15q11–q14 sequence corrects errors and gaps in the public access sequence to fully reveal large segmental duplications at breakpoints for Prader-Willi, Angelman, and inv dup(15) syndromes. Genome Biol. 2007;8:R114. doi: 10.1186/gb-2007-8-6-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makoff A, Flomen R. Common inversion polymorphisms and rare microdeletions at 15q13.3. Eur J Hum Genet. 2009;17:149–150. doi: 10.1038/ejhg.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, Krause V, Kumar RA, Grozeva D, Malhotra D, Walsh T, Zackai EH, Kaplan P, Ganesh J, Krantz ID, Spinner NB, Roccanova P, Bhandari A, Pavon K, Lakshmi B, Leotta A, Kendall J, Lee YH, Vacic V, Gary S, Iakoucheva LM, Crow TJ, Christian SL, Lieberman JA, Stroup TS, Lehtimäki T, Puura K, Haldeman-Englert C, Pearl J, Goodell M, Willour VL, Derosse P, Steele J, Kassem L, Wolff J, Chitkara N, McMahon FJ, Malhotra AK, Potash JB, Schulze TG, Nöthen MM, Cichon S, Rietschel M, Leibenluft E, Kustanovich V, Lajonchere CM, Sutcliffe JS, Skuse D, Gill M, Gallagher L, Mendell NR, Craddock N, Owen MJ, O’Donovan MC, Shaikh TH, Susser E, Delisi LE, Sullivan PF, Deutsch CK, Rapoport J, Levy DL, King MC, Sebat J Wellcome Trust Case Control Consortium. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Shen Y, Weiss LA, Korn J, Anselm I, Bridgemohan C, Cox GF, Dickinson H, Gentile J, Harris DJ, Hegde V, Hundley R, Khwaja O, Kothare S, Luedke C, Nasir R, Poduri A, Prasad K, Raffalli P, Reinhard A, Smith SE, Sobeih MM, Soul JS, Stoler J, Takeoka M, Tan WH, Thakuria J, Wolff R, Yusupov R, Gusella JF, Daly MJ, Wu BL. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet. 2009;46:242–248. doi: 10.1136/jmg.2008.059907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser N, Mechawar N, Jones I, Gochberg-Sarver A, Orr-Urtreger A, Plomann M, Salas R, Molles B, Marubio L, Roth U, Maskos U, Winzer-Serhan U, Bourgeois JP, Le Sourd AM, De Biasi M, Schröder H, Lindstrom J, Maelicke A, Changeux JP, Wevers A. Evaluating the suitability of nicotinic acetylcholine receptor antibodies for standard immunodetection procedures. J Neurochem. 2007;102:479–492. doi: 10.1111/j.1471-4159.2007.04498.x. [DOI] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- Myers S, Freeman C, Auton A, Donnelly P, McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- Ou Z, Kang SH, Shaw CA, Carmack CE, White LD, Patel A, Beaudet AL, Cheung SW, Chinault AC. Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet Med. 2008;10:278–289. doi: 10.1097/GIM.0b013e31816b4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnamenta AT, Wing K, Akha ES, Knight SJ, Bölte S, Schmötzer G, Duketis E, Poustka F, Klauck SM, Poustka A, Ragoussis J, Bailey AJ, Monaco AP International Molecular Genetic Study of Autism Consortium. A 15q13.3 microdeletion segregating with autism. Eur J Hum Genet. 2009;17:687–692. doi: 10.1038/ejhg.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak SE, Hinds DA, Koehler K, Nickel B, Patil N, Ballinger DG, Przeworski M, Frazer KA, Paabo S. Fine-scale recombination patterns differ between chimpanzees and humans. Nat Genet. 2005;37:429–434. doi: 10.1038/ng1529. [DOI] [PubMed] [Google Scholar]
- Rivera H, Zuffardi O, Gargantini L. Nonreciprocal and jumping translocations of 15q1-qter in Prader-Willi syndrome. Am J Med Genet. 1990;37:311–317. doi: 10.1002/ajmg.1320370304. [DOI] [PubMed] [Google Scholar]
- Robinson WP, Dutly F, Nicholls RD, Bernasconi F, Peñherrera M, Michaelis RC, Abeliovich D, Schinzel AA. The mechanisms involved in formation of deletions and duplications of 15q11–q13. J Med Genet. 1998;35:130–136. doi: 10.1136/jmg.35.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel AA, Brecevic L, Barnasconi F, Binkert F, Berthet F, Wuilloud A, Robinson WP. Intrachromosomal triplication of 15q11–13. J Med Genet. 1994;31:798–803. doi: 10.1136/jmg.31.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE, Schroer RJ, Novara F, De Gregori M, Ciccone R, Broomer A, Casuga I, Wang Y, Xiao C, Barbacioru C, Gimelli G, Bernardina BD, Torniero C, Giorda R, Regan R, Murday V, Mansour S, Fichera M, Castiglia L, Failla P, Ventura M, Jiang Z, Cooper GM, Knight SJ, Romano C, Zuffardi O, Chen C, Schwartz CE, Eichler EE. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, Cheung SW, Lanpher B, Nagl S, Herding HS, Nevinny-Stickel C, Immken LL, Patel GS, German JR, Beaudet AL, Stankiewicz P. A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat Genet. 2009;41:1269–1271. doi: 10.1038/ng.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkus ML, Lee MJ, Gault J, Logel J, Short M, Freedman R, Christian SL, Lyon J, Leonard S. A 2-base pair deletion polymorphism in the partial duplication of the alpha7 nicotinic acetylcholine gene (CHRFAM7A) on chromosome 15q14 is associated with schizophrenia. Brain Res. 2009;1291:1–11. doi: 10.1016/j.brainres.2009.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- Stassen HH, Bridler R, Hagele S, Hergersberg M, Mehmann B, Schinzel A, Weisbrod M, Scharfetter C. Schizophrenia and smoking: evidence for a common neurobiological basis? Am J Med Genet. 2000;96:173–177. [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietiläinen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Möller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Mühleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nöthen MM, Peltonen L, Collier DA, St Clair D, Stefansson K GROUP. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth-Fejel S, Magenis RE, Leff S, Brown MG, Comegys B, Lawce H, Berry T, Kesner D, Webb MJ, Olson S. Prenatal diagnosis of chromosome 15 abnormalities in the Prader-Willi/Angelman syndrome region by traditional and molecular cytogenetics. Am J Med Genet. 1995;55:444–452. doi: 10.1002/ajmg.1320550411. [DOI] [PubMed] [Google Scholar]
- van Bon BW, Mefford HC, Menten B, Koolen DA, Sharp AJ, Nillesen WM, Innis JW, de Ravel TJ, Mercer CL, Fichera M, Stewart H, Connell LE, Ounap K, Lachlan K, Castle B, Van der Aa N, van Ravenswaaij C, Nobrega MA, Serra-Juhé C, Simonic I, de Leeuw N, Pfundt R, Bongers EM, Baker C, Finnemore P, Huang S, Maloney VK, Crolla JA, van Kalmthout M, Elia M, Vandeweyer G, Fryns JP, Janssens S, Foulds N, Reitano S, Smith K, Parkel S, Loeys B, Woods CG, Oostra A, Speleman F, Pereira AC, Kurg A, Willatt L, Knight SJ, Vermeesch JR, Romano C, Barber JC, Mortier G, Pérez-Jurado LA, Kooy F, Brunner HG, Eichler EE, Kleefstra T, de Vries BB. Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome. J Med Genet. 2009;46:511–523. doi: 10.1136/jmg.2008.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman JA, Brunner HG. Understanding variable expressivity in microdeletion syndromes. Nat Genet. 2010;42:192–193. doi: 10.1038/ng0310-192. [DOI] [PubMed] [Google Scholar]
- Woodage T, Deng ZM, Prasad M, Smart R, Lindeman R, Christian SL, Ledbetter DH, Robson L, Smith A, Trent RJ. A variety of genetic mechanisms are associated with the Prader-Willi syndrome. Am J Med Genet. 1994;54:219–226. doi: 10.1002/ajmg.1320540308. [DOI] [PubMed] [Google Scholar]
- Wu T-C, Lichten M. Factors that affect the location and frequency of meiosis-induced double-strand breaks in Saccharomyces cerevisiae. Genetics. 1995;140:55–66. doi: 10.1093/genetics/140.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.