

Abstract
Over the past decade, a number of biocomputational tools have been developed to predict small RNA (sRNA) genes in bacterial genomes. In this study, several of the leading biocomputational tools, which use different methodologies, were investigated. The performance of the tools, both individually and in combination, was evaluated on ten sets of benchmark data, including data from a novel RNA-seq experiment conducted in this study. The results of this study offer insight into the utility as well as the limitations of the leading biocomputational tools for sRNA identification and provide practical guidance for users of the tools.
Keywords: bacteria, sRNA, noncoding RNAs, RNA-seq, bioinformatic predictions, Xenorhabdus nematophila
INTRODUCTION
In recent years, the expression of small RNAs (sRNAs) has been found to be widespread among bacteria. These regulatory RNAs belong to a broad range of classes, including but not limited to protein binding sRNAs, cis-encoded antisense sRNAs, trans-encoded base-pairing sRNAs, and sRNAs with intrinsic activity (Waters and Storz 2009; Liu and Camilli 2010). Even in the best-studied bacterial transcriptomes, the identities and functions of sRNAs are not fully understood. While experimental methods are critical for functional characterization of sRNAs (for review, see Sharma and Vogel 2009), computational methods for prediction of sRNAs, owing to their efficiency, can be a useful complement to experimental approaches.
A number of biocomputational tools have been developed over the last decade for the purpose of predicting sRNAs in bacterial genomes (for review, see Livny and Waldor 2007; Backofen and Hess 2010). While sRNAs have been identified within protein coding sequences, antisense to protein coding sequences, and within the untranslated regions between genes cotranscribed as part of an operon, many computational screens for sRNAs restrict their searches to intergenic regions of a genome. The biocomputational tools typically use one or more of four types of data to predict whether a genomic sequence corresponds to a sRNA: (1) primary sequence information such as transcription and regulatory signals, mono- and di-nucleotide frequency, and position of the sequence in relation to nearby genes; (2) secondary structure information such as the thermodynamic stability and minimum free energy of folding of a sequence; (3) primary sequence conservation, either in closely related genomes or in a pattern of conservation across a large number of genomes; and (4) secondary structure conservation by using pairwise or multiple sequence alignments to identify consensus secondary structures and their properties, particularly patterns of covariance suggestive of compensatory base pair mutations in conserved secondary structures. Biocomputational tools have been used to identify sRNAs in a range of bacteria, many of the computationally predicted sRNAs being subsequently validated through focused experiments. However, there has been a dearth of systematic comparisons of computational tools, and many of the approaches appear to suffer from low specificity when used for genome-wide screens. Further, there is little practical guidance for biologists who are deciding among and employing these tools.
In this study, four of the leading biocomputational tools, which use four different methodologies, were investigated: eQRNA (Rivas and Eddy 2001); RNAz (Washietl et al. 2005; Gruber et al. 2010); sRNAPredict3/SIPHT (Livny et al. 2006, 2008); and NAPP (Marchais et al. 2009). eQRNA identifies structural RNAs by searching for patterns of compensatory mutations consistent with a base-paired secondary structure (Rivas and Eddy 2001). RNAz identifies structural RNAs based on a combination of structural conservation and thermodynamic stability (Gruber et al. 2010). sRNAPredict3 identifies sRNAs based predominantly on regions of primary sequence conservation followed by transcription termination signals (Livny et al. 2006). NAPP (nucleic acid phylogenetic profiling) clusters noncoding sequences based on their conservation profiles across a large number of genomes and identifies novel noncoding RNAs by their inclusion in clusters enriched for known noncoding RNAs (Marchais et al. 2009). All four approaches use comparative genomics. The specific tools were chosen for this study based on the following criteria: the tool has been used in multiple studies for genome-wide prediction of sRNA genes, either a software implementation of the tool is available, or the authors have made results from their tool available for a variety of genomes, and the tools employ different methodologies relative to each other.
One of the major challenges to systematic comparison of various computational methods is that it is difficult to identify benchmark sets of bacterial sequences, especially intergenic regions, which are known not to contain sRNA genes. In the absence of such sets, assessing the specificity of a method is challenging since predictions that do not correspond to known sRNAs cannot be classified reliably as false positives or as yet to be verified sRNAs. To address this issue in part and to mitigate biases from particular sources of putative sRNA genes, the performance of the tools, both individually and in combination, was evaluated on ten sets of benchmark data drawn from a variety of sources, including experimentally confirmed sRNA genes, RNAs characterized in the Rfam database (Gardner et al. 2008), sRNAs suggested by previously published genome- tiling microarray experiments, sRNAs suggested by previously published RNA-seq studies, and sRNAs suggested by an RNA-seq experiment conducted in this study.
RESULTS
Benchmark data sets
Information on 776 putative small RNA transcripts was compiled from 10 data sources (Supplemental Table 1). Two of the data sources correspond to sets of experimentally validated sRNAs (Sittka et al. 2008; Huang et al. 2009), one is the Rfam database (Gardner et al. 2008), one is derived from the results of genome-tiling microarray experiments (Toledo-Arana et al. 2009), and six are derived from the results of RNA-seq experiments (Sittka et al. 2008; Liu et al. 2009; Yoder-Himes et al. 2009; Albrecht et al. 2010; Sharma et al. 2010). One of the data sets was generated by RNA-seq experiments conducted in this study using Xenorhabdus nematophila wild type and rpoS mutant strains (Vivas and Goodrich-Blair 2001). To enrich X. nematophila samples for small RNAs, samples were size-selected for RNAs 18–200 nucleotides in length, and rRNAs and tRNAs were targeted for depletion. Samples were submitted for sequencing to the Illumina Genome Analyzer II system, resulting in 2.6 gigabases of sequenced cDNA. Analysis of the sequencing data identified 97% of the reads mapping to the chromosome and 1% mapping to the plasmid of X. nematophila. Sequenced reads were processed into 57,447 discrete transcriptional units, 56% of which corresponded to protein coding genes, 31% of which were antisense to protein coding genes, 13% of which corresponded to intergenic regions, and <1% of which corresponded to rRNA or tRNA genes. Candidate small RNAs were identified as 219 transcriptional units in intergenic regions evincing sufficient expression, at least 100 sequencing reads on average across the entire extent of the transcriptional unit, and in samples from both strains. Of these 219 transcriptional units, 16% overlap candidate protein-coding ORFs identified by Glimmer3 (with length of at least 90 nucleotides and score at least 95) (Delcher et al. 2007), compared to the other six expression data sets used in this study, where, on average, 22% of putative sRNAs overlap candidate protein-coding ORFs identified by Glimmer3 (ranging from 15% for the Vibrio data set to 41% for the Helicobacter data set). These 219 transcriptional units comprised one of the 10 benchmark data sets.
Significance of predictions
Both eQRNA and RNAz have the useful property that they output a single value for each prediction that can be interpreted as the significance of the prediction. eQRNA scores alignments based on how well they fit a protein-coding model, a structural RNA model, or a “something else” model (Rivas and Eddy 2001). We use the bit score output by eQRNA that is a log-odds score corresponding to the probability that eQRNA's structural RNA model is favored over its two alternative models. For RNAz, its P score is a classification probability estimated by RNAz's support vector machine (Washietl et al. 2005). As a result, the performance of eQRNA and RNAz can be evaluated at different levels of significance.
With many predictive methods, a tension exists between sensitivity and precision. Increasing the number of predictions generally improves sensitivity at a cost to precision, whereas decreasing the number of predictions generally improves precision at a cost to sensitivity. The F-measure is a metric that accounts for both sensitivity and precision (van Rijsbergen 1979). Specifically, the F1 measure equally weights sensitivity and precision. The line graphs in Figure 1A illustrate the performance eQRNA and RNAz, as assessed by the F1-measure, at different levels of significance. Based on the 776 putative sRNAs from our 10 benchmark data sets, eQRNA's performance is optimal when a bit score cut-off of 3.5 is used, and RNA's performance is optimal when a class probability cut-off of 0.995 is used (Fig. 1A). The receiver operating characteristic (ROC) curves in Figure 1B illustrate the trade-offs between sensitivity and false positive rate, which is 1.0−specificity, at different levels of significance. In order to assess the tools “at their best,” i.e., when their performance is optimized on the benchmark data sets, we use a bit score cut-off of 3.5 for eQRNA and a class probability cut-off of 0.995 for RNAz throughout the remainder of this study.
FIGURE 1.

The performance of eQRNA as a function of its bit score and of RNAz as a function of its class probability score are shown when evaluated on 776 putative sRNAs. Each line represents 100 points, and each point represents the performance of a tool based on predictions at or above a score threshold. For eQRNA, 100 bit score thresholds between 0.0 and 25.0 were used. For RNAz, 100 class probability, P, thresholds between 0.5 and 1.0 were used. False positive rates were determined by rerunning each tool on shuffled alignments (see Materials and Methods). (A) The line graphs indicate the performance of eQRNA and RNAz, as determined by the F1-measure, at 100 different score thresholds. For eQRNA, the bit scores are shown on the lower x-axis, and for RNAz, the class probabilities are shown on the upper x-axis. eQRNA achieves maximum performance when predictions are restricted to those with a bit score ≥3.5. RNAz achieves maximum performance when predictions are restricted to those with a class probability ≥0.995. (B) The ROC curves illustrate the trade-offs between sensitivity and false positive rate at 100 different score thresholds for each of eQRNA and RNAz. The circle on each curve represents the point at which the F1-measure in A achieves a maximum value.
Ability of tools to identify putative sRNA genes
Four biocomputational tools were evaluated for their ability to identify putative sRNAs found in 10 benchmark data sets. Figure 2A illustrates the sensitivity of each of the tools, i.e., the percentage of putative sRNAs in each data set that was predicted by a tool. The sensitivities ranged from 2% (candidate sRNAs from Helicobacter RNA-seq experiments predicted by RNAz) to 71% (sRNAs from Escherichia data set predicted by NAPP). The mean sensitivity of eQRNA, RNAz, sRNAPredict3, and NAPP across the 10 data sets was 27%, 20%, 40%, and 49%, respectively, with standard deviations of 17%, 15%, 14%, and 21%. For five of the 10 data sets, NAPP achieved the highest sensitivity, for four of the 10 data sets, sRNAPredict3 achieved the highest sensitivity, and for one of the 10 data sets, RNAz achieved the highest sensitivity.
FIGURE 2.

The performance of four tools on the 10 benchmark data sets is illustrated. NAPP was not assessed on the Xenorhabdus data set since predictions from NAPP were unavailable for this genome. (A) The sensitivity of each tool is shown. Here, a sRNA in the data set is designated as correctly predicted by a tool if a prediction overlaps any part of the sRNA. (B) The precision of each tool is shown. (C) For those predictions that overlap a sRNA from a data set, the percentage that identify the correct strand of the sRNA is shown. Predictions from NAPP are omitted since no strand information is provided by the tool. (D) For sRNAs from the data sets that are correctly predicted, the percentage of their nucleotides that is correctly identified is shown.
The precisions of the tools were also investigated. However, our ability to quantify false positive predictions is limited by the quality and completeness of our data sets, so that the precision scores reported here are useful primarily for comparison of the relative precision among tools and should be used only as a lower bound on the absolute precisions of the tools. As illustrated in Figure 2B, the precisions ranged from 0% (candidate sRNAs from Burkholderia RNA-seq experiments predicted by eQRNA and RNAz) to 24% (candidate sRNAs from Vibrio RNA-seq experiments predicted by sRNAPredict3). The mean precision of eQRNA, RNAz, sRNAPredict3, and NAPP across the 10 data sets was 6%, 6%, 12%, and 4%, respectively, with standard deviations of 5%, 5%, 8%, and 4%. For nine of the 10 data sets, sRNAPredict3 achieved the highest precision, and for the Burkholderia data set, NAPP achieved the highest precision.
We then evaluated the ability of the tools to correctly identify the strand of a sRNA. For those predictions that correctly identified a sRNA from a data set, Figure 2C shows the percentage of predictions that also identify the correct strand of the sRNA. Results from NAPP are omitted from the strand analysis since NAPP does not report strand information for its predictions. sRNAPredict3 identifies the correct strand of sRNAs 80% of the time on average, and RNAz identifies the correct strand of sRNAs 76% of the time on average. eQRNA did not predict the correct strand of sRNAs at a rate significantly better than random.
The ability of the tools to predict the extent of a sRNA was also considered. For sRNAs correctly predicted by a tool, Figure 2D illustrates the percentage of nucleotides in the sRNA that were predicted. For eight of the 10 data sets, sRNAPredict3 outperformed the other methods, identifying 83% of nucleotides in a sRNA on average, and for two of the 10 data sets, eQRNA outperformed the other methods, identifying 72% of nucleotides in a sRNA on average. However, the results in Figure 2D should be taken in context since both sRNAPredict3 and eQRNA generally make substantially longer predictions, on average 174 nucleotides and 166 nucleotides, respectively, than RNAz and NAPP do, on average 119 nucleotides and 86 nucleotides, respectively.
In order to gauge whether the tools showed differential performance on Gram-positive bacteria and Gram-negative bacteria, we considered the tools' abilities to identify RNAs from these two classes of organisms. Since our 10 data sets are enriched for examples from Gram-negative bacteria, we restricted our focus to the data set corresponding to Rfam RNAs exclusive of rRNAs and tRNAs. The Rfam RNA data set consisted of 70 RNAs from Gram-positive bacteria and 62 RNAs from Gram-negative bacteria. eQRNA correctly identified 53% of RNAs from Gram-positive bacteria and 60% of RNAs from Gram-negative bacteria. RNAz correctly identified 16% of RNAs from Gram-positive bacteria and 32% of RNAs from Gram-negative bacteria. sRNAPredict3 correctly identified 56% of RNAs from Gram-positive bacteria and 56% of RNAs from Gram-negative bacteria. NAPP correctly identified 60% of RNAs from Gram-positive bacteria and 77% of RNAs from Gram-negative bacteria.
As an indication as to whether some tools are more likely than others to include short ORFs among their sRNA predictions, results from each program were compared to candidate protein-coding ORFs identified by the program Glimmer3 (Delcher et al. 2007). Glimmer3 was used to predict likely coding ORFs throughout the 14 genomes used in this study. In order to include possible short ORFs that may not yet be annotated, coding ORF predictions from Glimmer3 were considered if they were at least 90 nucleotides in length and had a score of at least 95. sRNA predictions from each program that overlap with a predicted ORF were then identified. For eQRNA, 14% of all sRNA predictions overlap with an ORF. For RNAz, 8% of all sRNA predictions overlap with an ORF. For sRNAPredict3, 15% of all sRNA predictions overlap with an ORF. For NAPP, 14% of all sRNA predictions overlap with an ORF. Since longer sRNA predictions are more likely to intersect ORFs than shorter sRNA predictions, these results should be taken in the context of the lengths of sRNA predictions, which on average are 166 nucleotides, 119 nucleotides, 174 nucleotides, and 86 nucleotides for eQRNA, RNAz, sRNAPredict3, and NAPP, respectively.
Assessment of tools in combination with each other
To assess the tools, we used both the F1 measure, which equally weights sensitivity and precision, and the F0.25 measure, which weights precision four times more than sensitivity. The motivation for including a measure that weights precision substantially more than sensitivity is that the biocomputational tools under consideration are often used for genome-wide screens. In some contexts, minimizing false positive predictions is more important than maximizing the number of sRNAs identified, particularly when computational predictions are followed up by more costly wet-lab experimentation.
Figure 3 illustrates the performance of each tool individually, as well as each pair of tools in combination, as assessed with the F1 measure (Fig. 3A) and the F0.25 measure (Fig. 3B). Based on the F1 measure, sRNAPredict3 outperforms the other tools individually for nine of the 10 data sets, whereas NAPP outperforms the other tools individually for the Burkholderia data set (Fig. 3A). Based on the F0.25 measure, sRNAPredict3 outperforms the other tools individually for nine of the 10 data sets, whereas NAPP outperforms the other tools individually for the Burkholderia data set (Fig. 3B).
FIGURE 3.

The F-measures for each tool individually (eQRNA, RNAz, sRNAPredict3, NAPP) as well as each pair of tools (eQRNA and RNAz, eQRNA and sRNAPredict3, RNAz and sRNAPredict3, eQRNA and NAPP, RNAz and NAPP, and sRNAPredict3 and NAPP) are shown for the 10 benchmark data sets. Predictions from pairs of tools correspond to overlapping predictions from both tools in the pair. NAPP was not assessed on the Xenorhabdus data set since predictions from NAPP were unavailable for this genome. (A) For each tool or pair of tools, the F1 measure, which weights sensitivity and precision equally, is shown. (B) For each tool or pair of tools, the F0.25 measure, which weights precision four times as much as sensitivity, is shown.
Using the F1 measure, sRNAPredict3's performance is generally comparable to or better than that of any pair of tools in combination, with few exceptions (Fig. 3A). sRNAPredict3 achieves a higher average F1 measure, 0.15, than any pair of tools in combination. Based on the F0.25 measure, however, the best performance is achieved by using a pair of tools in combination (Fig. 3B). The pair eQRNA and sRNAPredict3 in combination achieve the highest average F0.25 measure, 0.13, among all tools individually or in combination, but while using a combination of tools may lead to more precise results than an individual tool, there is no obvious set of tools whose combination consistently results in the best performance across the 10 data sets (Fig. 3B).
For the nine data sets that correspond to individual genomes, i.e., excluding the Rfam data set that is derived from many genomes, the tools showed the poorest performance on the Burkholderia data set, and the Burkholderia genome has the highest GC content (67% in the entire genome—63% in intergenic regions) among these genomes. To investigate if there is a relationship between the GC content of a genome and a tool's performance, the correlation was determined between each tool's F1 measure and the GC content of intergenic regions. Each tool's performance is negatively correlated with GC content. However, for eQRNA, RNAz, and NAPP, the correlation is statistically insignificant. sRNAPredict3's F1 measures have a correlation coefficient of −0.69 (one-tailed P-value of 0.02) with the GC content of intergenic regions.
Conservation of sRNAs
Each of the tools assessed in this study can only identify sRNAs whose sequences are conserved in other genomes. NAPP and SIPHT both consider conservation of a candidate noncoding RNA sequence in hundreds of other genomes. NAPP uses the profile of conservation across other genomes, while SIPHT uses the existence and significance of conservation in other genomes. In contrast, applications of eQRNA, RNAz, and sRNAPredict3 typically consider sequence conservation in only a handful of closely related genomes. Since the abilities of eQRNA, RNAz, and sRNAPredict3 to predict a sRNA gene may depend on the extent that the sRNA is conserved, we investigated the performance of each of these three tools when the tool identified sRNAs in different numbers of comparative genomes. We restricted our analysis to comparative genomes of organisms in the same genus as the reference genome's organism. For each of the three tools, we classified sRNA predictions into six groups: sRNAs predicted by the tool to occur in one, two, three, four, five, or more than five other genomes beyond the reference genome but corresponding to organisms from the same genus as the reference organism. The final group was not considered further because of an insufficient amount of data, i.e., many of the reference organisms used in the study did not have a sufficient number of close relatives whose genome sequences were publicly available. Figure 4A shows the performance of each of the three tools on the Rfam data set, as evaluated by the F1 measure, when the tool predicts sRNAs in various numbers of comparative genomes. As indicated in Figure 4A, the performance of each tool generally increases with the number of genomes predicted to contain sRNAs. This performance improvement stems from an increase in precision at a milder cost in sensitivity.
FIGURE 4.

(A) The performance of eQRNA, RNAz, and sRNAPredict3 on the Rfam data set is shown as a function of the number of comparative genomes predicted to contain sRNAs by the tool. (B) The average performance across all 10 data sets is shown for eQRNA, RNAz, and sRNAPredict3 as a function of the approximate evolutionary distance of the genomes used by the program for comparative analysis. Evolutionary distance between genomes is approximated by the dissimilarity of 16S rRNA sequences as measured by BLAST E-value. Each line in the figure corresponds to 11 data points, where each data point is the average performance of a program across all 10 data sets. For each genome in the 10 data sets, five out of 1350 bacterial genomes were randomly selected at the given evolutionary distance and used by a program as comparative genomes to make sRNA predictions.
Since each of the tools uses comparative genomics information in order to generate predictions, we investigated how the performance of a tool changes as more distantly related genomes are used for comparative analysis. To approximate evolutionary distance between two genomes, we used the dissimilarity of the genomes' 16S rRNA sequences as determined by BLAST's E-value. Two genomes whose 16S rRNA sequences have an E-value less than 10−300 when compared via BLAST normally correspond to organisms from the same genus. For each of the 14 genomes for which we make sRNA predictions throughout, we compared its 16S rRNA sequence to that from 1350 other bacterial genomes from RefSeq (Pruitt et al. 2005) and randomly chose five genomes at a given evolutionary distance to use as comparative genomes in making sRNA predictions. This process was repeated 11 times at different evolutionary distances. Figure 4B shows how the performance of a program changes as more distantly related genomes are used by the program to make sRNA predictions. eQRNA, RNAz, and sRNAPredict3 generally show poorer performance as more distantly related genomes are used for comparative analyses (Fig. 4B). All three programs achieve their best performance when genomes from the same genus are used (16S rRNA E-value less than 10−300). NAPP was not used because it requires a large number of comparative genomes across a broad range of evolutionary distances.
Runtime
The runtime of each of the tools was also evaluated. For eQRNA, assuming a pairwise alignment of length n is given, the computational complexity of the algorithm is dominated by the Inside algorithm (Baker 1979) used by eQRNA's RNA model and requires O(n3) time. For RNAz, assuming a multiple sequence alignment of length n is given, the computational complexity of the algorithm is O(n3). For sRNAPredict3, assuming coordinates of pairwise alignments and other genomic features such as terminators are given for a sequence of length n, the computational complexity of the algorithm is O(n). NAPP requires calculation and clustering of profiles for a large number of candidate sequences, so its computational complexity for an individual candidate can be viewed as the algorithm's complexity in generating predictions for all candidates, amortized over the number of candidates. For NAPP, assuming a conservation profile across y genomes is given for x candidate sequences (conserved noncoding elements), a distance matrix is computed in O(y·x2) time, and k-means clustering is performed in O(x·k·i) time, where k = 35 is the number of clusters (Marchais et al. 2009), and i is the number of iterations of the k-means algorithm. While k-means can be exponentially slow to converge in theory, in practice it converges quickly. Thus, assuming conservation profiles are given, the computational complexity of NAPP amortized over all candidates can be expressed as O(y·x). Actual wall-clock times for executing NAPP were unavailable. When screening 14 genomes, the average wall-clock time taken per genome by eQRNA, RNAz, and sRNAPredict3 when executed on a 4-node Intel Xeon 2.8GHz machine with 2GB memory was 696 min, 74 min, and 1 min, respectively.
Usability
eQRNA, RNAz, and sRNAPredict3 can be downloaded and run on a local machine. Each requires the use of auxiliary programs. eQRNA requires pairwise sequence alignments as input, RNAz requires multiple sequence alignments, and sRNAPredict3 requires pairwise sequence alignment information as well as transcription signal information such as terminator sites.
RNAz, SIPHT, and NAPP have web servers that enable access to the programs and/or their pre-computed predictions. The RNAz web server requires multiple sequence alignments as input (http://rna.tbi.univie.ac.at/cgi-bin/RNAz.cgi). SIPHT provides a web-interface to the sRNAPredict3 program (http://newbio.cs.wisc.edu/sRNA/). Currently, SIPHT enables predictions for 1912 replicons corresponding to 1022 genomes. NAPP has a web interface that enables access to a database of predictions currently corresponding to 1008 genomes (http://rna.igmors.u-psud.fr/NAPP/index.php).
DISCUSSION
Numerous biocomputational methods have been developed in recent years for predicting RNA genes in genome sequences. While these methods have proven useful for illuminating bacterial sRNA machinery in many applications, the methods also have a number of limitations. The ability of the methods to predict sRNAs is difficult to quantify, in part because our knowledge of sRNAs is incomplete, and characterizing with confidence a set of predictions as false positives is challenging. Different methods that predict sRNAs often give disjointed results. Further, many of the biocomputational tools either are not publicly available or have an inaccessible user interface. There is little guidance for biologists interested in choosing among and using these tools.
In this study, we assessed four biocomputational tools that have been used widely for sRNA prediction in bacteria. We restricted our investigation to tools whose results are publicly available and that have been used in multiple applications. The tools under consideration also employ different methodologies, which enable us to gain insight into the relative effectiveness of the different methods. The tools were assessed on 10 sets of data derived from experimentally verified sRNAs, noncoding RNAs reported in Rfam, putative small RNA transcripts identified by genome-tiling microarray experiments, and putative small RNA transcripts identified by RNA-seq experiments. We used data sets from disparate sources in order to minimize the biases that individual sources might have from overrepresentation of certain classes of noncoding RNAs.
We found that the biocomputational tools predicted 20%–49% of sRNAs in each data set on average, with precisions of 6%–12%. The modest fraction of sRNAs predicted by the tools with potentially substantial false positive rates highlights the challenge associated with computational identification of sRNAs and suggests that there is room for improvement of such tools. Though the number of sRNAs predicted by the tools is moderate, when the tools did identify a sRNA, they tended to perform well at also identifying the strand of the sRNA and the extent of the sRNA. Our results were generally consistent across the disparate data sets indicating that the results are not likely to be an artifact of the particular data sets used.
Among the tools assessed, NAPP identified the largest number of sRNAs from our benchmark data sets. However, the higher sensitivity of NAPP comes at the cost of a large number of predictions with potentially low precision. When considering multiple factors such as low false positive rates, ability to identify the correct strand of sRNAs, ability to identify the extent of nucleotides associated within each sRNA, and speed of execution, sRNAPredict3 generally had the best all around performance on our benchmark data sets. However, each of the tools has limitations. All four rely on comparative genomics information and are not effective at identifying orphan sRNAs. sRNAPredict3 uses information about rho-independent terminators, and, while its performance did not differ significantly when assessed on sRNAs from Gram-positive and Gram-negative organisms, its performance specifically on organisms that have a dearth of rho-independent terminators was not assessed. eQRNA and RNAz are based on structural properties of RNAs, and they will not predict genes that have little conserved structure. Also, sRNAPredict3 is designed specifically to identify sRNAs, whereas eQRNA and RNAz are designed to identify RNA structures more broadly, though we are assessing them here only on their ability to predict sRNAs. Finally, an important limitation is that the tools were executed in this study only with genomic sequences that do not contain protein-coding sequences, either sense or antisense. So our assessment of the tools included cis-encoded and trans-encoded sRNAs that do not overlap protein-coding sequences, but not other major classes of sRNAs such as cis-encoded sRNAs antisense to protein-coding genes.
For applications where low false positive rates are especially important, our results provide evidence that precision of the tools can be increased significantly by restricting them to sRNA predictions found in multiple related genomes. Further, combining predictions from different tools has potential to increase the precision of the generated predictions. The observation that combining different methods may boost performance is intriguing. It suggests that further studies on combining methodologies, either through meta-analyses of the results from existing tools or through new approaches that appropriately integrate components of the different methodologies, would be useful. In particular, there is a ripe opportunity for development of new biocomputational tools that incorporate data from RNA-seq experiments for the purpose of identifying sRNAs. Altogether, the results underscore that systematic detection of sRNA genes in bacterial genomes remains a rich and challenging problem.
MATERIALS AND METHODS
Genomic data
All genomes as well as genomic coordinates of annotated protein-coding genes, ribosomal RNA genes, and transfer RNA genes were downloaded from the NCBI (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/, 27 October 2010). All sequences not corresponding to annotated protein coding genes, ribosomal RNAs, or transfer RNAs were extracted from each reference genome and are denoted as intergenic regions (IGRs). When performing comparative genomics analyses on a reference genome, all available (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/, 27 October 2010) genomic sequences from the same genus as the reference genome but not from the same species were used. For each reference genome, a list of all RNAs in the genome was downloaded from the Rfam database version 10.0 (Gardner et al. 2008). RNAs annotated as ribosomal RNA or transfer RNA were removed from the lists of Rfam RNAs.
Biocomputational tools
To generate predictions with eQRNA (Rivas and Eddy 2001), first each IGR from the reference genome was aligned to each related genome using WU-BLASTN 2.0 (Gish, W. http://blast.wustl.edu). Only alignments at least 50 nucleotides in length with at least 65% identity and no more than 85% identity were retained. Longer sequences were analyzed in sliding windows 150 nucleotides in length, sliding by 50 nucleotides. Possible RNAs on both strands were scored using eQRNA, version 2.0.3c (ftp://selab.janelia.org/pub/software/qrna/). RNAs predicted in overlapping windows were merged into a single RNA prediction. When overlapping windows predicted RNAs on different strands, the strand identified by the most overlapping windows was designated as the strand for the predicted RNA. In the rare cases when a merged RNA prediction resulted from overlapping windows, half of which predicted one strand for the RNA and half of which predicted the opposite strand for the RNA, the strand for the predicted RNA was designated arbitrarily. When more than one comparative genome was available for generating eQRNA predictions, the union of sets of eQRNA predictions from different comparative genomes was reported. While eQRNA is designed to identify conserved RNA structures more broadly, it is assessed here only on its ability to predict sRNAs.
To generate predictions with RNAz (Gruber et al. 2010), first each IGR from the reference genome was aligned to each related genome using WU-BLASTN 2.0 (Gish, W. http://blast.wustl.edu). Only alignments at least 50 nucleotides in length were retained. Longer sequences were analyzed in sliding windows 150 nucleotides in length, sliding by 50 nucleotides, as was the case for eQRNA. Following the default parameters of RNAz, sequences from alignments with >25% gaps or >75% GC content were discarded. Similarly, consistent with the default parameters, if more than six sequences align with the reference IGR, a subset of six sequences was chosen with mean pairwise identity optimized to a target value of 80%. Multiple sequence alignments were then performed with ClustalW version 2.0.12 (Larkin et al. 2007). Possible RNAs were scored using RNAz, version 2.0pre (http://www.tbi.univie.ac.at/∼wash/RNAz/) (Gruber et al. 2010). While RNAz is designed to identify conserved RNA structures more broadly, it is assessed here only on its ability to predict sRNAs.
SIPHT is a computational tool built on the sRNAPredict approach (Livny et al. 2008). For available genomes, SIPHT sRNA predictions were downloaded from the SIPHT web interface (http://newbio.cs.wisc.edu/sRNA/). Independently, predictions were generated using the sRNAPredict3 tool (Livny et al. 2006). When we used similar parameters for sRNAPredict3 as were used for SIPHT, we obtained comparable results. Since the SIPHT web interface did not support predictions for all genomes corresponding to our benchmark data sets, specifically for Xenorhabdus nematophila, all results reported in our assessment correspond to predictions from sRNAPredict3 rather than SIPHT.
To generate predictions with sRNAPredict3 (Livny et al. 2006), first each IGR from the reference genome was aligned to each related genome using WU-BLASTN 2.0 (Gish, W. http://blast.wustl.edu) with parameters B and V set to 10,000. Alignments with an E-value less than or equal to 10−5 were retained. Transcriptional terminators were identified using the program TransTermHP (Kingsford et al. 2007), retaining candidate terminators predicted with at least 96% confidence, and using the program RNAMotif version 3.0.7 (03 April 2010) (Macke et al. 2001). Possible RNAs were scored using sRNAPredict3 (http://www.tufts.edu/sackler/waldorlab/sRNAPredict.html). The default parameters of sRNAPredict3 were used except that a minimum length of 50 nucleotides was used for sRNA predictions so as to be consistent with the other tools. It should be noted that predictions from eQRNA can be incorporated into the results output by sRNAPredict3. However, since eQRNA predictions do not affect predictions generated by sRNAPredict3, this feature of sRNAPredict3 was not utilized in our assessment.
Conserved noncoding elements identified by NAPP (Marchais et al. 2009) were supplied by the authors of the tool. Conserved noncoding elements were retained if they resided in clusters of conservation profiles that are enriched (P-value < 10−2) for known noncoding RNAs, as reported by Rfam (Gardner et al. 2008). Overlapping conserved noncoding elements were then merged into noncoding RNA predictions. Conserved noncoding elements from NAPP for Xenorhabdus nematophila were unavailable.
Bacterial strains and growth conditions
For sRNA isolation, X. nematophila wild-type strain HGB007 (ATCC 19061; wild type) and its rpoS::kan mutant derivative, HGB151 (Vivas and Goodrich-Blair 2001) were inoculated from −80°C frozen glycerol stocks into Luria–Bertani (LB) broth (Miller 1972) supplemented with 0.1% sodium pyruvate, 50 μg/mL ampicillin, and 50 μg/mL kanamycin. Cultures were grown overnight at 30°C on a tube roller, then subcultured 1:100 into flasks containing 30 mL LB with pyruvate and ampicillin and incubated at 30°C with shaking at 150 rpm.
sRNA isolation, tRNA depletion, and sRNA library construction
For each strain, cells from 1 mL of late exponential phase (OD600∼2.0) culture were collected by centrifugation at 10,000 × g. The mirVan miRNA Isolation Kit (Ambion) was used to isolate and enrich sRNAs, 3 mg of which were used for sRNA library construction. Libraries for sRNAs ranging from 18–200 bp were constructed by using the Small RNA Sample Prep Kit (Illumina) following the standard manufacturer's protocol (Illumina Small RNA Sample Preparation Guide). To reduce potential interference from tRNAs and 5S rRNAs, a stable RNA depletion protocol (Liu et al. 2009) was included. Briefly, 3′ and 5′ single strand RNA adapters were ligated to the sRNAs with TBE-urea gel purification after each ligation step. The gel-purified ligation product was mixed with 1 μl of Oligo Mix (5 pmol/μl of each oligonucleotide complementary to each X. nematophila tRNA and rRNA) (Supplemental Table 2) in 30 mL depletion buffer (50 mM Tris-HCl, pH 7.8; 300 mM KCl; 10 mM MgCl2; 10 mM DTT). The mixture was heated to 65°C for 5 min and slowly cooled to 37°C. Five units of RNase H (NEB) were added to the cooled mixture, and the reaction was kept at 37°C for 30 min. The depletion reaction was repeated once, followed by TBE-urea gel purification. cDNAs were synthesized from the tRNA- and 5S rRNA-depleted sRNA pool using the SRA RT primer (Illumina) that is specific to the 3′ adapter and SuperScript II reverse transcriptase (Invitrogen), and then PCR-amplified using Phusion Polymerase (Finnzymes) and primers GX1 and GX2 (Illumina) that are specific to 3′ and 5′ adapters, respectively. The amplification products were PAGE gel-purified and submitted to the Illumina Genome Analyzer II system for single read sequencing at the Tufts University Core Facility (Boston, MA).
Xenorhabdus nematophila RNA-seq analysis
The X. nematophila samples resulted in 65,584,055 sequence reads, each 40 nucleotides in length. Of these, 306,806 reads (0.5%) contained one or more ambiguous nucleotides and were discarded before further analysis. Using the program SSAHA2 (Ning et al. 2001), 65,277,249 reads (96.6%) mapped to the Xenorhabdus nematophila ATCC 19061 chromosome and 775,737 reads (1.2%) mapped to the Xenorhabdus nematophila ATCC 19061 XNC1_p plasmid with at least 95% identity. Of the reads mapping to the X. nematophila genome, 31,258,653 mapped uniquely to one region of the genome and were retained for further analysis. Sets of overlapping reads were merged into discrete transcriptional units. There were 57,447 transcriptional units that were identified from samples from both strains. To enable comparison of transcript expression levels from different samples, expression levels were normalized based on the total sequence reads for the sample.
Data sets
To evaluate the biocomputational tools, 10 sets of putative sRNA genes were collected from a variety of sources (see Supplemental Table 1 for the complete list). Altogether, the 10 data sets are comprised of 776 small RNA transcripts. For Escherichia coli, information on 79 experimentally confirmed sRNAs was downloaded from sRNAMap (Huang et al. 2009). For Salmonella, 64 validated sRNAs were used (Table 2 from Sittka et al. 2008). From genome-tiling microarray experiments in Listeria monocytogenes, 24 transcripts were used (Supplemental Table S2 from Toledo-Arana et al. 2009). From Rfam 10.0 (Gardner et al. 2008), information for 132 RNAs exclusive of ribosomal RNAs and transfer RNAs was obtained for Burkholderia cenocepacia AU 1054, Bacillus subtilis subsp. subtilis str. 168, Caulobacter crescentus CB15, Chlamydia trachomatis L2b/UCH-1/proctitis, Helicobacter pylori 26695, Listeria monocytogenes EGD-e, Pseudomonas aeruginosa PA01, Shewanella oneidensis MR-1, Staphylococcus aureus subsp. aureus N315, Streptomyces coelicolor A3(2), and Vibrio cholerae O1 biovar El Tor str. N16961. Candidate RNAs identified by a variety of RNA-seq experiments were also used: 119 candidate RNAs from Vibrio cholerae (transcripts denoted as “IGR” or “sRNA” in Supplemental Table 3 in Liu et al. 2009 merged into nonoverlapping transcripts and filtered to exclude transcripts within 30 nucleotides of an annotated protein-coding gene), 52 candidate RNAs from Salmonella (from Supplemental Table 3 in Sittka et al. 2008), 20 candidate RNAs from Chlamydia trachomatis (Supplemental Table 2 in Albrecht et al. 2010), 13 candidate RNAs from Burkholderia cenocepacia (Supplemental Table 5 in Yoder-Himes et al. 2009), 54 candidate RNAs from Helicobacter pylori (transcripts denoted as “sRNA” in Supplemental Table 13 in Sharma et al. 2010), and 219 candidate RNAs from Xenorhabdus nematophila, as identified in this study.
Evaluation metrics
The confusion matrix (Table 1) shows how, with complete knowledge about sRNA genes, computationally predicted RNAs would be identified as true positive or false positive predictions, and regions not computationally predicted to be RNAs would be identified as false negative or true negative predictions.
TABLE 1.
Confusion matrix

Of course, knowledge about sRNA genes is incomplete. The 10 benchmark data sets are used as surrogates for sets of actual sRNA genes—the left column in the confusion matrix (Table 1). Using the benchmark data sets as surrogates for sets of actual sRNA genes, we compute the sensitivity (also known as recall) of a biocomputational tool as its number of true positive predictions divided by the sum of its true positive and false negative predictions.
Unfortunately, estimating the false positive rate of a tool is more challenging since there are no large reliable sets of genomic sequences that are known not to contain sRNAs but have comparable properties to sequences containing sRNAs, i.e., there are no obvious surrogates for the final column in the confusion matrix (Table 1). To estimate false positive predictions generated by a tool, two imperfect approaches are used.
First, for eQRNA and RNAz, control sequences are generated that do not contain sRNAs. Since control sequences, by definition, contain no sRNAs, any RNA prediction made on a control sequence can reasonably be deemed spurious (a false positive). The challenge in generating control sequences is ensuring that they have properties similar to sRNA-containing sequences, but without actually containing sRNAs. Since eQRNA and RNAz rely on pairwise and multiple sequence alignments, respectively, control sequences are generated by shuffling columns of the sequence alignments. The control sequences have the same GC content and same alignment scores as actual sRNA containing sequences. However, any evidence of compensatory base changes in conserved RNA structures will be destroyed, and such covarying bases are a primary feature used by both eQRNA and RNAz for identifying RNA genes. To shuffle the pairwise sequence alignments for input to eQRNA, columns of the alignments were shuffled randomly with the criteria that the conservation and indel structures of the alignment be conserved, i.e., columns with a given level of conservation or with a given gap pattern are shuffled only with other columns that have the same level of conservation or the same gap pattern. Similarly, to shuffle the multiple sequence alignments for input to RNAz, a randomization approach was used that preserves the GC content of the aligned sequences as well as the indel and conservation patterns (Washietl and Hofacker 2004). After shuffling the alignments as described above, eQRNA and RNAz were rerun on all reference genomes, and any RNA predictions made by the programs were deemed false positives.
While shuffling alignments enables estimation of false positive rates, the approach has some limitations that should be noted. In addition to disrupting conserved RNA structures as it is intended to do, the shuffling also disrupts other motifs, such as regulatory sites, so that the control sequences are imperfect representations of actual non-sRNA containing sequences. Further, the approach used by tools like eQRNA and RNAz supports prediction of a broad range of conserved RNA structures, including noncoding RNA genes, rho-independent transcription terminators, certain transcriptional attenuators, and other cis-regulatory RNA structures. Many of the predictions from eQRNA and RNAz may correspond to conserved RNA structures, yet we designate them as false positives if they do not correspond to sRNA genes. Finally, the shuffling approach described above is not applicable to sRNAPredict3 and NAPP, which use primary sequence conservation rather than RNA structure conservation as a criterion for predicting sRNA genes.
As a second means to illuminate false positive predictions from the various tools, a measure of precision is used. Using the benchmark data sets as surrogates for sets of actual sRNA genes, we compute the precision (also known as positive predictive value) of a biocomputational tool as its number of true positive predictions divided by the sum of its true positive and false positive predictions. Here, predictions that do not correspond to sRNAs from the benchmark data are designated as false positives. These false positive designations are flawed in that benchmark data sets of sRNAs are certainly incomplete, so many predictions that do not correspond to sRNAs from the benchmark data may indeed correspond to previously uncharacterized sRNAs. Thus, the precision measure should be considered a lower bound on the actual precision of the tool. However, to the extent that the benchmark sRNAs are representative samples of all sRNAs in a genome, the measure is a meaningful metric for the relative precision of tools in comparison to one another.
Finally, F-measures are used to assess the biocomputational tools (van Rijsbergen 1979). The F-measure of a tool is a function of both its recall (sensitivity) and precision,
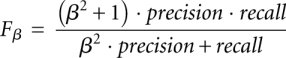 |
where β ≥ 0 is a parameter that designates the relative weight of recall relative to precision. When β = 1, recall and precision are equally weighted, and F1 is the harmonic mean of recall and precision. Both F1 and F0.25, which weights precision four times as much as recall, are used in this study.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Daniel Gautheret and Antonin Marchais for providing results from NAPP, and Daniel Gautheret for offering helpful comments on the manuscript. This work was supported by the National Science Foundation (MCB-0919808 to B.T.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2689811.
REFERENCES
- Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T 2010. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res 38: 868–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backofen R, Hess WR 2010. Computational prediction of sRNAs and their targets in bacteria. RNA Biol 7: 33–42 [DOI] [PubMed] [Google Scholar]
- Baker JK 1979. Trainable grammars for speech recognition. J Acoust Soc Am 65: S132 doi: 10.1121/1.2017061 [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23: 673–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, et al. 2008. Rfam: Updates to the RNA families database. Nucleic Acids Res 37: D136–D140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Findeiss S, Washietl S, Hofacker IL, Stadler PF 2010. RNAZ 2.0: Improved noncoding RNA detection. Pac Symp Biocomput 15: 69–79 [PubMed] [Google Scholar]
- Huang HY, Chang HY, Chou CH, Tseng CP, Ho SY, Yang CD, Ju YW, Huang HD 2009. sRNAMap: Genomic maps for small non-coding RNAs, their regulators, and their targets in microbial genomes. Nucleic Acids Res 37: D150–D154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsford CL, Ayanbule K, Salzberg SL 2007. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol 8: R22 doi: 10.1186/gb-2007-8-2-r22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- Liu JM, Camilli A 2010. A broadening world of bacterial small RNAs. Curr Opin Microbiol 13: 18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JM, Livny J, Lawrence MS, Kimball MD, Waldor MK, Camilli A 2009. Experimental discovery of sRNAs in Vibrio cholerae by direct cloning, 5S/tRNA depletion and parallel sequencing. Nucleic Acids Res 37: e46 doi: 10.1093/nar/gkp080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livny J, Waldor MK 2007. Identification of small RNAs in diverse bacterial species. Curr Opin Microbiol 10: 96–101 [DOI] [PubMed] [Google Scholar]
- Livny J, Brencic A, Lory S, Waldor MK 2006. Identification of 17 Pseudomonas aeruginosa sRNAs and prediction of sRNA-encoding genes in 10 diverse pathogens using the bioinformatics tool sRNAPredict2. Nucleic Acids Res 34: 3484–3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livny J, Teonadi H, Livny M, Waldor MK 2008. High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLoS ONE 3: e3197 doi: 10.1371/journal.pone.0003197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macke T, Ecker D, Gutell R, Gautheret D, Case DA, Sampath R 2001. RNAMotif—a new RNA secondary structure definition and discovery algorithm. Nucleic Acids Res 29: 4724–4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchais A, Naville M, Bohn C, Bouloc P, Gautheret D 2009. Single-pass classification of all noncoding sequences in a bacterial genome using phylogenetic profiles. Genome Res 19: 1084–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- Ning Z, Cox AJ, Mullikin JC 2001. SSAHA: A fast search method for large DNA databases. Genome Res 11: 1725–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR 2005. NCBI Reference Sequence (RefSeq): A curated nonredundant sequence database of genomics, transcripts, and proteins. Nucleic Acids Res 33: D501–D504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas E, Eddy SR 2001. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics 2: 8 doi: 10.1186/1471-2105-2-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma CM, Vogel J 2009. Experimental approaches for the discovery and characterization of regulatory small RNAs. Curr Opin Microbiol 12: 536–546 [DOI] [PubMed] [Google Scholar]
- Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermuller J, Reinhardt R, et al. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464: 250–255 [DOI] [PubMed] [Google Scholar]
- Sittka A, Lucchini S, Papenfort K, Charma CM, Rolle K, Binnewies TT, Hinton JCD, Vogel J 2008. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet 4: e1000163 doi: 10.1371/journal.pgen.1000163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, et al. 2009. The Listeria transcriptional landscape from saprophytism to virulence. Nature 459: 950–956 [DOI] [PubMed] [Google Scholar]
- van Rijsbergen CJ 1979. Information Retrieval. http://www.dcs.gla.ac.uk/Keith/Preface.html
- Vivas EI, Goodrich-Blair H 2001. Xenorhabdus nematophila as a model for host-bacterium interactions: rpoS is necessary for mutualism with nematodes. J Bacteriol 183: 4687–4693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Hofacker IL 2004. Consensus folding of aligned sequences as a new measure for the detection of functional RNAs by comparative genomics. J Mol Biol 342: 19–30 [DOI] [PubMed] [Google Scholar]
- Washietl S, Hofacker IL, Stadler PF 2005. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci 102: 2454–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Storz G 2009. Regulatory RNAs in bacteria. Cell 136: 615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder-Himes DR, Chain PSG, Zhu Y, Wurtzel O, Rubin EM, Tiedje JM, Sorek R 2009. Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc Natl Acad Sci 106: 3976–3981 [DOI] [PMC free article] [PubMed] [Google Scholar]