

Abstract
The diagnosis of malignant melanoma presents a clinical challenge and relies principally on histopathological evaluation. Previous studies have indicated that increased expression of the DEK oncogene, a chromatin-bound factor, could contribute to the development of melanoma and may be a frequent event in melanoma progression. Here, we investigated DEK expression by immunohistochemistry in a total of 147 melanocytic lesions, including ordinary nevi, dysplastic nevi, Spitz nevi, melanoma in situ, primary invasive melanomas, and metastatic melanomas. Most benign nevi (ordinary, dysplastic and Spitz nevi) were negative or exhibited weak staining for DEK with only 4 of 49 cases showing strong staining. Similar to benign nevi, melanoma in situ also demonstrated low levels of DEK expression. In contrast, the expression of DEK in primary invasive melanomas was significantly higher than benign nevi (p<0.0001). Moreover, DEK expression was significantly increased in deep melanomas (Breslow depth > 1mm) and metastatic melanomas as compared to superficial melanomas (Breslow depth ≤ 1mm) (p<0.05). Our findings indicate that DEK overexpression may be a frequent event in invasive melanomas, and further augmentation of DEK expression may be associated with the acquisition of ominous features such as deep dermal invasion and metastasis. These data suggest a role of DEK in melanoma progression.
Keywords: DEK, benign nevi, invasive melanomas, metastatic melanomas, melanoma progression
Introduction
Cutaneous malignant melanoma is the most lethal form of skin cancer, and its increasing incidence continues to pose a clinical challenge [1]. Successful treatment requires early diagnosis, yet confidently distinguishing malignant melanoma from other melanocytic lesions can sometimes prove difficult in practice [2–3]. This has prompted the search for markers in differentiating between benign and malignant melanocytic lesions.
One potential candidate is the oncogene DEK located on chromosome 6p. Originally discovered as a target of the [t(6;9)(p23;q34)] chromosomal translocation found in a subset of acute myelogenous leukemias [4], DEK has since been found to be overexpressed independent of the t(6;9) translocation in a number of human malignancies including malignant melanoma [10–11]. Focal amplifications containing the DEK locus have been described in retinoblastoma, bladder cancer, and most recently neuroendrocrine carcinoma of the lung; however, corresponding focal amplifications have not been identified in melanoma [5–7]. Rather, it is possible that DEK overexpression is mediated through gain of the short arm of chromosome 6, one of the most commonly observed cytogenetic abnormalities in melanomas [8–9]. Additionally, it is likely that overexpression of DEK in at least some melanomas occurs independently of chromosomal abnormalities, possibly through dysregulation of E2F transcription family [10–11]. Though its precise cellular function remains unclear, DEK has been implicated in numerous cellular processes including mRNA processing, cell-cell signaling, DNA damage repair, regulation of transcription, and modification of chromatin architecture [12–20]. Its participation in these disparate cellular activities is thought to be integrated at least in part via post-translational modification by phosphorylation, acetylation, and poly-ADP ribosylation [14–15, 21–22]. Two principle oncogenic properties of DEK overexpression have been described thus far: inhibition of senescence and inhibition of apoptosis [23–24]. We have recently found that DEK overexpression serves both functions in supporting melanoma tumorigenesis [10]. Specifically, DEK overexpression augments chemoresistance in melanoma by promoting the expression of the anti-apoptotic gene mcl-1 at the transcriptional level and additionally inhibits cellular senescence in a subset of metastatic melanoma cell lines.
The potential role of DEK in the pathogenesis of malignant melanoma is highlighted by two tissue microarray analyses that revealed DEK expression in 72% and 90% of primary and metastatic melanomas [10–11]. None of the few benign nevi analyzed demonstrated DEK expression by immunohistochemistry. However, a more rigorous analysis of DEK expression in a wider spectrum of melanocytic lesions has not been performed thus far. Specifically, the utility of DEK expression status in discriminating malignant melanoma from benign nevi remains undefined. Furthermore, given the known oncogenic functions and variable expression of DEK in primary and metastatic melanomas, it is possible that DEK expression may be associated with melanoma progression. We present here a detailed analysis of DEK expression in a broad spectrum of melanocytic lesions.
Materials and Methods
Case Selection
This study was approved by the Institutional Review Board at the University of Michigan Medical Center. After a search of the surgical pathology database in the Department of Pathology of the University of Michigan, a total of 147 specimens were collected, including 15 ordinary nevi, 17 dysplastic nevi, 17 Spitz nevi, 16 melanoma in situ, 46 primary invasive melanomas, and 36 metastatic melanomas. Among these cases, 32 metastatic melanomas (three individual cores per specimen) were analyzed on a tissue microarray slide while the remaining specimens were tested on the whole tissue sections. The staining patterns were consistent among the cores of the same lesion. Primary invasive melanomas were further segregated by Breslow depth into superficial (≤1 mm) or deep (>1 mm) melanomas. The clinical characteristics of the analyzed specimens are shown in Table 1. None of the tumors in primary deep melanomas and metastatic melanomas were derived from the same patient. None of the superficial melanomas developed local or distant metastases within the limitation of our follow-up period. The locations of the 36 metastatic melanomas were lymph nodes (27), skin and soft tissues (6), liver (1), small intestine (1) and spleen (1).
Table 1.
Patient demographic characteristics.
| n | Sex (M:F) | Age range (mean) | |
|---|---|---|---|
| Ordinary nevus | 15 | 3:12 | 14–60 (38) yrs |
|
|
|||
| Dysplastic nevus | 17 | 4:13 | 14–71 (35) yrs |
|
|
|||
| Spitz nevus | 17 | 7:10 | 1.6–48 (21) yrs |
|
|
|||
| Melanoma in situ | 16 | 8:8 | 45–89 (68) yrs |
|
|
|||
| Superficial melanoma | 23 | 14:9 | 28–87 (59) yrs |
|
|
|||
| Deep melanoma | 23 | 13:10 | 34–93 (61) yrs |
|
|
|||
| Metastatic melanoma | 36 | 21:15 | 20–86 (59) yrs |
Immunohistochemistry
Formalin-fixed paraffin-embedded 5-μm tissue sections were deparaffinized and pretreated with citrate buffer at pH 6.0. After antigen retrieval, the sections were incubated with mouse monoclonal anti-DEK antibodies (1:400 dilution; lot 610948; BD Transduction Laboratories, Franklin Lakes, NJ, USA) at room temperature for 30 min, followed by EnVision+ System horseradish peroxidase (HRP)-conjugated goat anti-mouse (Dako, Carpinteria, CA, USA) for 30 min. Sections were then treated with peroxidase substrate solution containing 0.01% hydrogen peroxide and 0.05% diaminobenzidine tetrahydrochloride (DAB, Dako) and conterstained with hematoxylin. Tonsil tissue served as positive control.
A nuclear stain in melanocytes was considered as positive for DEK. The stained slides were reviewed by a dermatopathologist (LM) and an independent researcher (FK). The percentage of melanocytes expressing DEK was scored as <25%, 25–50% or >50%. The staining intensity was recorded as weak or moderate/strong. High level DEK expression was defined as a moderate to strong staining in at least 25% of the tumor cells. Low level DEK expression was defined as a positive DEK staining with weak intensity.
Statistical Analysis
Statistical analysis was carried out using StatsDirect software (Version 2.7.8, StatsDirect, Cheshire, UK). The Fisher-Freeman-Halton test was applied to Table 3 to assess differences in DEK expression between various melanocytic lesions. A P-value < 0.05 was considered statistically significant.
Table 3.
Histological characteristics of primary invasive melanomas.
| Breslow depth range (mean) | Growth phase (radial:vertical) | Total cases (DEK-positive cases)
|
||||
|---|---|---|---|---|---|---|
| SSM | LM | ALM | NM | |||
| Superficial melanoma | 0.12–1.00mm (0.53) | 6:17* | 18 (12) | 5 (3) | 0 | 0 |
| Deep melanoma | 1.15–9.13mm (2.84) | 0:23 | 9 (9) | 2 (2) | 4 (3) | 8 (6) |
Among superficial melanomas, 11 of 17 with vertical growth phase and 3 of 6 with radial growth phase showed high level of DEK.
SSM: superficial spreading melanoma; LM: lentiginous melanoma; ALM: acral lentiginous melanoma; NM: nodular melanoma
Results
In normal skin, moderate levels of DEK expression were present in dermal lymphocytes as well as in basilar keratinocytes of the epidermis and the follicular epithelium (Figure 1A).
Figure 1.

Representative DEK immunohistochemistry in A) normal skin with DEK nuclear staining in dermal lymphocytes as well as basilar keratinocytes in epidermis and follicular epithelium, B) ordinary nevus lacking DEK expression, C) dysplastic nevus with weak DEK-reactivity, and D) DEK-negative Spitz nevus. (200×)
DEK expression seen in various melanocytic lesions is summarized in Tables 2–4. We did not observe a difference in DEK expression with age or gender within each category of the melanocytic lesions. In this series, most ordinary nevi were negative or expressed low levels of DEK (Table 2; Figure 1B). Three cases (20%) demonstrated strong DEK reactivity. All 17 dysplastic nevi showed none or low levels of DEK expression (Figure 1C). Sixteen of 17 Spitz nevi were DEK-negative or had weak DEK expression, with a single case displaying strong staining (Figure 1D). Overall, 8.2% (4/49) benign nevi presented with high levels of DEK expression. When positive, the superficial portion of the dermal melanocytes often demonstrated stronger staining than the deep dermal component (data not shown). Junctional melanocytes were often not stained or displayed weaker DEK reactivity as compared to dermal melanocytes. The overall expression of DEK was not statistically different between any of the benign nevi groups (ordinary, dysplastic or Spitz nevi).
Table 2.
Percentage and intensity of DEK staining in melanocytic lesions (see below for graphic illustration of DEK staining results).
| n | Negative | <25% and weak* | ≥25% and weak* | <25% and moderate/strong | ≥25% and moderate/strong† | |
|---|---|---|---|---|---|---|
| Ordinary nevus | 15 | 8 (53%) | 0 (0%) | 4 (27%) | 0 (0%) | 3 (20%) |
| Dysplastic nevus | 17 | 11 (65%) | 1 (6%) | 5 (29%) | 0 (0%) | 0 (0%) |
| Spitz nevus | 17 | 10 (59%) | 4 (24%) | 2 (12%) | 0 (0%) | 1 (6%) |
| Melanoma in situ | 16 | 11 (69%) | 2 (13%) | 2 (13%) | 0 (0%) | 1 (6%) |
| Superficial melanoma | 23 | 8 (35%) | 2 (9%) | 1 (4%) | 0 (0%) | 12 (52%) |
| Deep melanoma | 23 | 3 (13%) | 0 (0%) | 1 (4%) | 0 (0%) | 19 (83%) |
| Metastatic melanoma | 36 | 2 (6%) | 2 (6%) | 3 (8%) | 1 (3%) | 28 (78%) |
Low level of DEK expression: positive stain with weak intensity.
High level of DEK expression: positive stain in at least 25% of tumor cells with moderate to strong intensity.
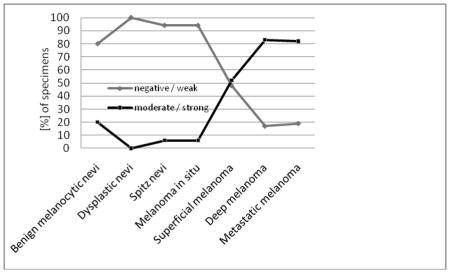
Table 4.
DEK expression and important prognostic factors in deep melanomas.
| Breslow depth
|
Ulceration | Angiolymphatic invasion | Neurotropism | Metastases (satellite/in-transit, nodal or distal) | |||
|---|---|---|---|---|---|---|---|
| 1.01 – 2 | 2.01 – 4 | >4 | |||||
| Negative or low level DEK (n=4) | 1 | 2 | 1 | 1 (25%) | 0 | 0 | 0 |
| High level DEK (n=19) | 9 | 7 | 3 | 8 (42%) | 4 (21%) * | 1 (5%) † | 8 (42%) ‡ |
1 patient with possible angiolymphatic invasion was not included.
this patient also had angiolymphatic invasion.
1 patient had both satellite metastasis and positive SLN.
Similarly to benign nevi, most cases of melanoma in situ showed negative or low levels of DEK expression with one case demonstrating strong staining (Figure 2A). However, upon acquiring features of invasion, DEK expression was greatly augmented. Thirty-one of 46 (67%) primary invasive (superficial and deep) melanomas displayed high levels of DEK, which was significantly more than all benign nevi combined (p<0.0001) or each individually (p<0.05 each) (Table 2; Figure 2B and 2C). In addition, there was a significant difference between melanoma in situ and superficial or deep melanomas (p=0.006 and p<0.0001, respectively), indicating the occurrence of dermal invasion may be associated with DEK upregulation. Among invasive melanomas, high levels of DEK were observed in a significantly higher proportion of deep melanomas (83%) than superficial melanomas (52%) (p<0.05). Melanoma tumor cells often demonstrated comparable DEK staining in superficial and deep dermal components without a stratified DEK expression seen in benign nevi (data not shown). No obvious difference in DEK levels was noted among various histological subtypes of deep melanomas (Table 3), although most desmoplastic melanomas did not express DEK (unpublished data). The levels of DEK expression appeared independent of further depth increments in deep melanomas (Table 4).
Figure 2.

Representative DEK immunohistochemistry in A) DEK-reactive melanoma in situ, B) primary superficial melanoma with DEK-positivity seen in in situ and invasive component, C) primary deep melanoma, and D) metastatic melanoma with strong DEK staining. (200×)
The association between DEK levels and important prognostic attributes was studied. In superficial melanomas, 11 of 17 (65%) tumors with vertical growth phase and 3 of 6 (50%) with radial growth phase displayed high levels of DEK (Table 3). Five of 9 superficial melanomas with dermal regression showed high levels of DEK expression. Interestingly, as shown in Table 4, all 19 deep melanomas that overexpressed DEK carried at least one adverse prognostic factor, including ulceration, angiolymphatic invasion, neurotropism, or associated metastases (satellite, in-transit, distal or nodal metastases). Besides one DEK-negative case (Breslow depth >4mm) that had ulceration, unfavorable prognostic indicators were not observed in the remaining 3 cases with low levels of DEK. Our findings suggest that high levels of DEK expression in deep melanomas may correlate with worse prognosis.
Consistent with previous reports [10–11], the majority (28/36, 78%) of metastatic melanomas were DEK-positive with high levels of expression (Table 2; Figure 2D). This staining pattern was not significantly different from deep melanomas.
Discussion
Alteration of chromosome 6p is one of the most consistent cytogenetic changes in melanomas [8–9]. In particular, gains of 6p have been associated with poor prognosis in ocular melanomas [25]. The chromatin architectural factor DEK is an oncogene located on the region of 6p22–23 [5–6]. We previously showed invariable overexpression of DEK in a genetically diverse set of 16 metastatic melanoma cell lines [10]. Furthermore, DEK reactivity by immunohistochemistry was observed in 72% and 90% of primary and metastatic melanomas using tissue microarrays, while 8 and 10 benign nevi, respectively, showed no detectable DEK expression [10–11]. In those studies, however, primary melanomas tested were mostly deep melanomas and benign melanocytic lesions were not well-represented. The current study expanded the previous findings and investigated DEK expression across a wide spectrum of benign and malignant melanocytic lesions comprising ordinary, dysplastic, and Spitz nevi, melanoma in situ, superficial and deep melanomas, as well as metastatic melanomas. This is the first study to correlate DEK levels in deep melanomas with histological prognostic factors to understand the role of DEK upregulation in melanoma progression. In this study, we took advantage of whole tissue sections to examine the spatial distribution of DEK expression in nevus or melanoma cells.
Consistent with earlier observations [10–11], our data showed that the majority of deep melanomas and metastatic melanomas were not only positive for DEK but also displayed high levels of expression. Unlike previously reported [10–11], 20–30% of benign nevi in our study demonstrated low levels of DEK expression with 8.2% showing strong reactivity. This discrepancy might be due to differences in antibody sensitivity and/or larger sample size of this study. Our results point to potential limitations of DEK immunohistochemistry in discriminating malignant melanocytic lesions from benign lesions. Although high levels of DEK was a more frequent event in deep melanomas and metastatic melanomas, its clinical utility should be exercised with caution given the potential overlap of DEK staining patterns in benign and malignant melanocytic lesions. Interestingly, we observed low levels of DEK expression in melanoma in situ. Additionally, we noted absence or low levels of DEK reactivity in a subset of junctional melanocytes for nevi or invasive melanomas as compared to dermal melanocytes. The etiology of this observation is unclear and it might be related to the role of DEK in the regulation of melanocytes migration in dermis.
One of the known functions of DEK in promoting melanoma oncogenesis is the inhibition of senescence. It has been demonstrated that benign nevi frequently contain oncogenic mutations in BRAF, yet remain arrested in a state of cellular senescence [26]. Indeed, rather than promoting proliferation, artificial expression of the oncogenic form of BRAF in normal human melanocytes leads to senescence in a process dubbed oncogene-induced senescence [27]. The events required to bypass this senescence response have not been well delineated. Given the anti-senescence properties of DEK described in keratinocytes and melanoma, it is conceivable that expression of DEK in melanocytes serves to circumvent oncogene-induced senescence and thus transform into melanoma either de novo or from pre-existing benign nevi. This would predict that the acquisition of DEK expression occurs prior to the transformation of nevi to melanomas. Indeed, our data showed low levels of DEK in some benign nevi and melanoma in situ. Upon acquiring invasive capability, superficial melanomas demonstrated higher DEK levels compared to benign nevi or melanoma in situ (p<0.05). It is possible that upregulation of DEK is initiated when vertical growth phase of melanoma occurs and continues to increase with invasion until plateauing in deep melanomas and their metastases when cellular senescence is bypassed.
The observation that DEK expression was significantly increased in deep and metastatic melanomas compared to superficial melanomas supports a role of DEK in melanoma progression. Moreover, we found that deep melanomas with high levels of DEK expression often presented with one or more poor prognostic features, such as angioinvasion, neurotropism, and the presence of metastases (including sentinel lymph node or other nodal involvement, and satellite, in-transit or distant metastases). It is possible that overexpression of DEK provides a dose-dependent effect that promotes advanced malignant features such as deep dermal invasion or subsequent metastasis. Likewise, DEK overexpression has recently been shown to be a negative prognostic factor in high grade neuroendrocrine carcinoma of the lung [7]. Interestingly, our previous in vitro study demonstrated that, in melanoma cell lines, long-term downregulation of DEK expression induced premature senescence whereas short-term downregulation of DEK bypassed melanoma chemoresistance [10]. Hence, DEK may be a relevant target for therapeutic intervention in deep melanomas and metastatic melanomas that overexpress the protein.
In summary, high level of DEK expression was less frequent in benign nevi. Most deep and metastatic melanomas displayed high levels of DEK. In deep melanomas, increased DEK expression was more commonly seen in cases having worse prognostic factors. Our results provide insight into DEK overexpression as an event for melanomagenesis. Our data support a role for DEK upregulation in melanoma progression. Future studies focusing on clinical outcomes may further confirm the association between DEK overexpression and poor prognosis in melanomas.
Acknowledgments
The authors thank Dr. Arul Chinnaiyan for providing laboratory support.
Footnotes
Disclosure/conflict of interest
The authors declare no conflict of interest.
References
- 1.Linos E, Swetter SM, Cockburn MG, Colditz GA, Clarke CA. Increasing burden of melanoma in the United States. J Invest Dermatol. 2009;129:1666–74. doi: 10.1038/jid.2008.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Troxel DB. Pitfalls in the diagnosis of malignant melanoma: findings of a risk management panel study. Am J Surg Pathol. 2003;27:1278–83. doi: 10.1097/00000478-200309000-00012. [DOI] [PubMed] [Google Scholar]
- 3.Troxel DB. Medicolegal aspects of error in pathology. Arch Pathol Lab Med. 2006;130:617–9. doi: 10.5858/2006-130-617-MAOEIP. [DOI] [PubMed] [Google Scholar]
- 4.von Lindern M, Fornerod M, van Baal S, et al. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell Biol. 1992;12:1687–97. doi: 10.1128/mcb.12.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans AJ, Gallie BL, Jewett MA, et al. Defining a 0. 5-mb region of genomic gain on chromosome 6p22 in bladder cancer by quantitative-multiplex polymerase chain reaction. Am J Pathol. 2004;164:285–93. doi: 10.1016/S0002-9440(10)63118-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orlic M, Spencer CE, Wang L, Gallie BL. Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosomes Cancer. 2006;45:72–82. doi: 10.1002/gcc.20263. [DOI] [PubMed] [Google Scholar]
- 7.Shibata T, Kokubu A, Miyamoto M, et al. DEK oncoprotein regulates transcriptional modifiers and sustains tumor initiation activity in high-grade neuroendocrine carcinoma of the lung. Oncogene. 2010 doi: 10.1038/onc.2010.217. [DOI] [PubMed] [Google Scholar]
- 8.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 9.Santos GC, Zielenska M, Prasad M, Squire JA. Chromosome 6p amplification and cancer progression. J Clin Pathol. 2007;60:1–7. doi: 10.1136/jcp.2005.034389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khodadoust MS, Verhaegen M, Kappes F, et al. Melanoma proliferation and chemoresistance controlled by the DEK oncogene. Cancer Res. 2009;69:6405–13. doi: 10.1158/0008-5472.CAN-09-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carro MS, Spiga FM, Quarto M, et al. DEK Expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle. 2006;5:1202–7. doi: 10.4161/cc.5.11.2801. [DOI] [PubMed] [Google Scholar]
- 12.Soares LM, Zanier K, Mackereth C, Sattler M, Valcarcel J. Intron removal requires proofreading of U2AF/3′ splice site recognition by DEK. Science. 2006;312:1961–5. doi: 10.1126/science.1128659. [DOI] [PubMed] [Google Scholar]
- 13.Mor-Vaknin N, Punturieri A, Sitwala K, et al. The DEK nuclear autoantigen is a secreted chemotactic factor. Mol Cell Biol. 2006;26:9484–96. doi: 10.1128/MCB.01030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kappes F, Fahrer J, Khodadoust MS, et al. DEK is a poly(ADP-ribose) acceptor in apoptosis and mediates resistance to genotoxic stress. Mol Cell Biol. 2008;28:3245–57. doi: 10.1128/MCB.01921-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gamble MJ, Fisher RP. SET and PARP1 remove DEK from chromatin to permit access by the transcription machinery. Nat Struct Mol Biol. 2007;14:548–55. doi: 10.1038/nsmb1248. [DOI] [PubMed] [Google Scholar]
- 16.Alexiadis V, Waldmann T, Andersen J, et al. The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes Dev. 2000;14:1308–12. [PMC free article] [PubMed] [Google Scholar]
- 17.Kappes F, Scholten I, Richter N, Gruss C, Waldmann T. Functional domains of the ubiquitous chromatin protein DEK. Mol Cell Biol. 2004;24:6000–10. doi: 10.1128/MCB.24.13.6000-6010.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ko SI, Lee IS, Kim JY, et al. Regulation of histone acetyltransferase activity of p300 and PCAF by proto-oncogene protein DEK. FEBS Lett. 2006;580:3217–22. doi: 10.1016/j.febslet.2006.04.081. [DOI] [PubMed] [Google Scholar]
- 19.Fu GK, Grosveld G, Markovitz DM. DEK, an autoantigen involved in a chromosomal translocation in acute myelogenous leukemia, binds to the HIV-2 enhancer. Proc Natl Acad Sci U S A. 1997;94:1811–5. doi: 10.1073/pnas.94.5.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sammons M, Wan SS, Vogel NL, et al. Negative regulation of the RelA/p65 transactivation function by the product of the DEK proto-oncogene. J Biol Chem. 2006;281:26802–12. doi: 10.1074/jbc.M600915200. [DOI] [PubMed] [Google Scholar]
- 21.Kappes F, Damoc C, Knippers R, et al. Phosphorylation by protein kinase CK2 changes the DNA binding properties of the human chromatin protein DEK. Mol Cell Biol. 2004;24:6011–20. doi: 10.1128/MCB.24.13.6011-6020.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cleary J, Sitwala KV, Khodadoust MS, et al. p300/CBP-associated factor drives DEK into interchromatin granule clusters. J Biol Chem. 2005;280:31760–7. doi: 10.1074/jbc.M500884200. [DOI] [PubMed] [Google Scholar]
- 23.Wise-Draper TM, Allen HV, Thobe MN, et al. The human DEK proto-oncogene is a senescence inhibitor and an upregulated target of high-risk human papillomavirus E7. J Virol. 2005;79:14309–17. doi: 10.1128/JVI.79.22.14309-14317.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wise-Draper TM, Allen HV, Jones EE, et al. Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol. 2006;26:7506–19. doi: 10.1128/MCB.00430-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Namiki T, Yanagawa S, Izumo T, et al. Genomic alterations in primary cutaneous melanomas detected by metaphase comparative genomic hybridization with laser capture or manual microdissection: 6p gains may predict poor outcome. Cancer Genet Cytogenet. 2005;157:1–11. doi: 10.1016/j.cancergencyto.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 27.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]