

Abstract
Highly active antiretroviral therapy (HAART) has greatly reduced the morbidity and mortality from HIV infection, but AIDS continues to be a serious health problem worldwide. Despite enormous efforts to develop a vaccine, there is still no cure, and alternative approaches including gene therapy should be explored. Here we developed and compared combinatorial foamy virus (FV) anti-HIV vectors that also express a mutant methylguanine methyltransferase (MGMTP140K) transgene to increase the percentage of gene modified cells after transplantation. These FV vectors inhibit replication of HIV-1 and also the simian immunodeficiency virus/HIV-1 (SHIV) chimera that can be used in monkey AIDS gene therapy studies. We identified a combinatorial FV vector that expresses 3 anti-HIV transgenes and inhibits viral replication by over 4 logs in a viral challenge assay. This FV anti-HIV vector expresses an HIV fusion inhibitor and two shRNAs targeted to HIV-1 tat and rev, and can be produced at high titer (3.8×107 transducing units/ml) using improved helper plasmids suitable for clinical use. Using a competitive repopulation assay, we show that human CD34+ cells transduced with this combinatorial FV vector efficiently engraft in a mouse xenotransplantation model, and that the percentage of transduced repopulating cells can be increased after transplantation.
INTRODUCTION
AIDS continues to be a serious health problem in the United States and worldwide. Highly active antiretroviral therapy (HAART) has greatly reduced viral loads and reduced morbidity and mortality from AIDS, 1 but the side effects can be severe2 and the emergence of drug resistance is a problem. Despite substantial efforts to develop a vaccine, 3-5 there is still no cure and alternative therapies need to be explored. One approach to cure AIDS has been by transplantation with genetically modified cells that inhibit HIV replication. Many synthetic genes have been developed that can inhibit replication of HIV-1 using a gene therapy approach using T lymphocytes or hematopoietic stem cells (HSCs) (reviewed in 6). One limitation in targeting mature lymphocytes for gene therapy, the predominant host cell for HIV-1 infection, is the limited number and persistence of engrafted gene-modified T cells observed in AIDS gene therapy studies. 7 The ability of HSCs to reconstitute the entire hematopoietic system, including T lymphocytes, macrophages, dendritic cells and microglial cells, suggests that they are ideal targets for AIDS gene therapy. However, the inability to efficiently deliver anti-HIV transgenes to HSCs has been a major roadblock in clinical trials. In a phase I clinical trial using bone marrow-derived CD34+ cells transduced with a Moloney leukemia virus (MLV) vector containing a Rev response element (RRE) decoy, short term marking rates of 0.003-0.01% and long-term marking rates below 0.001% were obtained. 8,9 In another study where mobilized CD34+ cells were used as targets, the average long-term marking rates with an MLV vector containing a ribozyme were 0.001% to 0.01%.10 Thus, methods for more efficient and potentially safer gene transfer of anti-HIV transgenes to HSCs are needed. Efficient gene delivery to T cells has been achieved using lentiviral vectors that express an antisense transcript to HIV env. 11 However, producing lentiviral vectors with other transgenes that target HIV packaging functions has reduced titers, 12,13 which affects their utility, although redesign of packing plasmids may overcome this obstacle.
Foamy viruses (FVs) are non-pathogenic, 14-16 integrating retroviruses with several properties that distinguish them from gamma- or lentiviruses. FV vectors have improved from early replication competent vectors to third generation vectors that do not contain detectable replication competent retroviruses. 17,18 FV vectors can efficiently transduce pluripotent murine 19 and human repopulating cells assayed in a murine xenotransplantation model. 20,21 Importantly, foamy vectors efficiently transduce repopulating cells in the canine model, 22 which has more closely reflected efficacy in clinical trials. Significant silencing of FV vector transgenes in murine, canine or human repopulating cells was not observed in these studies. FV vectors also have a distinct integration profile from gamma and lentiviral vectors, 23 which may reduce their risk for leukemia in a gene therapy setting.
Here we developed combinatorial anti-HIV FV vectors for AIDS gene therapy that also contain the P140K mutant of the O6-methylguanine-DNA methyltransferase (MGMT) gene to allow for in vivo selection after HSC transplantation. MGMT confers resistance to methylating agents such as temozolomide as well as to nitrosoureas such as bis-chloroethyl-nitrosourea (BCNU) and allows for efficient selection at the stem cell level in large animal models (reviewed in 24). The MGMT protein is expressed in normal human tissues, so in order to enhance selection, O6-benzylguanine (O6BG) is used to inactivate endogenous MGMT. The mutant P140K MGMT transgene confers resistance to O6BG25,26 so that an alkylating agent such as BCNU or temozolomide used in conjunction with O6BG allows for efficient selection of transduced hematopoietic repopulating cells in vivo. 27-29
We evaluated the ability of combinatorial FV anti-HIV vectors that also contain a MGMT transgene to inhibit HIV-1 and also SHIV (simian-human immunodeficiency virus) replication. SHIV chimeras contain the HIV-1 env, rev, tat, and vpu genes on a background of SIVmac and readily infect macaques. 30 The SHIV-macaque model is an excellent preclinical setting to test AIDS gene therapy strategies, since transgenes that target the HIV-1 components of SHIV can be assessed in this model. We also evaluated the engraftment of FV-transduced human CD34+ cells and MGMT-mediated in vivo selection of human hematopoietic repopulating cells using an anti-HIV FV vector in a mouse xenotransplantation assay.
RESULTS
FV anti-HIV vectors
The foamy vector plasmid FV-SMPGW contains the P140K version of MGMT for in vivo selection driven by a spleen focus-forming virus (SFFV) promoter, and also an enhanced green fluorescent protein (EGFP) reporter gene driven from the human phosphoglycerate kinase (PGK) promoter, as a platform to carry anti-HIV transgenes (Figure 1). To enhance MGMT and EGFP expression in vivo, a safety-modified woodchuck post-transcriptional regulatory element that does not express the partial X ORF31 was used. We chose this configuration because in previous experiments in the canine and macaque model we have observed efficient MGMT-based selection at the stem cell level using the SFFV promoter in lentiviral vectors (manuscript under review, H.P. Kiem and G.D. Trobridge), and have also observed efficient EGFP expression from the PGK promoter in both myeloid and lymphoid lineages in large animal models. 22,32 The FV vectors we developed contain several safety features including a U3-deleted LTR and two engineered stop codons to eliminate FV gag gene expression from the cis-acting region (CAR) required for efficient gene transfer. The FV vector system does not encode the foamy transcriptional transactivator Tas (formerly called Bel-1) which is required for transcription from the LTR, 33,34 so these FV vectors are self-inactivating.
Figure 1. FV vectors.
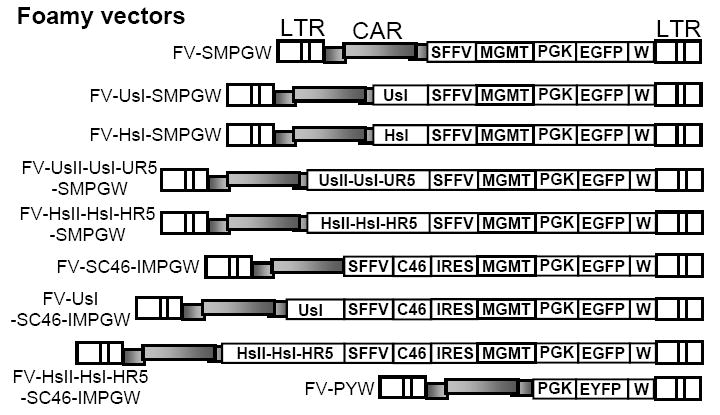
The FV vector FV-SMPGW contains an SFFV promoter (S) driving expression of the P140K mutant of MGMT (M) and also contains a human PGK promoter (P) driving expression of EGFP (G). This vector also contains a safety modified WPRE element (W). The following anti-HIV transgenes were added to this vector; a human U6 Pol III promoter that expresses an shRNA targeted to tat/rev at site I (FV-UsI-SMPGW), a human H1 Pol III promoter driven shRNA targeted to tat/rev at site I (FV-HsI-SMPGW), a combinatorial cassette with H1 promoter driving shRNAs targeted to tat/rev at site I and site II and to human CCR5 at bp 1023 (FV-HsIHsIIHR5-SMPGW). A combinatorial cassette with the U6 promoter driving shRNAs targeted to tat/rev at site I and site II and to human CCR5 at bp 741 (FV-UsIUsIIHR5-SMPGW), the transmembrane-localized C46 HIV envelope fusion inhibitor expressed from the SFFV promoter with an IRES expressing MGMT (FV-SC46-IMPGW), the U6 driven tat/rev site I shRNA combined with the C46-expressing vector (FV-U6sI-SC46-IMPGW) and the combinatorial H1 driven cassette with shRNAs targeted to tat/rev at site I and site II and to human CCR5 at bp 1023 combined with the C46-expressing vector (FV-HsII-HsI-HR5-SC46-IMPGW). The control FV vector FV-PYW has the human PGK promoter driving EYFP (Y). The long terminal repeats (LTR) and foamy cis-acting region (CAR) are also indicated.
The anti-HIV transgenes we used target HIV-1 genes that are also in the chimeric SHIV. We used this approach so that in future studies these FV vectors could be assessed in a pigtailed macaque model for AIDS gene therapy. Effective vectors in this model could then be tested in clinical studies since the anti-HIV transgenes would not change. Rapid escape of HIV-1 has been demonstrated for single shRNA therapeutics, 35 so combinatorial cassettes were developed in an attempt to reduce the potential for escape from a single anti-HIV shRNA. We evaluated transgenes that target a site I (sI) or a second site II (sII) in HIV-1 tat and rev, 36,37, which are also in SHIV, inhibit fusion mediated by the HIV-1 Env, which is also in SHIV, or target CCR5 expression, which affects macrophage-tropic strains of HIV-1 and SHIV. Homozygous CCR5-negative individuals resist HIV-1 infection without apparent immune dysfunction, 38,39 so CCR5 is a clinically-relevant target. We first compared CCR5-specific shRNAs using the psiCHECK system and identified two effective shRNAs target sites in CCR5 (Supplemental Figure 1). Two shRNA combinatorial cassettes were synthesized that expressed shRNAs using either human U6 Pol III promoters, or H1 Pol III promoters. A previous study had shown that the U6 promoter, which has a higher expression level than the H1 promoter, may reduce the fitness of transduced primary human T lymphocytes. 40 The two cassettes were introduced into FV-SMPGW to create FV-HsIHsIIHR5-SMPGW, which has two tat/rev shRNAs and a CCR5 shRNA each driven by a separate H1 promoter; and FV-UsIUsIIUR5-SMPGW, which has two tat/rev shRNAs and a different CCR5 shRNA each driven by a separate U6 Pol III promoter (Figure 1). We also evaluated a membrane-anchored C46 peptide that contains a partial ORF from the HIV envelope transmembrane subunit spanning from aa 628 to 673 of gp41. 41 For the C46 transgene, the signal peptide of the low-affinity nerve growth factor receptor (LNGFR) directs translocation into the endoplasmic reticulum, and the human immunoglobulin G2 (IgG2) hinge fused to the truncated CD34 membrane-spanning domain serves as a scaffold to anchor the peptide to the cell surface. High level expression of the C46 fusion inhibitor is required for a potent antiviral effect, 42 so we expressed C46 from the strong SFFV promoter and expressed MGMT from an encephalomyocarditis virus internal ribosome entry site (IRES). We chose this configuration because expression from an IRES leads to lower levels of expression than the transgene expressed from the promoter 5’ to the IRES. 43 Thus, higher levels of C46 expression should be obtained relative to MGMT. Following selection with BCNU and O6BG, cells expressing lower levels of MGMT would have a selective disadvantage, and cells surviving BCNU selection might have even higher levels of C46 than unselected cells.
Improved FV helper plasmids
Previously described helper constructs contain significant overlapping regions between the Gag, Pol and Env helper constructs. 18,21 Ideally, there would be no overlap between helper plasmids so that recombination between helper plasmids that may potentially lead to a replication competent retrovirus would be reduced. Towards this goal, we generated foamy Gag, Pol and Env helper constructs (Figure 2) that do not have significant overlap (pFMYGagCO and pFMYPol) or that have very limited overlap (31 nt of 100% identity for pFMYPol and pFMYEnv) between adjacent helper genes. The pFMYGagCO helper has the terminal 259 aa codons of the Gag ORF replaced with a codon-optimized gag sequence mutated to eliminate identity to the 5’ region of the Pol helper plasmid. The identity between the 3’ region of pol and the 5’ region of env was reduced by introducing a total of 4 silent mutations in this overlapping region. We found that these helpers gave similar titers to previously published helper constructs, 18,21 typically approximately 1×106 transducing units (TU)/ml unconcentrated and 4×107 TU/ml after 100-fold concentration for FV-SMPGW and, as expected, do not generate detectable replication-competent retrovirus. Importantly, the addition of the anti-HIV transgene cassettes evaluated here had a negligible impact on titers. For example, we were able to produce the combinatorial vector FV-HshIIHshIHshR5-SC46-IMPGW at a concentrated titer of 3.8×107 TU/ml. For two combinatorial vectors, FV-HshIIHshIHshR5-SMPGW and pFV-UsI-SC46-IMPGW, we compared titers to lentiviral vectors with identical transgene cassettes (Supplemental Fig. 2). The titers of lentiviral vectors that include combinatorial anti-HIV cassettes are reduced 14.4-fold and 150-fold, respectively, for the two combinatorial cassettes. Titers of the FV vectors with identical cassettes were not adversely affected.
Figure 2. Improved FV helper constructs.

A map of the wt HFV infectious clone pHSRV13 is shown above the foamy Gag, Pol and Env helper constructs that were generated by high-fidelity PCR from the wt HFV plasmid pHSRV13. To eliminate overlap between the Gag and Pol helper plasmids the carboxyl terminus 259 codons of the Gag ORF was replaced with a codon-optimized (CO) sequence (indicated by stripes) to create the pFV-GagCO helper plasmid. The overlap between the pFV-Pol and pFV-Env helper plasmids was reduced to only 31 bp of 100% identity by incorporating a total of four silent mutations indicated by crosses in the expanded view of the overlap between pol and env. All 3 foamy helper plasmids are driven by a cytomegalovirus (CMV) promoter. LTR is long terminal repeat. The foamy accessory proteins Tas (foamy transcriptional transactivator) and Bet are not present in the replication-incompetent FV vector system.
MGMT transgene expression and in vitro selection from FV vectors
We incorporated the P140K mutant MGMT25 into our vectors to allow us to increase the percentage of protected cells after transplantation. The MGMT cassette could be used to expand protected gene-modified cells in an AIDS gene therapy. We evaluated the expression of MGMT by immunofluorescence after transduction by the vector FV-SMPGW, which expresses both MGMT and EGFP, and compared expression to the control vector FV-PGW which only expresses EGFP (Figure 3a). MGMT expression was clearly higher in EGFP-positive transduced cells than in EGFP-negative untransduced cells, and only in the vector that contained MGMT.
Figure 3. FV vector MGMT expression and selection.
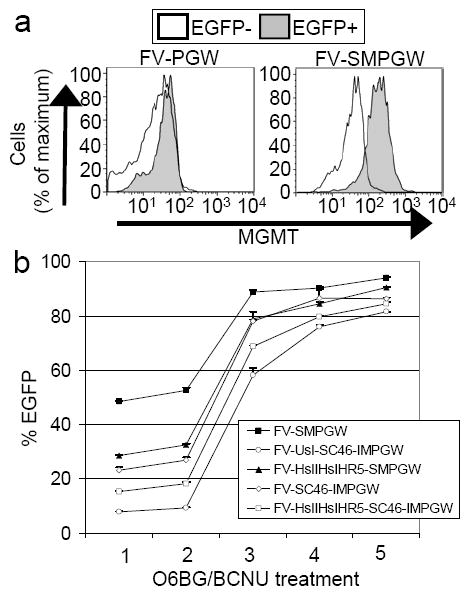
(a) Human HT1080 cells were transduced with the MGMT-containing foamy vector FV-SMPGW and the control vector FV-PGW and untransduced EGFP negative (EGFP-) and transduced EGFP positive (EGFP+) cells were gated and analyzed using an MGMT antibody conjugated to R-Phycoerythrin. Transduced (EGFP+) cells express the MGMT transgene for the FV-SMPGW vector but not the control FV-PGW vector that does not contain an MGMT transgene. (b). CEM.NKR-R5 lymphocytes were transduced with the foamy vectors that contain MGMT and EGFP and then selected multiple times with O6BG and BCNU. After MGMT-mediated selection the percentage of transduced cells increases as evidenced by an increase in the percentage of EGFP-expressing cells.
To confirm the function of the MGMT cassettes, we transduced CEM.NKR-R5 lymphocytes with FV vectors that contain both MGMT and EGFP at MOIs that gave between 5 and 50% EGFP-expressing cells and performed several rounds of selection with O6BG and BCNU (Figure 3b). As expected over the course of the O6BG and BCNU treatments, the percentage of EGFP-expressing cells increased, resulting in over 80%-EGFP expressing cells for all vectors tested after 5 rounds of selection. Efficient selection occurred regardless of whether MGMT was expressed directly from the SFFV promoter (vectors FV-SMPGW, FV-HshIIHshIHshR5-SMPGW) or if it was expressed from an internal ribosomal entry site (IRES) on a mRNA driven by the SFFV promoter (vectors FV-SC46-IMPGW, FV-U6shI-SC46-IMPGW, FV-HshIIHshIHshR5-SC46-IMPGW).
Foamy combinatorial anti-HIV vectors potently inhibit SHIV infection
We compared the ability of FV vectors with different anti-HIV transgene cassettes to inhibit SHIV infection in vitro. SHIV chimeras contain the HIV-1 env, rev, tat, and vpu genes on a background of SIVmac and can be used to test AIDS gene therapy strategies in a macaque model. We employed a challenge assay where transduced lymphocytes were exposed to SHIV and the supernatants were tested for virus using an assay that allows quantitation of SHIV infectious units as blue foci on transduced cells (blue cell focus-forming unit, BCFU). 44,45 This approach allowed us to compare the ability of each vector to inhibit SHIV replication over a period of 3 weeks using a highly quantitative assay for infectious virions produced from FV-transduced cell populations. CEM.NKR-CCR5 lymphocytes were transduced with the different FV vectors, then selected with O6BG and MGMT to approximately 80% EGFP-expressing cells. To directly compare the relative ability of each transgene cassette to inhibit replication, these FV-transduced polyclonal populations were then enriched by FACS to over 95% EGFP-expressing cells prior to challenge to minimize the effects from residual unprotected cells. We reasoned that sorting gene-modified cells to a similar and very high level would minimize the effects of different levels of unprotected cells, and would allow for a better comparison of the relative efficacy of the different vectors.
In the first experiment, vectors that expressed the C46 alone, the U6-driven site I shRNA, the triple H1-driven shRNA cassette, the triple U6-driven cassette, or a combination of the U6-driven site I shRNA and the C46 transgene were compared in an in vitro infection assay where FV-transduced lymphocytes were challenged in triplicate at a relatively low MOI (0.05) (Figure 4a). There is an additional difference between the H1 and U6-driven cassettes; the H1-driven site I and site II tat/rev shRNAs are shifted -1 bp relative to the U6 shRNAs in order to provide a canonical adenosine as the first transcribed nucleotide. 46 For this in vitro SHIV challenge the virus is an X4 isolate, so R5-specific shRNAs would not have contributed to inhibition in this assay. Both the C46 transgene and the triple H1-driven cassette potently inhibited SHIV replication. Importantly, in these experiments FV-transduced cells were in continuous culture for over 5 weeks after vector exposure, so the antiviral efficacy observed was due to stable transgene expression. The combination of the U6-driven site I shRNA and the C46 transgene provided potent inhibition of replication. The FV vector with only the U6-driven site I shRNA also inhibited replication, but by day 21 the viral titers had increased dramatically in comparison to the triple H1-driven cassette or the C46 transgene. Interestingly, the H1-driven combinatorial cassette more potently inhibited SHIV infection than the U6-driven cassette.
Figure 4. Inhibition of SHIV replication.
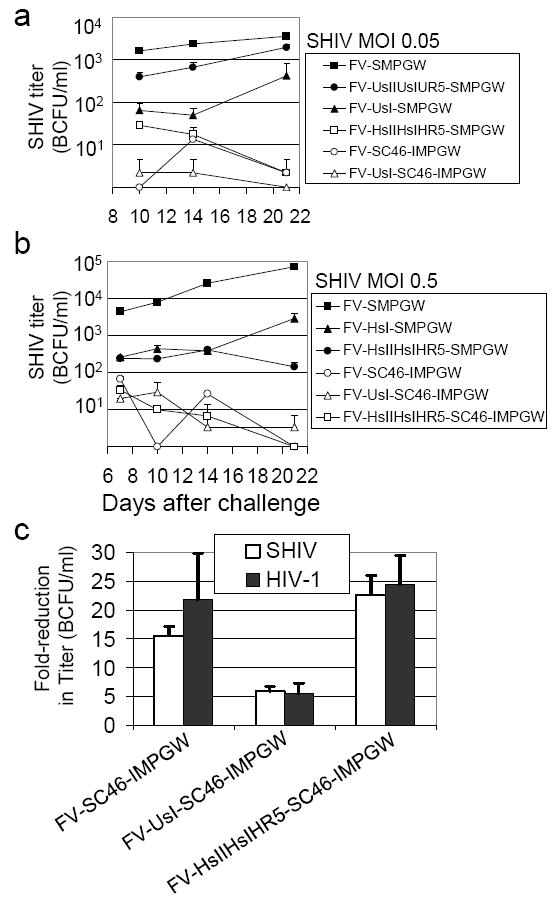
(a) Human CEM.NKR-R5 lymphocytes were transduced with foamy vectors, selected with O6BG and BCNU then sorted to over 95% EGFP-expressing cells and challenged in triplicate with SHIV KU-1 at an MOI of 0.05. At the indicated times after challenge the titer of SHIV in the supernatant was determined by MAGI-CCR-5 assay (BCFU is blue cell focus-forming unit). Zero titers are plotted on the X axis. (b) Human CEM.NKR-CCR5 lymphocytes cells were transduced with the indicated vectors but the challenge virus MOI was increased to 0.5 (c) The three best anti-HIV vectors were evaluated for SHIV and HIV-1 inhibition using a single-cycle replication assay where MAGI-CCR-5 cells were transduced with the indicated vectors, sorted to over 98% EGFP-positive, then challenged and tested for virus replication 48 hours after challenge. The fold-reduction in detected infectious units is reported for each vector relative to the control vector FV-SMPGW that does not contain an anti-HIV transgene.
Based on these results we combined the C46 transgene with the most potent shRNA cassette, HsIIHsIHR5, to create FV-HsIIHsIHR5-SC46-IMPGW. We also created a vector with a single H1-driven site I shRNA, FV-HsI-SMPGW, to compare its effectiveness to the combinatorial HsIIHsIHR5 cassette. We then repeated the challenge assay using three independently generated FV-transduced populations that were O6BG/BCNU-selected, sorted to over 95% EGFP-expressing cells, and then challenged at a 10-fold higher MOI (0.5) (Figure 4b). At this higher challenge dose, SHIV replicated to over 7×104 BCFU/ml in cells transduced with the control FV-SMPGW vector by day 21 after challenge. The triple H1-driven cassette still potently inhibited replication over 180-fold by day 21 after challenge, and at this last timepoint the inhibition of SHIV replication was significantly higher than the vector that expressed only a single site I shRNA driven by an H1 promoter. At the higher challenge dose, the C46 transgene again potently inhibited SHIV replication, with over 960-fold reduction at day 14 and by day 21 over 4 logs (no infectious units were detected). FV vectors that contained the C46 in addition to either the single U6-driven site I shRNA or the triple H1-driven HsIIHsIHR5 cassette also potently inhibited replication over 4 logs by day 21 after challenge. The C46 transgene by itself potently inhibited SHIV replication in this assay, and we could not observe additional effects mediated by either the U6-driven sI shRNA or the triple H1-driven cassette, even at this higher MOI.
Comparison of FV anti-HIV vectors’ ability to inhibit HIV-1 and SHIV using a single-cycle infection assay
We next compared the ability of the three best vectors to inhibit SHIV and HIV-1 using a highly quantitative single-cycle infection assay. In this assay the indicator MAGI-CCR-5 cell line was transduced with FV anti-HIV vectors, and after 14 days the cells were sorted by flow cytometry to over 98% percent EGFP-expressing cells and infected with approximately 1000 transducing units of either HIV-1 or SHIV. The FV-transduced and challenged MAGI-CCR-5 cells were then evaluated for viral replication 48 hours after infection. The MAGI-CCR-5 cell line detects both SHIV and HIV-1 infection, so this approach allowed us to directly compare the relative ability of our three best vectors to inhibit SHIV and HIV-1 infection during only one round of infection. For each FV anti-HIV vector the fold-reduction in infected cells is reported relative to cells transduced with the control vector FV-SMPGW (Figure 4c). The FV vector expressing C46 provided approximately 15-20-fold inhibition of viral replication. In this assay we observed a dramatic reduction of infected cells as determined by activation of the LTR-driven β-galactosidase transgene in the indicator cell line, and thus most C46-expressing cells had no evidence of infection. However, we can not rule out that some cells, possibly ones with lower C46 expression, may have been infected. The FV vector that also contained the U6-driven inhibited infection to a lesser extent and the FV vector with the triple H1-driven HsIIHsIHR5 cassette and C46 provided approximately 23-fold protection. For all three vectors the difference between inhibition of SHIV and inhibition of HIV was not significant (p>0.05) as would be expected, since these vectors target HIV-1 tat, rev and/or env that are in both HIV-1 and SHIV.
Copy number analysis and analysis of recombination
We determined the average proviral vector copy number in the transduced human lymphocytes used for the above SHIV challenge experiments by real-time PCR. We found that all anti-HIV vectors were similar in copy number which varied by less than 3.3-fold (Supplemental Figure 3a). Interestingly, we found that the vectors with U6-driven cassettes had the lowest copy numbers. Recombination events during reverse transcription have previously been described for lentiviral vectors with tandem Pol III promoters. 47 We evaluated potential recombination in our foamy vectors using PCR primers that flanked the shRNA expression cassettes (Supplemental Figure 3b,c). We found that the tandem triple U6 vector had efficiently recombined, which likely explains the lack of antiviral activity we observed for this vector. For the two FV vectors that contained tandem triple H1-driven cassettes, we observed evidence of partial recombination, with products corresponding to a single R5 shRNA, two shRNAs, and the intact triple H1-driven cassette. Despite this partial recombination, the triple H1-driven cassette inhibited replication (Figure 4), and we could detect expression of both the sI and sII shRNAs, see below. As expected, there was no evidence of recombination for vectors with single shRNA cassettes.
Evaluation of anti-HIV transgene expression from FV anti-HIV vectors
We evaluated C46 expression by transducing HT1080 cells with either the control vector FV-SMPGW or the anti-HIV vector FV-SC46-IMPGW that expresses the C46 fusion inhibitor. After vector exposure, cells were cultured for over three weeks and C46 expression was evaluated by immunofluorescence. High-level C46 expression was readily detected in transduced EGFP-positive cells in the FV-SC46-IMPGW vector but not in untransduced EGFP-negative cells, nor in the FV-SMPGW control vector which does not contain the C46 transgene (Figure 5a).
Figure 5. Anti-HIV transgene expression.
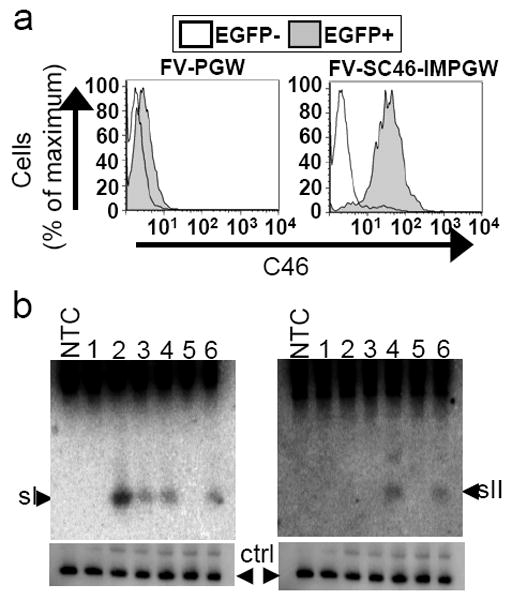
(a) C46 expression. Human HT1080 cells were transduced with the foamy vector FV-SC46-IMPGW and the control vector FV-SMPGW and untransduced EGFP negative (EGFP-) and transduced EGFP positive (EGFP+) cells were gated and analyzed for C46 expression using a primary antibody specific for C46, and a secondary antibody conjugated to phycoerythrin. Transduced (EGFP+) cells express the C46 transgene for the FV-SC46-IMPGW vector but not the control FV-SMPGW vector that does not contain the C46 transgene. (b) siRNA expression. CEM.NKR-CCR5 cells were transduced with the following vector constructs and controls. NTC is non-transduced control of CEM.NKR-CCR5 cells, 1=FV-SMPGW, 2=FV-UsI-SMPGW, 3=FV-HsI-SMPGW, 4=FV-HsIIHsIHR5-SMPGW, 5=FV-SC46-SMPGW, 6=FV-HsIIHsIHR5-SMPGW. The cells were selected using O6BG and BCNU and then sorted, total RNA was extracted and analyzed by Northern blot. The top left panel was hybridized to a site I tat/rev probe, and the top right was hybridized to a site II tat/rev probe. In the bottom panels U6 snRNA was probed as a control for RNA loading. The detected site I and site II siRNAs are approximately 21 nucleotides.
We also evaluated the expression of siRNAs in transduced CEM.NKR-CCR5 lymphocytes by northern blot analysis. We were able to detect siRNA expression for both the site I and the site II tat/rev of all FV vectors that contained a corresponding shRNA, while siRNA expression was not detected from the vectors FV-SMPGW or FV-SC46-IMPGW that do not contain an shRNA cassette (Figure 5b). RNA loading was evaluated using a probe to U6 snRNA. 48 In the vectors that expressed both the site I and site II tat/rev shRNAs, both siRNA species could be detected. Expression of the site I tat/rev siRNAs was higher from the U6 Pol III promoter than from the H1 Pol III promoter, as has been observed for lentiviral vectors expressing CCR5 shRNAs. 40 We could not detect expression of CCR5 siRNAs by northern blot and were also unable to detect a reduction in the level of CCR5 expression by immunostaining (data not shown). It is unclear whether this is due to the relative location of the CCR5 shRNA cassette on the vector (3’ to the sII and sI cassettes) or if it is specific to the CCR5 shRNA. However, we had shown that this CCR5 site could be targeted by shRNA, at least when transfected using the psiCHECK system (see Supplemental Figure 1). Thus, in the two vectors that also contained an H1-driven R5 shRNA cassette, we were unable to detect CCR5-specific siRNA expression or knockdown of CCR5 as determined by antibody staining. This cannot be easily explained by recombination, since the R5 shRNA expression cassette was the 3’ shRNA and recombination would be expected to leave this cassette intact. However, as expected based on the SHIV and HIV-1 challenge results, all FV vectors stably expressed the tat/rev siRNAs after MGMT selection.
Engraftment of FV-transduced human CD34+ cells in NOD-SCID IL2Rγnull mice
To translate an AIDS gene therapy approach to the clinic, it is important to determine if hematopoietic repopulating cells transduced with proposed anti-HIV vectors can efficiently expand and also engraft. We did not observe large differences in the percentage of human CEM.NKR-R5 lymphocytes obtained after transduction with the various anti-HIV cassettes, or in the expansion of these cells during O6BG and BCNU selection (see Figure 3). However, to address this issue more carefully, we transduced human CD34+ cells and evaluated potential toxicity in progenitors and also engraftment in the NOD-SCID IL2Rγ null mouse model which allows for robust engraftment of human SCID repopulating cells (SRCs). 49 For these studies we created a control vector FV-PYW (Figure 1) that expressed only the enhanced yellow fluorescent protein (EYFP). Human CD34+ cells were transduced with either the FV-HsIIHsIHR5-SC46-IMPGW vector or the FV-PYW vector, and the percentage of EGFP or EYFP expressing cells in colony forming units (CFUs) was compared to the percentage of EGFP-expressing cells in corresponding liquid cultures in 5 replicates of 100 CFUs each. We found that the percentage of EGFP-expressing (FV-HsIIHsIHR5-SC46-IMPGW) CFUs was similar to the percentage of transduced cells in liquid cultures; the percentage of EGFP expressing cells in liquid culture was 0.72-fold lower than the percentage of EGFP expressing CFUs. This was similar to what we observed for the control EYFP-expressing FV-PYW control, where the percentage of EYFP expressing cells in liquid culture was 0.77-fold lower than EYFP expressing CFUs. When the fold-differences between the percentage of transduced cells in liquid culture versus progenitors was compared between the 5 replicates for the two vectors (mean of 0.72-fold versus 0.77-fold) the difference was not statistically significant (p=0.86, Students T-test). In these studies the size of control EYFP and anti-HIV EGFP colonies were variable, but there was no obvious difference in overall colony size for the FV-HsIIHsIHR5-SC46-IMPGW progenitors and the control FV-PYW progenitors. Thus, there was no evidence that expression of the anti-HIV transgene cassette altered the growth of human CD34-derived progenitors.
To assess the effects of the MGMT and anti-HIV cassettes on engraftment, we performed a competitive repopulation experiment in NOD-SCID IL2Rγ null mice. The EYFP protein differs only by 5 aa with EGFP and can be easily discriminated by flow cytometry and, thus, is an excellent marker for this purpose. We performed this competitive repopulation transplant in a cohort of 5 mice using human CD34+ cells transduced with our combinatorial vector FV-HsIIHsIHR5-SC46-IMPGW (arm 1) or the control vector FV-PYW (arm 2). In this experiment, two different vector stock preparations were used for each vector, and the order of injection of each arm was alternated to control for the potential effect of these variables on engraftment. One animal died prior to the first analysis at 5 weeks, and engraftment and marking were analyzed in the BM of the remaining 4 mice at 5 weeks after transplantation. Engraftment was high, and the marking levels in vivo closely matched what we observed in the human CD34+ cultures pre-transplant (Figure 6). An MOI of 2 based on transduction of human HT1080 cells was used for both the experimental arm FV-HsIIHsIHR5-SC46-IMPGW and the control arm FV-PYW. Marking was significantly higher for the FV-PYW control arm in pre-transplantation liquid cultures and in the BM at 5 weeks after transplantation. However, there was no significant difference in the marking levels in liquid cultures pre-transplantation and the marking in the BM at 5 weeks for either the control vector FV-PYW (p=0.35) or for the combinatorial anti-HIV vector FV-HsIIHsIHR5-SC46-IMPGW (p=0.15). These data show that the anti-HIV and MGMT cassettes in the combinatorial FV vector FV-HsIIHsIHR5-SC46-IMPGW do not interfere with the engraftment capacity of human CD34+ SRCs.
Figure 6. Engraftment and MGMT-mediated in vivo selection of FV-transduced SRCs.
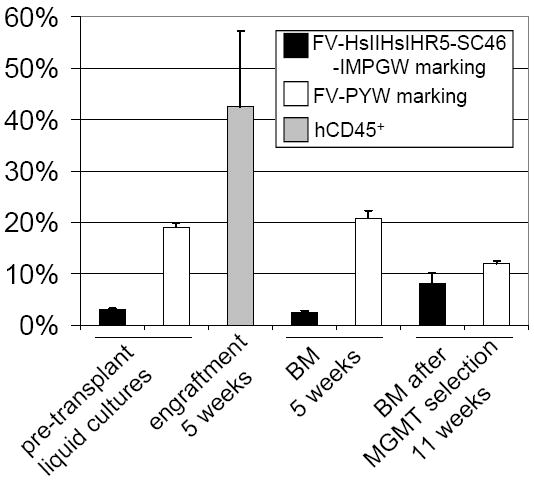
The mean marking for the FV EGFP-expressing anti-HIV vector and the control EYFP-expressing vector is shown in pre-transplant liquid cultures and CFU assays, and in human CD45+ cells in mouse BM 5 weeks after transplantation as determined by flow cytometry. The mean engraftment of human CD45+ cells as determined by flow cytometry is also shown in mouse BM at 5 weeks. Error bars represent the standard error from four transplanted mice. Mice were treated with O6BG and BCNU at 7 and 10 weeks after infusion of SRCs and the percentage of EGFP and EYFP expressing CD45+ cells in BM at 11 weeks after transplantation is shown.
Expansion of FV-transduced human SRCs
At 7 and 10 weeks after infusion of FV-transduced cells, we treated the NOD-SCID IL2Rγ null mice with O6BG and BCNU to determine if we could increase the percentage of gene-modified cells in vivo. Following these two treatments the percentage of EGFP expressing cells increased significantly (p<0.05) for the anti-HIV MGMT vector FV-HsIIHsIHR5-SC46-IMPGW (Figure 6), and marking with the control vector FV-PYW decreased as expected. The final marking level was 8.2% for the FV-HsIIHsIHR5-SC46-IMPGW experimental arm. It should be noted that if this vector was used without a control arm, the marking would be expected to be twice as high, approximately 16%. In summary, human SRCs transduced with a potent combinatorial FV anti-HIV vector can be expanded after transplantation using the MGMT transgene.
DISCUSSION
Here we developed FV vectors with combinatorial anti-HIV cassettes that confer potent protection from both HIV-1 and SHIV infection and demonstrated their efficacy for HSC gene transfer. These vectors also express an EGFP reporter gene for facile detection of transduced cells in vivo, and an MGMT cassette to allow for selection of protected cells after transplantation.
FV vectors have been developed for gene therapy approaches and have several advantages, including their large packaging capacity18 and their ability to efficiently transduce hematopoietic repopulating cells. 19,20,22 FV vectors are particularly advantageous for AIDS gene therapy because foamy retrovirus has low nucleotide sequence homology with HIV, 50 so mobilization of foamy vector proviruses in transduced cells by wild-type HIV infection will not be a consideration, 51 Also, expression of anti-HIV transgenes that target HIV packaging functions and greatly reduce HIV-1-based lentiviral titers, 8,13 do not adversely affect foamy vectors (52 and our data). The ability to efficiently deliver anti-HIV transgenes that cannot be delivered by HIV-1-based lentiviral vectors is a key advantage of FV vectors for AIDS gene therapy. We observed that FV vector titers were not adversely affected by transgene cassettes that greatly reduced lentiviral titers.
Previous studies have shown that a U6-driven shRNA that targets tat and rev can protect from SIV or HIV-1 infection when introduced using foamy vectors. 52,53 However, protection from single shRNAs can be rapidly overcome by single base pair mutations. 35 We observed an increase in viral replication at later timepoints when a FV vector with a single tat/rev shRNA was used to inhibit SHIV replication, but observed potent inhibition in vitro when this shRNA was combined with an additional tat/rev shRNA. When we combined our combinatorial H1-driven shRNA cassette with the C46 fusion inhibitor, we observed a >20-fold reduction in a single cycle infection assay, and a >10,000-fold reduction in a challenge assay that evaluates multiple cycles of virion production and infection. In both assays, cells were transduced at relatively low MOIs and maintained stable resistance to viral infection. Even after MGMT-mediated selection and extensive culture, transduced lymphocytes remained highly protected from SHIV infection. We did observe efficient recombination between tandem shRNA cassettes with three U6 Pol III promoters, and partial recombination when using three H1 promoters. We combined a total of six transgenes, four anti-HIV transgenes, MGMT, and EGFP into the vector FV-HsIIHshIHR5-SC46-IMPGW and were able to demonstrate stable expression and function for 5 of the 6 of these transgenes. However, we could not detect expression or function for the H1-driven CCR5 shRNA in this vector, or in the FV-HsIIHsIHR5-SMPGW vector. Despite the absence of detectable R5-specific siRNA expression, this combinatorial vector that expressed three anti-HIV transgenes potently inhibited both SHIV and HIV-1 infection. Future combinatorial shRNA FV vector designs should take these findings into consideration, and it may be that expression of multiple shRNAs from a miRNA48 may overcome the above limitations.
A previous study showed that high-level expression of shRNAs from the U6 Pol III promoter impaired the ability of transduced lymphocytes to expand, and that expression of a CCR5 shRNA from a H1 Pol III promoter was well tolerated in macaque HSCs. 54 We thus developed an anti-HIV cassette where three shRNAs were each driven by three H1 promoters and found that this cassette potently inhibited SHIV and HIV-1 replication and did not impair engraftment of human SRCs. Ectopic expression of the C46 fusion inhibitor had previously been shown not to induce major toxic effects in transplanted murine hematopoietic cells. 55 Interestingly, we found that FV vectors with H1-driven cassettes were effective, showing that higher level expression was not necessary for a potent antiviral effect. Human SRCs transduced with FV vectors that express the P140K mutant form of MGMT and a combinatorial anti-HIV cassette were able to efficiently engraft NOD-SCID IL2Rγ null mice. The competitive repopulation assay demonstrated that this FV vector engrafted as efficiently as the control FV vector, when compared to the relative pre-transplant marking in CD34+ cells. Thus, expression of FV vector-delivered H1-driven tat/rev shRNAs does not appear to compromise SRC engraftment. The mouse xenotransplantation model may overestimate transduction of primate HSCs, 56 so future studies to evaluate transduction of nonhuman primate long-term repopulating cells with FV vectors will be of great value in moving this approach to the clinic. However, the NOD-SCID IL2Rγ null competitive repopulation model we have described here should be an excellent way to compare potential off-target effects of anti-HIV cassettes including cassettes that mediate RNAi.
The FV vectors we describe here target steps of the HIV-1 replication cycle that are also used by SHIV, so they can be evaluated in the highly relevant macaque SHIV model, which has played a crucial role in the development of anti-HIV treatment strategies. 57-59 In this model, MGMT-mediated selection could be used to increase the percentage of FV-transduced cells prior to challenge to determine if macaques are protected from simian AIDS, towards establishing a proof-of-concept for AIDS gene therapy. These FV vectors should also be effective for clinical studies given the potent inhibition of HIV-1 that we have demonstrated. The improved FV helper plasmids we describe here are well suited for future clinical applications. After removing the PGK-EGFP cassette from our FV vectors, they may be particularly effective in a clinical setting of AIDS lymphoma. After autologous transplant with FV-transduced cells, treatment of patients with O6BG and BCNU would not only have an anti-leukemic effect, but should also increase the number of cells protected from HIV-1.
MATERIALS AND METHODS
FV MGMT vectors and helper plasmids
The FV vector plasmid pFV-SMPGW was constructed by synthesizing an LTR-fusion promoter with the human cytomegalovirus immediate early promoter (CMV) and the FV LTR R and U5 region, in addition to the cis-acting gag region required for efficient foamy replication, followed by a multiple cloning site, and a 3’ LTR with a deleted FV U3 region and the R and U5 region. This dsDNA fragment was synthesized by Blue Heron Biotechnology (Bothell, WA) using GeneMaker technology according to the sequence specified and inserted into a 2-kb plasmid backbone derived from pUC19 from the SapI to BsmBI site. A 2.3-kb FV CAR with the 3’ region of pol and the 5’ region of env required for efficient gene transfer18,60 was PCR-amplified from the FV infectious plasmid pHSRV13 (kind gift from A. Dusty Miller, Fred Hutchinson Cancer Research Center) using the primers 5’-TTCGAATTAAGGCTATGGATTTGGCCATGGGAC-3’ and 5’- CAAGGTGTATACTATGAACCCCATCCGGCGCGCC-3’. The PCR amplification was performed with Phusion high fidelity DNA polymerase from New England Biolabs (Ipswich, MA) using 5 cycles at 50°C annealing temperature and 25 cycles at a 60°C annealing temperature following the manufacturer’s protocol. This FV CAR was inserted into the synthesized vector at an engineered multiple cloning site. A transgene cassette including the SFFV promoter, P140K MGMT mutant, and a PGK promoter driving enhanced green fluorescent protein and a modified woodchuck post-transcriptional regulatory element was transferred from the previously described lentiviral vector RSC-SMPGW32 to create the FV vector plasmid pFV-SMPGW. To create pFV-PYW, the SFFV-MGMT cassette was removed and the EGFP was replaced with enhanced yellow fluorescent protein (EYFP) from the lentiviral vector plasmid pRRL.sin.cPPT.Pgk.EYFP.pre (kind gift from Luigi Naldini, San Raffaele Telethon Institute for Gene Therapy).
The foamy helper plasmids were generated by PCR amplification of the foamy structural genes gag, pol and env from pHSRV13 using Phusion enzyme with the following primers: pFMYGag, 5’-CGGCCGGCCACCATGGCTTCAGGAAGTAATGTTGAAGAATATG-3’ and 5’-TTTAGTCTCTTTGGTCTCCGCCGGAAGCGGC-3’, pFMYPol, 5’-CGGCCGGCCACCATGAACCCTCTGCAGCTGTTACAGCCGCT-3’ and 5’-TTTACTCATTCTTTTCCAAATGATCCATTGTTGCAGTGTC-3’ and pFMYEnv 5’-CGGCCGGCCACCATGGCCCCACCCATGACACTGCAACAATGGATCAT-3’ and 5’-AGGCTACTGATTCTTCTTTTTCGTAGGAATC-3’. After PCR amplification the genes were subcloned into the NotI sites of pCMVβ replacing the β-galactosidase gene. pFMYGagCO was constructed by replacing a 795-bp fragment from the NsiI site in Gag to the 3’ NotI site after the Gag ORF, with a synthesized fragment with an identical Gag ORF, but with the carboxyl terminus 259 Gag codons optimized for expression in human cells.
Construction of foamy anti-HIV vectors
The anti-HIV shRNA transgenes were inserted into the FV vector plasmids at a SacII site in the vector multiple cloning site 3’ to the foamy CAR and 5’ to the MGMT-EGFP transgene cassette. pFV-UsI-SMPGW contains a human U6 Pol III promoter driving expression of a previously described site I shRNA targeted to tat/rev. 36,37 Plasmids that contain U6 promoters driving expression of shRNAs targeting two sites in human CCR5 were constructed and tested using the psiCHECK (fPromega, Madison, WI) shRNA system following the manufacturer’s protocols. A three-shRNA cassette with three H1 Pol III promoters each driving expression of a shRNA that targets site I, site II HIV-1 tat/rev, 36,37 or a 21-bp CCR5 shRNAs targeting bp 1023-1043 of human CCR5 was synthesized and subcloned into the pFV-SMPGW plasmid, creating pFV-HsIIHshIHR5-SMPGW. For the H1-driven cassette HsIIHsIHR5, we modified the previously described site I and site II tat/rev shRNAs and the CCR5 1024 shRNA by shifting the shRNAs -1 bp, so the start site would be an adenosine, the canonical first nucleotide used by the H1 promoter. 46 pFV-UsIIUsIUR5-SMPGW was made by inserting a synthesized cassette with three U6 promoters, driving expression of a site I tat/rev shRNA, a site II tat/rev shRNA, and a CCR5 shRNAs designed to bp 741-761 of human CCR5 into the pFV-SMPGW plasmid. The single H1-driven shRNA foamy vector plasmid pFV-HsI-SMPGW was constructed by deleting the other two H1-driven shRNA cassettes out of pFV-HsIIHsIHR5-SMPGW using unique restriction sites engineered between the H1 promoter-shRNA cassettes. The C46 anti-HIV peptide contains a partial ORF from the HIV envelope protein transmembrane subunit spanning from aa 628 to 673 of gp41. 41 To create pFV-SC46-IMPGW, the C46 transgene was amplified by PCR using primers 5’-CCAGGTACCGCTCTAGCGCTACCGGTC-3’ and 5’-CCAGGTACCGCTGGAGCTCTCCGGATCAG-3’, and the PCR product was introduced into the Acc65 I sites of pFV-SMPGW to create pFV-SC46-PGW. The IRES-MGMT was then introduced by ligating a FseI to BsiWI fragment of MP71.C46.IRES2.MGMT.Pre (kind gift of Chris Baum, Hannover Medical School, Hannover, Germany) to create pFV-SC46-IMPGW. pFV-UsI-SC46-IMPGW and pFV-HsIIHshIHR5-SC46-IMPGW were constructed by combining pFV-SC46-IMPGW with either pFV-UsI-SMPGW or pFV-HsIIHshIHR5-SMPGW using standard cloning techniques. The sequences of foamy vector, helper plasmids and transgene cassettes are provided in Supplemental Table 1.
Cell culture
Human 174xCEM cells (Cat# 272), 61 CEM.NKR-CCR5 (Cat# 4376), 62 and MAGI-CCR-5 cells45 (Cat# 3522) were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, from Drs. Peter Cresswell, Alexandra Trkola and Julie Overbaugh, respectively. 174xCEM cells and CEM.NKR-CCR5 cells were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM). MAGI-CCR-5 cells and human embryonic kidney 293 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM). All cultures were supplemented with 10% heat-inactivated (56°C for 30 minutes) fetal bovine serum (FBS; HyClone, Logan, UT), unless otherwise stated, with 100 U/ml of penicillin, and 100 μg/ml of streptomycin, and were incubated at 37°C in a 5% CO2 atmosphere. Human CD34+ cells were collected from volunteers as part of an institutional review board approved protocol, isolated by magnetic beads (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s instructions, and stored in liquid nitrogen until use.
Preparation of FV vector stocks
Foamy vector stocks were produced using either calcium phosphate transfection or by polyethylenimine (PEI) transfection. Calcium phosphate transfection was performed essentially as previously described, 18 except that 15 μg of the vector plasmid was used with 15 μg of the foamy Gag helper pFMYGagCO plasmid, 6 μg of the Pol helper plasmid pFMYPol, and 1.5 μg of the Envelope helper plasmid pFMYEnv. For PEI transfection, 63 14 μg of the vector, 14 μg of the foamy Gag helper pFMYGagCO, 5.6 μg of the Pol helper pFMYPol, and 1.4 μg of the Env helper pFMYEnv were mixed in 2 ml of serum-free medium. 105 μl of 1 μg/μl PEI was added to the diluted DNA dropwise and the solution was immediately vortexed for 10 seconds and incubated at room temperature for 15 minutes. The DNA/PEI mix was then added evenly over a 10-cm tissue culture dish. The next day at approximately 16 hours after transfection the medium was replaced with medium supplemented with 2 mM HEPES. Vector was harvested at 72 hours after transfection and concentrated 100-fold as previously described. 18 For the NOD-SCID IL2Rγ null mouse studies, 5% DMSO was added to the vector stocks and frozen at -80°C. These frozen vector preparations were thawed, and the DMSO was removed by dialysis with IMDM using a Microcon Ultracel YM-50 Centrifugal Filter (Millipore, Billerica, MA) just prior to use.
Preparation of HIV and SHIV challenge virus
The following viruses and infectious virus plasmids were obtained through the AIDS Research and Reference Reagent Program from the following investigators. SHIV KU-1 was obtained (Cat# 3441) from Dr. Opendra Narayan and Dr. Sanjay Joag. HIV-1 89.664 (Cat#1966) was obtained from Dr. Ronald Collman. SHIV KU-1 and HIV 89.6 were amplified by passage on 174xCEM cells, 0.22 μm filtered, and stored frozen at -80°C prior to use. Virus preparations were titered using MAGI-CCR-5 cells as previously described, 44 and the number of β-galactosidase positive foci was enumerated by microscopy.
Anti-S/HIV assays and MGMT-mediated selection in vitro
For in vitro challenge assays, CEM.NKR-CCR5 cells were transduced with FV vectors at an MOI between 2 and 5 and selected using O6BG and BCNU to over 80% EGFP-positive cells, then sorted to over 95% EGFP-positive cells prior to challenge. O6BG and BCNU (Sigma, St. Louis, MO) were made up at stock concentrations of 50 mM in 100% DMSO, and 10 mM in 10% ethanol, respectively, and stored frozen at -80°C until use. O6BG and BCNU were used at final concentrations of 50 μM and 10 μM, respectively, for the selection of FV-transduced CEM.NKR-CCR5 cells. Cells were exposed to O6BG for one hour before BCNU was added. For challenge cells were first plated overnight at a density of 2×106 cells/ml, and then the next day plated for challenge at 2×104 cells in 100 μl in a 48-well plate in triplicate. Challenge virus was added at an MOI of 0.05 or 0.5. During the 3 week challenge, cells were passaged at a ratio of 1:3 into fresh medium every 2 to 3 days. Medium containing infectious SHIV was removed from the infected cultures at the indicated times and centrifuged at 2000g for 5 minutes to pellet cells. Supernatant containing virus was then added to MAGI-CCR-5 cells to determine the titer as described above.
For the single cycle HIV-1 and SHIV assay, the HIV-1 and SHIV indicator MAGI-CCR-5 cell line was transduced in triplicate with FV anti-HIV vectors at an MOI of 3. The cells were sorted by flow cytometry for EGFP expression to over 98% EGFP-expressing cells. For each vector, polyclonal cell lines were plated and infected with approximately 1000 transducing units of either the HIV-1 isolate HIV89.6 or the SHIV isolate KU-1. The MAGI-CCR-5 cells were then evaluated for SHIV and HIV-1 replication 48 hours after infection as described above. For each anti-HIV vector, the fold-reduction in infected cells was calculated as the number of infected β-galactosidase-expressing foci relative to the number of infected β-galactosidase-expressing foci for the control vector RSC-SMPGW.
MGMT and C46 immunostaining
HT1080 cells were transduced with either FV-PGW or FV-SMPGW at an MOI of 0.5 and cultured for over two weeks. 1×106 cells were trypsinized and washed twice with Dulbecco’s modified phosphate buffered saline (D-PBS). The cells were fixed with 2% paraformaldehyde in D-PBS at room temperature for 20 minutes and then permeabilized with 0.1% Triton X-100 in D-PBS at room temperature for 5 minutes. The cells were washed in D-PBS, resuspended in D-PBS with 5% nonfat milk, then mixed with 0.6 μg of MGMT primary antibody (Kamiya Biomedicial, Seattle, WA) and incubated at 4°C overnight. The cells were then washed in D-PBS, resuspended in D-PBS with 5% nonfat milk, then mixed with 1 μl of goat anti-mouse secondary antibody conjugated to R-Phycoerythrin (Dako, Carpinteria, CA), and incubated at room temperature for 45 minutes. The cells were then washed twice in D-PBS and analyzed by flow cytometry.
For C46 staining, 1×106 cells were trypsinized and washed twice with D-PBS, then blocked with D-PBS with 5% nonfat milk on ice for 10 minutes. The volume was brought to 100 μl, and 1 μg of 2F5 antibody was added (Polymun Scientific, Vienna, Austria) and incubated on ice for 30 minutes. The cells were washed twice with D-PBS, then incubated on ice for 30 min in 200 μl of a 1:80 dilution of 0.5 mg/ml goat anti-human secondary antibody conjugated to R-phycoerythrin (Jackson ImmunoResearch, West Grove, PA) in D-PBS. The cells were washed twice with D-PBS and analyzed by flow cytometry.
Analysis of copy number and recombination
Relative marking levels in foamy transduced CEM.NKR-CCR5 lymphocytes was analyzed by quantitative real-time PCR assay. We amplified 300 ng DNA in duplicate with a wpre-specific primer/probe combination (5’-CCTCCTTGTATAAATCCTGGTTG-3’ and 5’-GGTTGCGTCAGCAAACACAG-3’; probe, 5’-GAGGAGTTGTGGCCCGTTGTCTAMRA-3’. A β-globin primer/probe combination (5’CCTATCAGAAAGTGGTGGCTGG-3’ and 5’-TTGGACAGCAAGAAAGTGAGCTT-3’; probe, 5’TGGCTAATGCCCTGGCCCACAAGTATAMRA-3’ was used to adjust for equal loading of DNA per reaction against the standard with no vector. Reactions were run using ABI master mix (Applied Biosystems, Branchburg, NJ) on the ABI 7500 Real Time PCR system under the following thermal cycling conditions: 50°C for 2 minutes and 95°C for 10 minutes, then 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. To analyze potential recombination of the shRNA cassettes 200 ng genomic DNA or 0.1 ng vector plasmid DNA was PCR amplified using Platinum Taq polymerase (Invitrogen, Carlsbad, CA) and the oligonucleotide primers 5’-AGCATCCTGTTCCAAAATATGTG-3’ and 5’-TTTGGTATTTTTCCATGCCTTG-3’. The PCR amplification was performed using 30 cycles under the following conditions: 30 sec of denaturation at 98°C, 30 sec of annealing at 58°C and 1 min of extension at 72°C. Products were analyzed by 2% agarose gel electrophoresis and ethidium bromide staining.
Detection of siRNA expression by northern blot
FV vector-transduced CEM.NKR-CCR5 lymphocytes were selected using O6BG and BCNU to over 80% EGFP-positive cells, then sorted to over 95% EGFP-positive cells, and total RNA was isolated using STAT-60 (Tel-Test, Friendswood, TX) reagent according to the manufacturer’s recommendations. Ten μg of total RNA was run on a 10% TBE-Urea Ready Gel (Bio-Rad Laboratories, Hercules, CA), together with a size marker, and then electro-transferred to a Hybond N+ membrane (GE Healthcare, Piscataway, NJ). The membrane was baked at 80°C for 2 hours and then prehybridized for 3 hours at 37°C in QuikHyb hybridization buffer (Stratagene, La Jolla, CA) with 100 μg/ml final concentration of salmon sperm DNA. The membrane was then hybridized overnight in QuikHyb hybridization buffer at 37°C with 5 pmol of oligonucleotide DNA, probe-end labeled with γ -[32P]-ATP using 10 units of polynucleotide kinase, (Invitrogen, Carlsbad, CA). The shRNA probes used were: tat/rev siteI 5’-GCGGAGACAGCGACGAAGAG-3’, tat/rev siteII 5’-GCCTGTGCCTCTTCAGCTAC-3’. After hybridization the membrane was then briefly washed in 6x SSPE buffer (20X SSPE is 3 M NaCl, 0.2 M NaH2PO4, 0.02 M EDTA, pH 7.4), then exposed using a Molecular Dynamics Storage Phosphor Screen (GE Healthcare, Piscataway, NJ) for 1 week and analyzed using a Typhoon Trio Variable Mode Imager (GE Healthcare) and ImageQuant TL (GE Healthcare) software. To evaluate RNA loading, the blots were stripped with 1% sodium dodecyl sulfate at 85°C for 30 minutes and washed, then hybridized as described above using a U6 snRNA probe 5’-TATGGAACGCTTCTCGAATT-3’.
Xenotransplantation studies in the NOD-SCID IL2Rγnull mouse model using a competitive repopulation approach
Mice were housed at the Fred Hutchinson Cancer Research Center (FHCRC) under conditions approved by the American Association for Accreditation of Laboratory Animal Care. Study protocols were approved by the Institutional Animal Care and Use Committee. Human CD34+ cells were thawed and cultured in transduction medium (IMDM with 10% heat-inactivated FBS, 100 ng/ml each of Flt-3 ligand, stem cell factor [SCF], thrombopoietin [TPO], interleukin 3, interleukin-6 and granulocyte colony-stimulating factor [G-CSF]) overnight before vector addition. For each experimental arm of the competitive repopulation assay, 7.5×105 human CD34+ cells were added to one well of a 6-well plate pretreated with CH-296 fibronectin fragment (Takara, New York, NY) at 2μg/cm2 and exposed overnight to vector at an MOI of 2 in transduction medium. The following morning, cells were washed with D-PBS, and cells from each well were resuspended in 200 μl of D-PBS and injected by tail vein into previously irradiated NOD-SCID IL2Rγ null mice (# 005557, The Jackson laboratory, Bar Harbor, Maine). The mice were irradiated with 250 cGy from a 137Cs source at 89.5 cGy/min using a Mark I series 30 JL Shepherd irradiator the day before infusion. To determine the transduction efficiency, CD34+ cells were cultured in transduction medium, and the percentage of EGFP positive cells was determined by flow cytometry on day 3 and on day 10 after vector exposure. Bone marrow aspirates were obtained from the femur using a 28-gauge needle. BM cells were incubated in hemolytic buffer, then washed with D-PBS with 2% FBS and incubated at 4C for 30 min with Phycoerythrin-labeled mouse anti-human CD45-specific antibody (BD Pharmingen Cat. #555483) or isotype ctrl (BD Pharmingen Cat. #555749), and then washed twice in D-PBS with 2% FBS and analyzed by flow cytometry. For in vivo selection, mice were treated with 5 mg/kg O6BG and 7.5 mg/kg BCNU at 7 weeks and 10 weeks post-transplant by intraperitoneal injection. O6BG was prepared at a stock concentration of 1 mg/ml in 40% PEG-400, and 60% D-PBS. BCNU was prepared in 10% ethanol at a stock concentration of 10mM. O6BG was diluted to 0.2 mg/ml in 0.9% saline and administered by intraperitoneal injection in split doses at 1 hour and at 45 minutes prior to BCNU administration.
Supplementary Material
Acknowledgments
This work was supported in part by grants AI063959, AI061839, DK077806, DK56465, AI42552 from the National Institutes of Health, Bethesda, MD. H.P.K. is a Markey Molecular Medicine Investigator. We would like to thank Tulin Okbinoglu, David Dickerson, Christina Ironside and John Ngo for technical assistance. We thank Martin Wohlfahrt for advice and assistance with the detection of siRNA expression. We also acknowledge the assistance of Bonnie Larson and Helen Crawford in preparing the manuscript.
References
- 1.Yeni PG, Hammer SM, Carpenter CC, Cooper DA, Fischl MA, Gatell JM, et al. Antiretroviral treatment for adult HIV infection in 2002: updated recommendations of the International AIDS Society-USA Panel (Review) JAMA. 2002;288:222–235. doi: 10.1001/jama.288.2.222. erratum appears in JAMA. 2003 Jan-Feb;11(1):32. [DOI] [PubMed] [Google Scholar]
- 2.Carr A. Toxicity of antiretroviral therapy and implications for drug development (Review) Nature Reviews Drug Discovery. 2003;2:624–634. doi: 10.1038/nrd1151. [DOI] [PubMed] [Google Scholar]
- 3.Klausner RD, Fauci AS, Corey L, Nabel GJ, Gayle H, Berkley S, et al. Medicine. The need for a global HIV vaccine enterprise. Science. 2003;300:2036–2039. doi: 10.1126/science.1086916. [DOI] [PubMed] [Google Scholar]
- 4.Sekaly RP. The failed HIV Merck vaccine study: a step back or a launching point for future vaccine development? J Exp Med. 2008;205:7–12. doi: 10.1084/jem.20072681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cold shower for AIDS vaccines (Editorial) Nat Med. 2007;13:1389–1390. doi: 10.1038/nm1207-1389. [DOI] [PubMed] [Google Scholar]
- 6.Rossi JJ, June CH, Kohn DB. Genetic therapies against HIV (Review) Nat Biotechnol. 2007;25:1444–1454. doi: 10.1038/nbt1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Lunzen J, Glaunsinger T, Stahmer I, von BV, Baum C, Schilz A, et al. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Molecular Therapy. 2007;15:1024–1033. doi: 10.1038/mt.sj.6300124. [DOI] [PubMed] [Google Scholar]
- 8.Bauer G, Selander D, Engel B, Carbonaro D, Csik S, Rawlings S, et al. Gene therapy for pediatric AIDS (Review) Ann NY Acad Sci. 2000;918:318–329. doi: 10.1111/j.1749-6632.2000.tb05501.x. [DOI] [PubMed] [Google Scholar]
- 9.Kohn DB, Bauer G, Rice CR, Rothschild JC, Carbonaro DA, Valdez P, et al. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34(+) cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood. 1999;94:368–371. [PubMed] [Google Scholar]
- 10.Amado RG, Mitsuyasu RT, Rosenblatt JD, Ngok FK, Bakker A, Cole S, et al. Anti-human immunodeficiency virus hematopoietic progenitor cell-delivered ribozyme in a phase I study: myeloid and lymphoid reconstitution in human immunodeficiency virus type-1-infected patients. Hum Gene Ther. 2004;15:251–262. doi: 10.1089/104303404322886101. [DOI] [PubMed] [Google Scholar]
- 11.Humeau LM, Binder GK, Lu X, Slepushkin V, Merling R, Echeagaray P, et al. Efficient lentiviral vector-mediated control of HIV-1 replication in CD4 lymphocytes from diverse HIV+ infected patients grouped according to CD4 count and viral load. Molecular Therapy. 2004;9:902–913. doi: 10.1016/j.ymthe.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Bahner I, Sumiyoshi T, Kagoda M, Swartout R, Peterson D, Pepper K, et al. Lentiviral vector transduction of a dominant-negative Rev gene into human CD34+ hematopoietic progenitor cells potently inhibits human immunodeficiency virus-1 replication. Molecular Therapy. 2007;15:76–85. doi: 10.1038/sj.mt.6300025. [DOI] [PubMed] [Google Scholar]
- 13.Mautino MR, Morgan RA. Potent inhibition of human immunodeficiency virus type 1 replication by conditionally replicating human immunodeficiency virus-based lentiviral vectors expressing envelope antisense mRNA. Hum Gene Ther. 2000;11:2025–2037. doi: 10.1089/10430340050143444. [DOI] [PubMed] [Google Scholar]
- 14.Falcone V, Schweizer M, Neumann-Haefelin D. Replication of primate foamy viruses in natural and experimental hosts (Review) Current Topics in Microbiology & Immunology. 2003;277:161–180. doi: 10.1007/978-3-642-55701-9_7. [DOI] [PubMed] [Google Scholar]
- 15.Hooks JJ, Gibbs CJ., Jr The foamy viruses (Review) Bacteriological Reviews. 1975;39:169–185. doi: 10.1128/br.39.3.169-185.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flugel RM. Spumaviruses: a group of complex retroviruses (Review) J Acquir Immune Defic Syndr. 1991;4:739–750. [PubMed] [Google Scholar]
- 17.Trobridge G, Vassilopoulos G, Josephson N, Russell DW. Gene transfer with foamy virus vectors. Methods Enzymol. 2002;346:628–648. doi: 10.1016/s0076-6879(02)46082-x. [DOI] [PubMed] [Google Scholar]
- 18.Trobridge G, Josephson N, Vassilopoulos G, Mac J, Russell DW. Improved foamy virus vectors with minimal viral sequences. Molecular Therapy. 2002;6:321–328. doi: 10.1006/mthe.2002.0672. [DOI] [PubMed] [Google Scholar]
- 19.Vassilopoulos G, Trobridge G, Josephson NC, Russell DW. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood. 2001;98:604–609. doi: 10.1182/blood.v98.3.604. [DOI] [PubMed] [Google Scholar]
- 20.Josephson NC, Trobridge G, Russell DW. Transduction of long-term and mobilized peripheral blood-derived NOD/SCID repopulating cells by foamy virus vectors. Hum Gene Ther. 2004;15:87–92. doi: 10.1089/10430340460732481. [DOI] [PubMed] [Google Scholar]
- 21.Josephson NC, Vassilopoulos G, Trobridge GD, Priestly GV, Wood BL, Papayannopoulou T, et al. Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors. Proc Natl Acad Sci USA. 2002;99:8295–8300. doi: 10.1073/pnas.122131099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiem H-P, Allen J, Trobridge G, Olson E, Keyser K, Peterson L, et al. Foamy virus-mediated gene transfer to canine repopulating cells. Blood. 2007;109:65–70. doi: 10.1182/blood-2006-04-016741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trobridge GD, Miller DG, Jacobs MA, Allen JM, Kiem H-P, Kaul R, et al. Foamy virus vector integration sites in normal human cells. PNAS. 2006;103:1498–1503. doi: 10.1073/pnas.0510046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trobridge G, Beard BC, Kiem H-P. Hematopoietic stem cell transduction and amplification in large animal models. Hum Gene Ther. 2005;16:1355–1366. doi: 10.1089/hum.2005.16.1355. [DOI] [PubMed] [Google Scholar]
- 25.Crone TM, Goodtzova K, Edara S, Pegg AE. Mutations in human O6-alkylguanine-DNA alkyltransferase imparting resistance to O6-benzylguanine. Cancer Res. 1994;54:6221–6227. [PubMed] [Google Scholar]
- 26.Xu-Welliver M, Kanugula S, Pegg AE. Isolation of human O6-alkylguanine-DNA alkyltransferase mutants highly resistant to inactivation by O6-benzylguanine. Cancer Res. 1998;58:1936–1945. [PubMed] [Google Scholar]
- 27.Zielske SP, Reese JS, Lingas KT, Donze JR, Gerson SL. In vivo selection of MGMT(P140K) lentivirus-transduced human NOD/SCID repopulating cells without pretransplant irradiation conditioning. J Clin Invest. 2003;112:1561–1570. doi: 10.1172/JCI17922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neff T, Horn PA, Peterson LJ, Thomasson BM, Thompson J, Williams DA, et al. Methylguanine methyltransferase-mediated in vivo selection and chemoprotection of allogeneic stem cells in a large-animal model. J Clin Invest. 2003;112:1581–1588. doi: 10.1172/JCI18782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neff T, Beard BC, Peterson LJ, Anandakumar P, Thompson J, Kiem H-P. Polyclonal chemoprotection against temozolomide in a large-animal model of drug resistance gene therapy. Blood. 2005;105:997–1002. doi: 10.1182/blood-2004-08-3169. [DOI] [PubMed] [Google Scholar]
- 30.Joag SV, Li Z, Foresman L, Stephens EB, Zhao LJ, Adany I, et al. Chimeric simian/human immunodeficiency virus that causes progressive loss of CD4+ T cells and AIDS in pig-tailed macaques. J Virol. 1996;70:3189–3197. doi: 10.1128/jvi.70.5.3189-3197.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schambach A, Bohne J, Baum C, Hermann FG, Egerer L, von Laer D, et al. Woodchuck hepatitis virus post-transcriptional regulatory element deleted from X protein and promoter sequences enhances retroviral vector titer and expression. Gene Ther. 2006;13:641–645. doi: 10.1038/sj.gt.3302698. [DOI] [PubMed] [Google Scholar]
- 32.Trobridge GD, Beard BC, Gooch C, Wohlfahrt M, Olsen P, Fletcher J, et al. Efficient transduction of pigtailed macaque hemtopoietic repopulating cells with HIV-based lentiviral vectors. Blood. 2008;111:5537–5543. doi: 10.1182/blood-2007-09-115022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keller A, Partin KM, Lochelt M, Bannert H, Flugel RM, Cullen BR. Characterization of the transcriptional trans activator of human foamy retrovirus. J Virol. 1991;65:2589–2594. doi: 10.1128/jvi.65.5.2589-2594.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lochelt M, Zentgraf H, Flugel RM. Construction of an infectious DNA clone of the full-length human spumaretrovirus genome and mutagenesis of the bel 1 gene. Virology. 1991;184:43–54. doi: 10.1016/0042-6822(91)90820-2. [DOI] [PubMed] [Google Scholar]
- 35.Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003;77:11531–11535. doi: 10.1128/JVI.77.21.11531-11535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li MJ, Bauer G, Michienzi A, Yee JK, Lee NS, Kim J, et al. Inhibition of HIV-1 infection by lentiviral vectors expressing Pol III-promoted anti-HIV RNAs. Molecular Therapy. 2003;8:196–206. doi: 10.1016/s1525-0016(03)00165-5. [DOI] [PubMed] [Google Scholar]
- 37.Lee NS, Dohjima T, Bauer G, Li H, Li MJ, Ehsani A, et al. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat Biotechnol. 2002;20:500–505. doi: 10.1038/nbt0502-500. [DOI] [PubMed] [Google Scholar]
- 38.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 39.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 40.An DS, Qin FX, Auyeung VC, Mao SH, Kung SK, Baltimore D, et al. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Molecular Therapy. 2006;14:494–504. doi: 10.1016/j.ymthe.2006.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Egelhofer M, Brandenburg G, Martinius H, Schult-Dietrich P, Melikyan G, Kunert R, et al. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J Virol. 2004;78:568–575. doi: 10.1128/JVI.78.2.568-575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hermann FG, Martinius H, Egelhofer M, Giroglou T, Tonn T, Roth SD, et al. Protein scaffold and expression level determine antiviral activity of membrane-anchored antivital peptides. Hum Gene Ther. 2009;20:325–336. doi: 10.1089/hum.2006.158. [DOI] [PubMed] [Google Scholar]
- 43.Ngoi SM, Chien AC, Lee CG. Exploiting internal ribosome entry sites in gene therapy vector design (Review) Current Gene Therapy. 2004;4:15–31. doi: 10.2174/1566523044578095. [DOI] [PubMed] [Google Scholar]
- 44.Kimpton C, Walton A, Gill P. A further tetranucleotide repeat polymorphism in the vWF gene. Hum Mol Genet. 1992;1:287. doi: 10.1093/hmg/1.4.287. [DOI] [PubMed] [Google Scholar]
- 45.Chackerian B, Long EM, Luciw PA, Overbaugh J. Human immunodeficiency virus type 1 coreceptors participate in postentry stages in the virus replication cycle and function in simian immunodeficiency virus infection. J Virol. 1997;71:3932–3939. doi: 10.1128/jvi.71.5.3932-3939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuschl T. Expanding small RNA interference. Nat Biotechnol. 2002;20:446–448. doi: 10.1038/nbt0502-446. [DOI] [PubMed] [Google Scholar]
- 47.ter Brake O, ’t Hooft K, Liu YP, Centlivre M, von Eije KJ, Berkhout B. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Molecular Therapy. 2008;16:557–564. doi: 10.1038/sj.mt.6300382. [DOI] [PubMed] [Google Scholar]
- 48.Aagaard L, Zhang J, von Eije KJ, Li H, Saetrom P, Amarzguioui M, et al. Engineering and optimization of the miR-106b cluster for ectopic expression of multiplexed anti-HIV RNAs. Gene Ther. 2008;15:1566. doi: 10.1038/gt.2008.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 50.Flugel RM, Rethwilm A, Maurer B, Darai G. Nucleotide sequence analysis of the env gene and its flanking regions of the human spumaretrovirus reveals two novel genes. EMBO J. 1987;6:2077–2084. doi: 10.1002/j.1460-2075.1987.tb02473.x. erratum appears in EMBO J 1990 Nov;9(11):3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bukovsky AA, Song JP, Naldini L. Interaction of human immunodeficiency virus-derived vectors with wild-type virus in transduced cells. J Virol. 1999;73:7087–7092. doi: 10.1128/jvi.73.8.7087-7092.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor JA, Vojtech L, Bahner I, Kohn DB, Laer DV, Russell DW, et al. Foamy virus vectors expressing anti-HIV transgenes efficiently block HIV-1 replication. Molecular Therapy. 2008;16:46–51. doi: 10.1038/sj.mt.6300335. [DOI] [PubMed] [Google Scholar]
- 53.Park J, Nadeau P, Zucali JR, Johnson CM, Mergia A. Inhibition of simian immunodeficiency virus by foamy virus vectors expressing siRNAs. Virology. 2005;343:275–282. doi: 10.1016/j.virol.2005.08.038. [DOI] [PubMed] [Google Scholar]
- 54.An DS, Donahue RE, Kamata M, Poon B, Metzger M, Mao SH, et al. Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. PNAS. 2007;104:13110–13115. doi: 10.1073/pnas.0705474104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schambach A, Schiedlmeier B, Kuhlcke K, Verstegen M, Margison GP, Li Z, et al. Towards hematopoietic stem cell-mediated protection against infection with human immunodeficiency virus. Gene Ther. 2006;13:1037–1047. doi: 10.1038/sj.gt.3302755. [DOI] [PubMed] [Google Scholar]
- 56.Mezquita P, Beard B, Kiem H-P. NOD/SCID repopulating cells contribute only to short-term repopulation in the baboon. Gene Ther. 9999 doi: 10.1038/gt.2008.108. prepublished online 19 June 2008. [DOI] [PubMed] [Google Scholar]
- 57.Levy JA. The value of primate models for studying human immunodeficiency virus pathogenesis (Review) Journal of Medical Primatology. 1996;25:163–174. doi: 10.1111/j.1600-0684.1996.tb00013.x. [DOI] [PubMed] [Google Scholar]
- 58.Nathanson N, Hirsch VM, Mathieson BJ. The role of nonhuman primates in the development of an AIDS vaccine (Review) AIDS. 1999;13(Suppl A):S113–S120. [PubMed] [Google Scholar]
- 59.Warren J. Preclinical AIDS vaccine research: survey of SIV, SHIV, and HIV challenge studies in vaccinated nonhuman primates. Journal of Medical Primatology. 2002;31:237–256. doi: 10.1034/j.1600-0684.2002.02010.x. [DOI] [PubMed] [Google Scholar]
- 60.Heinkelein M, Dressler M, Jarmy G, Rammling M, Imrich H, Thurow J, et al. Improved primate foamy virus vectors and packaging constructs. J Virol. 2002;76:3774–3783. doi: 10.1128/JVI.76.8.3774-3783.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salter RD, Howell DN, Cresswell P. Genes regulating HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics. 1985;21:235–246. doi: 10.1007/BF00375376. [DOI] [PubMed] [Google Scholar]
- 62.Trkola A, Matthews J, Gordon C, Ketas T, Moore JP. A cell line-based neutralization assay for primary human immunodeficiency virus type 1 isolates that use either the CCR5 or the CXCR4 coreceptor. J Virol. 1999;73:8966–8974. doi: 10.1128/jvi.73.11.8966-8974.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. PNAS. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Collman R, Balliet JW, Gregory SA, Friedman H, Kolson DL, Nathanson N, et al. An infectious molecular clone of an unusual macrophage-tropic and highly cytopathic strain of human immunodeficiency virus type 1. J Virol. 1992;66:7517–7521. doi: 10.1128/jvi.66.12.7517-7521.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.