

Abstract
The Drosophila immune deficiency (IMD) pathway mobilizes c-Jun N-terminal kinase (JNK), caspase, and nuclear factor-κB (NF-κB) modules to counter infection with Gram-negative bacteria. Dredd is an essential caspase in the IMD pathway, and it is widely established that NF-κB activation depends on Dredd. More recent cell culture studies suggested a role for Dredd in the activation of dJNK (Drosophila JNK). However, there are no epistatic or mechanistic data on the involvement of Dredd in dJNK activation. More importantly, there is no in vivo evidence to demonstrate a physiological requirement for Dredd in the IMD/dJNK pathway. We performed a comprehensive analysis of the role of Dredd in the IMD/dJNK pathway, and we demonstrated that Dredd is essential for the activation of IMD/dJNK in cell culture. We positioned Dredd activity at an early point of the IMD/dJNK pathway and uncovered a series of interactions between Dredd and additional proximal IMD pathway molecules. Mechanistically, we showed that the caspase activity inhibitor p35 blocked dJNK activation and the induction of dJNK-dependent genes in cell culture and in vivo. Most importantly, we demonstrated that dredd mutant flies are completely inhibited in their ability to activate dJNK or express dJNK-responsive target genes after bacterial infection in vivo. In conclusion, we established Dredd as an essential component of the IMD pathway required for the full activation of IMD/dJNK in cell culture and in vivo. Our data enhance our appreciation of Dredd-dependent IMD signal transduction events.
Keywords: Caspase, Drosophila, Innate Immunity, Jun N-terminal kinase (JNK), Signal Transduction, Dredd, IMD
Introduction
The innate immune system is an essential first line of defense against microbial invaders in all multicellular organisms (1). A range of autoimmune, neurological, and cancerous diseases originates from dysfunctions in innate immune response pathways, which underlines the importance of this aspect of immunity. Innate immunity is mediated by germ line-encoded gene products that activate an immediate and potent immune response (2, 3). Studies on the Toll pathway established Drosophila melanogaster as a powerful tool to study immune signal transduction pathways. Initially, Toll was described in axis formation in the early fly embryo (4). Subsequent studies demonstrated that Toll mediates immune responses to fungal and Gram-positive bacterial infections (5–7). This groundbreaking discovery initiated the search for and subsequent discovery of Toll-like receptors in mammals (8–11). Similar to Drosophila Toll, mammalian Toll-like receptors activate nuclear factor-κB (NF-κB) transcription factors and thereby play a fundamental role in the regulation of an appropriate immune response to infection (12).
The Drosophila immune deficiency (IMD)2 pathway is another example of the evolutionary conservation of innate immune responses across distantly related species (13). The IMD pathway is an immune response pathway in flies with significant parallels to the human tumor necrosis factor (TNF) pathway. Both pathways signal through conserved NF-κB, c-Jun N-terminal kinase (JNK), and caspase modules. Detection of bacterial diaminopimelic acid-containing peptidoglycan (PGN) by the peptidoglycan recognition proteins LC and LE (PGRP-LC and PGRP-LE) activates the IMD pathway (14–18). Activation of the pathway leads to the initiation of a signal transduction cascade, mediated by the Imd, Fas-associated death domain (dFADD), TAK1-binding protein 2 (dTAB2), and inhibitor of apoptosis 2 (dIAP2) proteins (19–26). The detailed mechanisms of the early IMD signal transduction are not fully understood. Recent data indicated that immune challenge triggers the caspase-mediated cleavage of Imd and that cleavage and subsequently ubiquitination of the Imd protein are essential for IMD/Rel activation (27).
IMD pathway initiation leads to downstream activation of the Drosophila TGF-β-activated kinase 1 (TAK1) ortholog, dTAK1 (28, 29). dTAK1 mediates the induction of two divergent cascades, which culminate in dJNK (JNK ortholog) and Relish (Rel, p105 NF-κB homolog) activation (28, 30, 31). More specifically, dTAK1 activates a kinase cascade of MAPK kinase 4 (dMKK4) and MAPK kinase 7 (dMKK7) that results in the transient phosphorylation of dJNK (32, 33). Phospho-dJNK activates a subset of immune-responsive AP1-dependent target genes (33, 34). In addition to dMKK4/7 activation, dTAK1 also activates the Drosophila I-κ kinase (I-κ kinaseβ/ird5 and I-κ kinaseγ/kenny) complex, which is required for the initiation of the IMD/Rel arm (35–38). Rel is a composite protein with an N-terminal NF-κB transcription factor domain and an auto-inhibitory C-terminal ankyrin repeat domain (39, 40). Active I-κ kinase contributes to the IMD/Rel transcriptional response through the phosphorylation of the NF-κB domain of Rel (41). Rel activation also requires the endoproteolytic separation of the NF-κB domain from the C-terminal ankyrin repeat domain. The liberated phospho-NF-κB transcription factor domain translocates to the nucleus and induces the prolonged expression of a broad cohort of pro-immune genes such as antimicrobial peptides. Proteolytic cleavage of Rel requires the protein Dredd, which is often considered to be the Drosophila homolog of caspase-8 (42, 43). dredd loss-of-function flies are highly susceptible to infection with Gram-negative bacteria, and they fail to induce Rel cleavage and subsequently fail to express Rel-dependent genes upon infection (44). Given that Rel is cleaved at a caspase consensus cleavage site, and caspase inhibitors block Rel cleavage, the current model suggests that Dredd cleaves Rel (43).
In contrast to the extensive molecular, genetic, and cell biological studies in IMD/Rel activation, the IMD/dJNK arm remains relatively understudied. To advance our understanding of IMD/dJNK activation, our laboratory recently performed a whole genome RNAi screen for IMD/dJNK modifiers in a Drosophila tissue culture cell line (45). We found that RNAi-mediated depletion of dredd resulted in a loss of PGN-dependent phosphorylation of dJNK in cell culture. Our observations are in line with a previous tissue culture study that indicated a requirement for Dredd in the phosphorylation of dJNK through the IMD pathway (46). These results suggest a general requirement for Dredd in the activation of the IMD/dJNK arm in the Drosophila S2 cell line. However, there are no data on the involvement of Dredd in the IMD/dJNK transcriptional response to PGN stimulation; the epistatic relationship of Dredd and additional IMD/dJNK members remains unexplored, and follow-up experiments to elucidate the mechanism of Dredd-mediated dJNK activation have not been performed. Most importantly, the cell culture data remain entirely unsubstantiated in vivo.
To address these questions, we asked if Dredd is essential for IMD/dJNK activation in cell culture and in vivo. We show that PGN-dependent phosphorylation of dJNK and induction of dJNK-dependent response genes require Dredd in cell culture experiments. We demonstrate that Dredd interacts with early IMD pathway members and functions upstream of dTAK1 in the activation of dJNK in the Drosophila macrophage-like S2 cell line. We show that the expression of the caspase inhibitor p35 effectively blocks signal transduction to dJNK. In agreement with our cell culture data, we demonstrate that p35 inhibits phosphorylation of dJNK and activation of dJNK-dependent response genes after bacterial challenges in vivo. Most importantly, we show for the first time that dredd mutant flies fail to phosphorylate dJNK or express dJNK-dependent response genes after immune challenge. Our in vitro and in vivo results clearly establish a fundamental requirement for Dredd in IMD/dJNK activation.
EXPERIMENTAL PROCEDURES
S2 Cell Culture
The Drosophila embryonic macrophage-like S2 cell line was cultured at 25 °C in HyQ TNM-FH medium (HyClone) supplemented with 10% heat-inactivated fetal bovine serum, 50 units/ml penicillin, and 50 μg/ml streptomycin (Invitrogen). Serum-free S2 cells were cultured in SFX-INSECT medium (HyClone) supplemented with 50 units/ml penicillin and 50 μg/ml streptomycin (Invitrogen). Cells were treated with 5 μg of PGN (InvivoGen) to induce the IMD pathway.
Drosophila Husbandry
All Drosophila fly stocks were cultured on standard cornmeal medium at 25 °C. UASp35/CyO flies were purchased from the Bloomington Drosophila stock center. The dreddB118 and the yolkGAL4 fly stocks have been described elsewhere (29, 44). For infection studies, flies were stabbed with a sharpened tungsten needle dipped in a pellet of an overnight Escherichia coli DH5α culture. Flies were then recovered at 25 °C for the indicated times depending on the experimental approach before further analysis. For caspase activity studies, UASp35/CyO flies were crossed to yolkGAL4 flies, and the progeny were separated by gender 3–5 days after hatching.
Expression Plasmids
The p35 expression plasmid has been described previously (47). The pMT-HAdTAK1CA plasmid was generated by amplifying the genomic region of dTAK1 lacking the kinase inhibitory domain. dTAK1CA has been described previously (46). The primers used were as follows: dTAK1 forward 5′-CACCGAATTCATGGCCACAGCATCGC-3′ and dTAK1 reverse 5′-TTAATCTAGACTACGTGTATTCCAGG-3′. dFadd, dIAP2, Dredd, Imd, and p35 expression plasmids were generated by cloning the respective coding regions into pENTR/D-TOPO (Invitrogen). Each construct was then recombined with pAMW (6×Myc) or pAHW (3×HA) following the manufacturer's recommendations in a Gateway LR clonase reaction (Invitrogen). DreddC408A (DreddCA) has been described previously (42). For transient transfections, S2 cells were seeded at 1 × 106 cells per ml, and 2 μg of plasmid DNA was delivered into the cells with Cellfectin II (Invitrogen) following the manufacturer's recommendations. Cells were incubated overnight at 25 °C and analyzed the next day. For stable cell transfections, 3 ml of S2 cells (3 × 106 cells) were co-transfected with plasmid DNA and a hygromycin B resistance selection plasmid (pCoHygro, Invitrogen) at a ratio of 19:1. After 3 days, transfection medium was replaced with fresh medium containing hygromycin B (300 μg/ml, Sigma). The process was repeated over a period of 3 weeks for selection of stable transfected cell lines. Cells transfected only with a hygromycin B resistance selection plasmid were used as a control cell line where indicated. Copper-sulfate (CuSO4, pMT)-dependent plasmids were induced by the addition of 500 μm CuSO4 and incubated at 25 °C for the indicated times. Cells treated with the CuSO4 solvent double distilled H2O were used as a control where indicated.
RNAi Treatment
The dsRNA used in this study has been described previously (48). S2 cells were seeded at 2.5 × 105 cells per ml and incubated with 10 μg/ml dsRNAs at 25 °C for 3 days before analysis. Stable cell lines were transfected with 10 μg/ml dsRNAs with the Cellfectin II reagent (Invitrogen) according to the manufacturer's recommendations. Cells were incubated at 25 °C for 3 days prior to further analysis. dsRNA targeting CG11318 was applied as control dsRNA in all dsRNA-dependent assays.
Immunoprecipitation
1 × 106 S2 cells were transfected with the appropriate expression plasmids as described above. Cells were centrifuged at 1000 × g for 3 min and then lysed in 200 μl of lysis buffer (50 mm HEPES (pH 7.5), 10 mm EDTA (pH 8), 50 mm KCl, 50 mm NaCl, 1 mm MgCl2, 0.1% Nonidet P-40 protease inhibitors (inhibitor mixture tablets from Roche Applied Science), phosphatase inhibitors (Sigma, phosphatase inhibitor mixture)) for 10 min at 4 °C. After clearing the sample of cell debris and cell wall residues by centrifugation at 21,000 × g for 10 min at 4 °C, rabbit anti-Myc (Sigma, 1:500) or mouse anti-HA (Sigma, 1:500) was added to the supernatant, and samples were rocked at 4 °C overnight. The next day, protein G-Sepharose beads (Amersham Biosciences) were added and incubated with the supernatant for 1 h at 4 °C. Beads were pelleted by centrifugation at 300 × g for 30 s and washed in lysis buffer three times. After discarding the supernatant, beads were resuspended in 2× sample buffer. Prior to analysis by Western blot, all samples were boiled for 5 min at 95 °C.
Western Blotting and Protein Quantification
For protein analysis, 1 × 106 S2 cells or five flies were lysed in 2× sample buffer and boiled for 5 min at 95 °C, and 5–10 μl were analyzed on an 8–10% SDS-polyacrylamide gel. Proteins were separated by electrophoresis and transferred to a nitrocellulose membrane by semi-dry transfer. Membranes were blocked in blocking buffer (LI-COR Biosciences) for 1 h and probed with mouse anti-HA (Sigma, 1:5000), rabbit anti-Myc (Sigma, 1:5000), mouse anti-Myc (Sigma, 1:5000), rabbit anti-JNK (Santa Cruz Biotechnology, 1:4000), mouse anti-phospho-JNK (Cell Signaling, 1:2000), mouse anti-Rel110 (undiluted hybridoma supernatant (49)), and rabbit anti-phospho-Rel (1:300 (50)) overnight. Membranes were then incubated with secondary antibodies conjugated to Alexa Fluor 750 or Alexa Fluor 680 (Invitrogen, 1:10,000) for 1 h and visualized with an LI-COR Aerius automated infrared imaging system. Plate-based quantitative analysis was carried out as described previously (51). Briefly, 1 × 106 cells per ml were plated in a 96-well plate in a total volume of 150 μl of serum-free media. Cells were then fixed in 3.7% formaldehyde (Sigma) and solubilized in 0.1% Triton X-100. Rabbit anti-JNK and mouse anti-phospho-JNK primary antibodies were used to stain proteins, which were visualized with Alexa Fluor 750- or 680-coupled secondary antibodies using the LI-COR Aerius automated infrared imaging system.
Reverse Transcription PCR (RT-PCR)
For RT-PCR, total RNA was isolated from 1 × 106 S2 cells or 10 flies using TRIzol (Invitrogen) according to manufacturer's recommendations. To eliminate DNA residues, RNA was treated with DNase I (Invitrogen). Superscript III (Invitrogen) was used to generate cDNA using 3 μg (S2 cells) or 5 μg (flies) of RNA and random primers (Invitrogen), according to the manufacturer's instructions. Transcript amplification was performed in an Eppendorf PCR machine using the TaqDNA polymerase (New England Biolabs) and the following primers: p35 forward 5-CCCAGACGGTTATTCGAGA-3′ and p35 reverse 5′-GCCCCAGTTCGATTCTGTAG-3′. Samples were then analyzed on a 1% agarose gel.
Quantitative Real Time PCR
cDNA was prepared as described above. Transcript amplification was monitored with PerfeCTa SYBR Green FastMix (Quanta Biosciences) using an Eppendorf realplex 2 PCR machine and the following primers: actin forward 5′-TGCCTCATCGCCGACATAA-3′ and actin reverse 5′-CACGTCACCAGGGCGTAAT-3′; att forward 5-AGTCACAACTGGCGGAC-3′ and att reverse 5′-TGTTGAATAAATTGGCATGG-3′; dipt forward 5′-ACCGCAGTACCCACTCAATC-3′ and dipt reverse 5′-ACTTTCCAGCTCGGTTCTGA-3′; puckered forward 5′-GCCACATCAGAACATCAA-3′ and puckered reverse 5′-CCGTTTTCCGTGCATCTT-3′; mmp-1 forward 5-ACGACTCCATCTGCAAGGAC-3′ and mmp-1 reverse 5′-GGAGATGAGCTGTGGGTA-3′. To quantify relative expression values, all samples were normalized to the actin expression level and quantified using the ΔΔCT method.
RESULTS
Dredd Is Required for IMD/dJNK Activation in Cell Culture
Numerous reports demonstrated that Dredd is required for proteolytic activation of Rel (43, 44, 49). However, two recent studies in Drosophila tissue culture cells suggest a separate requirement for Dredd in the phosphorylation of dJNK through the IMD pathway (45, 46). To explore this requirement further, we analyzed how RNAi-mediated depletion of dredd influences activation of dJNK in the Drosophila macrophage-like S2 cell line. To activate the IMD pathway, we incubated S2 cells with commercial preparations of PGN. Treatment of S2 cells with PGN resulted in Rel cleavage, Rel phosphorylation, and a transient phosphorylation of dJNK (Fig. 1A). All three events were fully blocked upon depletion of dredd (Fig. 1A).
FIGURE 1.

Dredd is required for IMD/dJNK activation in cell culture. A, Western blot analysis of lysates from S2 cells incubated with control dsRNA (1st to 3rd lanes) or dredd dsRNA (4th to 6th lanes) and stimulated with PGN for the indicated times. Lysates were probed with antibodies that detect Rel-110 (1st panel), phospho-Rel (P-Rel, 2nd panel), phospho-dJNK (P-dJNK, 3rd panel), and total dJNK (4th panel). In contrast to control cells, stimulation with PGN does not induce Rel cleavage and does not induce phosphorylation of Rel or dJNK protein in dredd-depleted cells. Molecular weights are indicated to the right of each panel. IB, immunoblot. B–E, quantitative real time PCR analysis of control cells and dredd-depleted S2 cells, stimulated with PGN and recovered for the indicated times. The relative expression (rel. expr.) levels for attacin, diptericin, puckered, and mmp-1 are standardized to actin levels. Values of control cells and dredd-depleted cells at the indicated time points after PGN stimulation are reported relative to unstimulated control cells and dredd-depleted cells, respectively. Measurements for each transcript are presented as a box plot to graphically illustrate the results of three independent experiments. Comparison of control cells and dredd-depleted cells show a strong reduction of attacin, diptericin, puckered, and mmp-1 expression levels in dredd-depleted cells.
Although these observations validate a general requirement for Dredd in PGN-mediated phosphorylation of dJNK, there are no data on the involvement of Dredd in the dJNK component of the IMD pathway transcriptional response to PGN. To address this question, we examined the expression of the dJNK-responsive transcripts puckered (puc) and matrix metalloproteinase-1 (mmp-1) in control S2 cells or S2 cells pretreated with dredd dsRNA and incubated with PGN for various periods. Differences in passage numbers of our S2 cells caused minor variability in the relative induction of PGN-responsive transcripts in replicate assays. Nonetheless, each transcript showed a stereotypical and reproducible response to PGN. For example, stimulation of S2 cells with PGN repeatedly resulted in a gradual induction of the IMD/Rel-dependent antimicrobial peptides attacin (att) and diptericin (dipt) (Fig. 1, B and C, respectively). In contrast, IMD/dJNK activation resulted in a rapid and transient induction of puc and mmp-1 (Fig. 1, D and E, respectively). As expected, we detected a considerable drop in the PGN-mediated induction of att and dipt in S2 cells treated with dredd dsRNA (Fig. 1, B and C). Likewise, depletion of dredd greatly decreased the expression of the dJNK-dependent transient response genes puc and mmp-1 (Fig. 1, D and E). Thus, our cell culture data confirm a role for Dredd in the activation of dJNK through the IMD pathway and establish an essential role for Dredd in the dJNK arm of the IMD pathway transcriptional response.
Dredd Acts Upstream of dTAK1 in dJNK Phosphorylation in Cell Culture
A previous study indicated that Dredd acts upstream of dTAK1 in the phosphorylation of Rel (46). However, this study did not describe the epistatic relationship of Dredd with dTAK1 in the activation of dJNK. To address this question, we generated an S2 cell line that inducibly expresses a constitutively active HA-tagged dTAK1 variant (pMT-HAdTAK1CA). dTAK1CA encodes a truncated dTAK1 protein that lacks the kinase inhibitory domain and therefore is considered constitutively active. We detected HAdTAK1CA protein within 100 min of induction (Fig. 2A, 1st panel). When we probed the same samples with a phospho-dJNK-specific antibody, we detected dJNK phosphorylation at a similar time point (Fig. 2A, 2nd panel). In addition, we detected a parallel induction of puc expression in the same experimental samples (Fig. 2B). Furthermore, simultaneous depletion of dMKK4 and dMKK7 from HAdTAK1CA-expressing cells resulted in a loss of PGN-dependent phosphorylation of dJNK (Fig. 2C, 2nd and 5th lanes). Thus, we are confident that our cell culture system reliably reproduces key features of dTAK1-dependent activation of dJNK in the IMD pathway.
FIGURE 2.
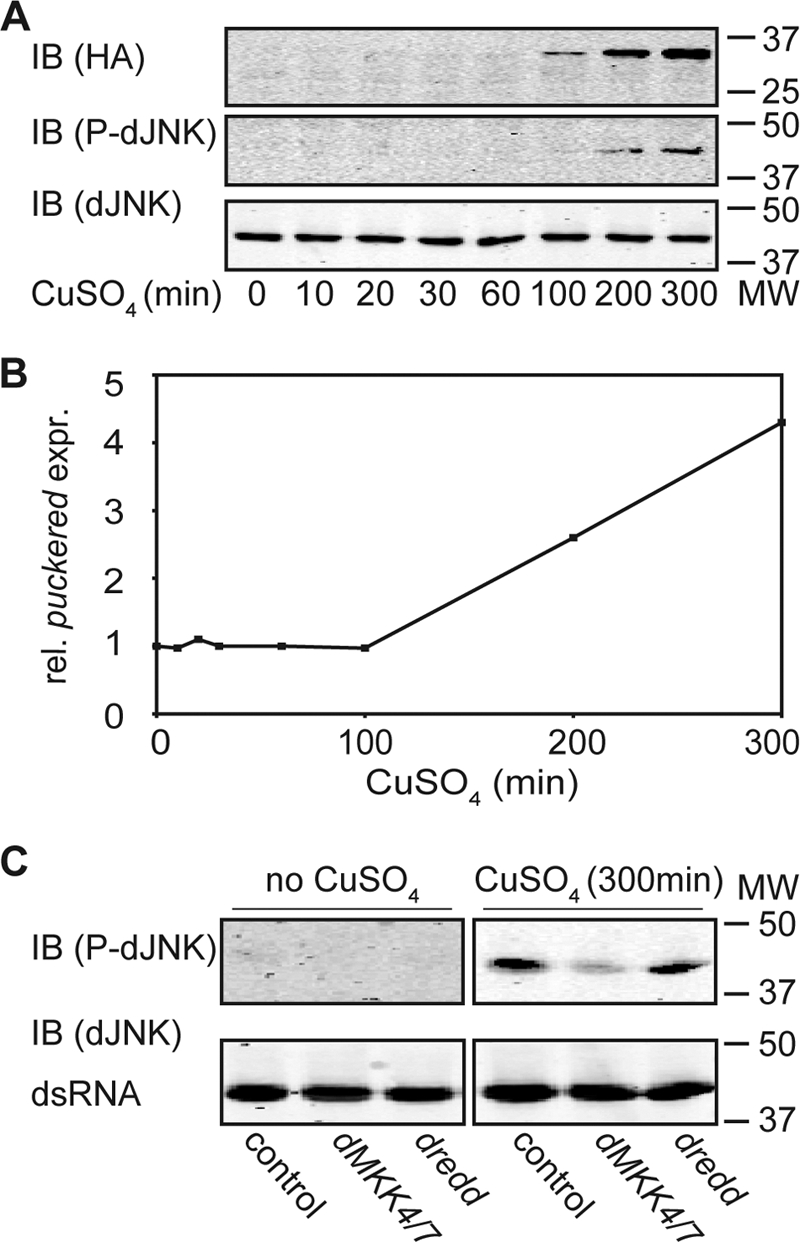
Dredd acts upstream of dTAK1 in dJNK phosphorylation in cell culture. A, Western blot analysis of lysates from S2 cells that inducibly express a pMT-HATAK1CA and incubated with CuSO4 for the indicated times. Protein levels are visualized with HA- (1st panel), P-dJNK- (2nd panel), and total dJNK (3rd panel)-specific antibodies. Induction of the pMT-HATAK1CA expression plasmid in response to CuSO4 results in phosphorylation of the dJNK protein. Molecular weights are indicated to the right of each panel. IB, immunoblot. B, quantitative real time PCR analysis of the same samples described in A. The relative expression levels for puckered were standardized to actin levels. Values of puckered expression at the indicated time points after CuSO4 stimulation are reported relative to the unstimulated pMT-HATAK1CA sample. Induction of pMT-HATAK1CA in response to CuSO4 induces the expression of puckered. C, Western blot analysis of lysates from S2 cells stably transfected with a pMT-HATAK1CA expression plasmid not incubated with CuSO4 (1st to 3rd lanes) or incubated with CuSO4 (4th to 6th lanes) for the indicated times. Cells were incubated with control dsRNA (1st and 4th lanes), with dMKK4/7 dsRNA (2nd and 5th lanes), or with dredd dsRNA (3rd and 6th lanes). Protein levels were visualized with P-dJNK- (1st panel) and total dJNK (2nd panel)-specific antibodies. Western blot is representative of three independent experiments. In contrast to control cells, depletion of dMKK4/7 shows a loss of phosphorylation of dJNK although depletion of dredd does not change relative P-dJNK:total dJNK levels compared with control dsRNA-treated cells. Molecular weights are indicated to the right of each panel.
We then asked if Dredd is required up- or downstream of dTAK1 for the activation of dJNK. To address this question, we depleted dredd by RNAi in HAdTAK1CA-expressing cells and analyzed whole cell lysates for phospho-dJNK by Western blot. In contrast to dMKK4/dMKK7-depleted cells, we did not detect a change of dJNK phosphorylation when dredd was depleted (Fig. 2C, 3rd and 6th lanes). Instead, phospho-dJNK levels remained at a level similar to control cells. These data indicate that Dredd acts upstream of dTAK1 and that Dredd is required for the transduction of a phospho-relay through the IMD pathway to dJNK.
Dredd Interacts with Early IMD Pathway Components
Our observation that Dredd acts upstream of dTAK1 in the IMD pathway suggests an interaction of Dredd with proximal IMD pathway members. To explore this possibility, we undertook a detailed examination of potential interactions between Dredd, Imd, dFADD, and dIAP2. For these experiments, we generated Myc- or HA-tagged expression plasmids for Imd, dFADD, dIAP2, and Dredd (Fig. 3A). We then probed potential protein-protein interactions in reciprocal co-immunoprecipitation assays performed in S2 cell lysates. We initially probed for a potential interaction between dIAP2 and dFADD. For these studies, we performed anti-Myc immunoprecipitations on whole cell lysates prepared from S2 cells that were co-transfected with HAIAP2 HAdlAP2 and MycdFADD expression plasmids. We confirmed expression of the respective constructs (Fig. 3A) and determined whether HAdIAP2 co-precipitates with MycdFADD (Fig. 3B). We detected a strong co-precipitation of HAdIAP2 with MycdFADD (Fig. 3B, 6th lane). In contrast, we did not observe precipitation of HAdIAP2 in the absence of MycdFADD (Fig. 3B, 4th lane). In the reciprocal approach, we detected a specific co-precipitation of MycdFADD with HAdIAP2 (Fig. 3C, 6th lane). These data indicate a molecular interaction between dIAP2 and dFADD.
FIGURE 3.

Dredd interacts with early IMD pathway components. A, Western blot analysis of lysates from S2 cells transfected with the indicated HA- (left panel) or Myc (right panel)-tagged expression plasmids. Protein levels were visualized with HA- (upper left panel) and Myc (upper right panel)-specific antibodies. Control lysates from nontransfected S2 cells were loaded where indicated. dJNK was visualized as a loading control (lower left and right panel). Molecular weights are indicated to the left of each panel. * marks an unspecific band. IB, immunoblot. B and C, Western blot analysis of lysates from S2 cells transfected with HAdIAP2 and MycdFADD as indicated. Protein levels of input and immunoprecipitated (I.P.) samples were visualized with HA- (upper panel) or with Myc (middle panel)-specific antibodies. dJNK was visualized as a loading control (lower panel). 1st to 3rd lanes show lysates of the input samples, and 4th to 6th lanes show the same samples after immunoprecipitation with a Myc- (B) or an HA (C)-specific antibody. HAdIAP2 co-immunoprecipitates with MycdFADD. Molecular weights are indicated on the right of each panel. IB, immunoblot. D and E, Western blot analysis of lysates from S2 cells transfected with HAdFADD and MycDredd as indicated. Protein levels of input and immunoprecipitated samples were visualized with HA- (upper panel) or with Myc (middle panel)-specific antibodies. dJNK was visualized as a loading control (lower panel). 1st to 3rd lanes show lysates of the input samples, and 4th to 6th lanes show the same samples after immunoprecipitation with a Myc- (D) or an HA (E)-specific antibody. MycDredd co-immunoprecipitates with HAdFADD. Molecular weights are indicated to the right of each panel. F and G, Western blot analysis of lysates from S2 cells transfected with HAdFADD and MycImd as indicated. Protein levels of input and immunoprecipitated samples were visualized with HA- (upper panel), or with Myc (middle panel)-specific antibodies. dJNK was visualized as a loading control (lower panel). 1st to 3rd lanes show lysates of the input samples, and 4th to 6th lanes show samples immunoprecipitated with a Myc- (F) or HA (G)-specific antibody. HAdFADD co-immunoprecipitates with MycImd. Molecular weights are indicated to the right of each panel. H, Western blot analysis of lysates from S2 cells transfected with HAdIAP2 and MycDredd as indicated. Protein levels of input and immunoprecipitated samples were visualized with HA- (upper panel) or with Myc (middle panel)-specific antibodies. dJNK was visualized as a loading control (lower panel). 1st to 3rd lanes show lysates of the input samples, and 4th to 6th lanes show the same samples after immunoprecipitation with an HA-specific antibody. MycDredd co-immunoprecipitates with HAdIAP2. Molecular weights are indicated to the right of each panel. I, Western blot analysis of lysates from S2 cells transfected with HAdIAP2, MycdFADD, Myc (empty destination vector), and MycDredd as indicated. Protein levels of input and immunoprecipitated samples were visualized with HA- (upper panel) or with Myc (middle panel)-specific antibodies. dJNK was visualized as a loading control (lower panel). 1st to 5th lanes show lysates of the input samples, and 6th to 10th lanes show the same samples after immunoprecipitation with an HA-specific antibody. Dredd does not compete with dFADD for binding to dIAP2. Molecular weights are indicated to the right of each panel. Asterisk marks unspecific bands, MycDredd is indicated with a closed arrowhead, and MycdFADD is indicated with an open arrowhead. J, network of interactions between Dredd and additional early IMD signaling molecules based on data shown in B–H.
We then tested all potential pairwise interactions between Imd, dFADD, dIAP2, and Dredd. Our results are presented in Fig. 3, D–H. We identified co-immunoprecipitations of dFADD with Dredd (Fig. 3, D and E). As anticipated, we detected the reported interactions between Imd and dFADD (Fig. 3, F and G) (25). In addition, we showed that Dredd co-precipitates with dIAP2 (Fig. 3H). We did not observe co-immunoprecipitations of Dredd or dIAP2 with Imd under our experimental conditions. Our findings are summarized in Fig. 3J and describe a robust network of physical interactions among proximal IMD pathway molecules. In all cases tested, we only detected immunoprecipitation of the full-length proteins shown in Fig. 3A. Importantly, these findings establish Dredd as a central element of the proximal signaling complex.
As immunoprecipitation of dIAP2 results in the co-purification of Dredd or dFADD in pairwise assays, we asked if Dredd competes with dFADD for interaction with dIAP2. For these studies, we followed the precipitation of mycdFADD by HAdIAP2 in the presence or absence of equal amounts of mycDredd. We found that immunoprecipitation of HAdIAP2 led to the purification of roughly equal amounts of mycdFADD in the absence (Fig. 3I, 8th and 9th lanes) or presence of competing amounts of mycDredd (Fig. 3I, 10th lane). These data suggest that Dredd does not compete with dFADD for binding to dIAP2.
Baculovirus p35 Inhibits Dredd-dependent Activation of dJNK in Cell Culture
We then asked if the baculovirus pan-caspase inhibitor p35 blocks PGN-dependent phosphorylation of dJNK. To this end, we generated an S2 cell line that constitutively expresses p35 (Fig. 4A). We determined the extent to which p35 blocks PGN-mediated phosphorylation of dJNK in a quantitative plate-based assay. PGN-mediated phosphorylation of dJNK was markedly impaired in S2 cells that express p35 compared with control S2 cells (Fig. 4B). The residual phosphorylation of dJNK in cells that express p35 is likely a consequence of the fact that stable S2 cell lines are not clonal, and the expression levels of transgenic constructs vary across cells in a given population.
FIGURE 4.

Baculovirus p35 inhibits dJNK activation in cell culture. A, agarose gel electrophoresis of RT-PCR products amplified from RNA extracted from control S2 cells (1st and 3rd lanes) or S2 cells that were stably transfected with a baculovirus p35 expression plasmid (2nd and 4th lanes) using p35-specific primers. RT-PCR was performed without (1st and 2nd lanes) or with (3rd and 4th lanes) reverse transcriptase (RT) enzyme. p35 is only expressed in stably transfected cells. B, in cell Western (ICW) analysis of control cells (upper row of each plate) and cells stably transfected with baculovirus p35 (lower row of each plate) and stimulated with PGN for the indicated times. Protein levels were visualized with total dJNK- (lower plate) and P-dJNK (upper plate)-specific antibodies. In contrast to control cells, stimulation with PGN induced lower levels of dJNK phosphorylation in p35-expressing cells. C–F, quantitative real time PCR analysis of control cells and S2 cells stably transfected with baculovirus p35 stimulated with PGN and recovered for the indicated times. The relative expression levels for attacin (C), diptericin (D), mmp-I (E), and puckered (F) are standardized to actin levels. Values of control cells and p35-expressing cells at the indicated time points after PGN stimulation are reported relative to unstimulated control cells and p35-expressing cells, respectively. Measurements for each transcript are presented as a box plot to graphically illustrate the results of three independent experiments. Comparison of control cells and p35-expressing cells shows a reduction of attacin, diptericin, mmp-I, and puckered expression levels in p35-expressing cells. G, Western blot analysis of lysates from control cells (1st to 3rd lanes) and cells stably transfected with baculovirus p35 (4th to 6th lanes) and stimulated with PGN for the indicated times. Protein levels are visualized with HA- (upper panel), P-dJNK- (middle panel), and total dJNK (lower panel)-specific antibodies. HAp35 is only detectable in cell stably transfected with the HAp35 expression construct (upper panel). The asterisk marks full-length HAp35, and the dot marks a potential truncated variant of HAp35. In contrast to control cells, stimulation with PGN induces lower levels of dJNK phosphorylation in p35-expressing cells (middle panel). Western blot is representative for the results of three independent experiments. Molecular weights are indicated to the right of each panel. IB, immunoblot. H, Western blot analysis of lysates from S2 cells transfected with HAp35, MycDredd, and MycDreddCA as indicated. Protein levels of input and immunoprecipitated (I.P.) samples were visualized with HA- (upper panel), or with Myc (middle panel)-specific antibodies. dJNK was visualized as a loading control (lower panel). 1st to 5th lanes show lysates of the input samples, and 6th to 10th lanes show the same samples after immunoprecipitation with an HA-specific antibody. HAp35 co-immunoprecipitates MycDredd, and to a lesser extent MycDreddCA. Molecular weights are indicated to the right of each panel. I, Western blot analysis of lysates from S2 cells treated with the indicted dsRNAs and stimulated with PGN for the indicated times. Protein levels are visualized with Rel-110- (upper panel), P-dJNK- (middle panel), and total dJNK (lower panel)-specific antibodies. PGN treatment of control cells (1st and 2nd lanes of each panel) results in Rel cleavage, indicated by the loss of Rel-110 (3rd lane, upper panel) and dJNK phosphorylation (3rd lane, middle panel). In contrast to control cells, stimulation with PGN does not induce Rel cleavage or dJNK phosphorylation in dredd-depleted cells (4th lane of each panel). Depletion of the caspases drice, dcpI, damm, decay, dronc, and strica does not affect Rel cleavage or dJNK phosphorylation (5th to 10th lanes of each panel). Molecular weights are indicated to the left of each panel.
We expanded our studies to determine whether p35 affects the IMD/dJNK-responsive transcriptional pathway. For these experiments, we generated an S2 cell line that constitutively expresses an HA-tagged p35 variant. We consistently found that PGN-dependent transcriptional levels of the Rel-responsive antimicrobial peptides dipt and att and the induction of dJNK-responsive transcripts mmp-1 and puc were reduced in S2 cells that express HAp35 compared with control S2 cells (Fig. 4, C–F).
In line with our observations with untagged p35, we detected considerably less phosphorylation of dJNK in response to PGN in cells that stably express HAp35 compared with control cells (Fig. 4G, upper panel). Caspase inhibition by p35 requires the caspase-mediated cleavage of the p35 reactive site loop and the formation of a stable p35-caspase complex that consists of a 25-kDa cleavage product of p35 and a mature caspase (52, 53). In our assays, we did not detect processing of p35 to the 25-kDa product typically found in complex with inhibited caspases. This finding prompted us to ask if HAp35 and Dredd form a molecular complex. In co-immunoprecipitation assays, we showed that immunoprecipitation of HAp35 co-precipitates MycDredd and to a lesser extent a proteolytically inactive mycDredd variant (Fig. 4H, 9th and 10th lanes). In both cases, the co-purified caspase corresponded to the full-length variant. We consider these findings noteworthy, as p35 typically interacts with processed, mature caspases and the established paradigm for p35 action suggests that it acts as a suicide inhibitor of proteolytically active caspases.
We then expanded our studies to test all Drosophila caspases for involvement in IMD/dJNK activation. To this end, we depleted each of the seven caspases from S2 cells individually and monitored subsequent IMD pathway responses to PGN exposure (Fig. 4I). We analyzed whole cell lysates by Western blot analysis with Rel-, phospho-dJNK-, and total dJNK-specific antibodies. As anticipated, we detected Rel cleavage and dJNK phosphorylation in S2 cells or S2 cells treated with a control dsRNA in response to PGN treatment (Fig. 4I, upper and middle panel, first 3 lanes). Both events were fully blocked in cells depleted of dredd (Fig. 4I, 4th lane). In contrast, we were unable to detect inhibition of either PGN-dependent Rel cleavage or dJNK phosphorylation in cells depleted of any other caspase (Fig. 4I, 5th to 10th lanes).
In summary, our data demonstrate that Dredd is the sole essential caspase in the IMD pathway, that HAp35 interacts with Dredd in S2 cells, and that HAp35 blocks the PGN-dependent dJNK phosphorylation and transcriptional response. However, given the atypical nature of the p35-Dredd interactions, we cannot definitively conclude that p35 prevents the induction of IMD/dJNK responses by specifically inhibiting the caspase activity of Dredd.
Baculovirus p35 Inhibits dJNK Activation in Vivo
To date, the involvement of Dredd in IMD/dJNK activation is completely unexplored in vivo. IMD pathway activity is typically monitored in vivo by following the immune responses of flies that were pierced with a sterile needle dipped in pellets of Gram-negative bacteria. In this system, the fat body is a major site of antimicrobial peptide expression. Consistent with a requirement for Dredd in IMD/Rel activation, expression of p35 blocks the challenge-dependent induction of dipt in adult fat bodies (19). We used the GAL4-UAS binary expression system to monitor the effects of p35 on the IMD/dJNK pathway (54). Specifically, we crossed yolk-GAL4 transgenic flies with UAS-p35 transgenic flies to generate yolk-GAL4/UAS-p35 progeny. As yolk-GAL4 expression is restricted to female fat bodies, p35 expression is likewise restricted to the fat bodies of female yolk-GAL4/UAS-p35 flies. The use of a female-specific driver enabled us to use male flies as isogenic controls in all our experiments. We initially examined the expression of p35 in male and female flies by RT-PCR analysis. As expected, female flies expressed p35 at a very high level, whereas male flies showed a weak expression of p35 that likely resulted from leaky expression from the upstream activating sequence elements (Fig. 5A). We then monitored the infection-dependent phosphorylation of dJNK in male and female yolk-GAL4/UAS-p35 flies. Male flies showed a transient increase in phospho-dJNK levels in response to bacterial challenge. In contrast, the relative levels of dJNK phosphorylation remained unchanged in female flies (Fig. 5B). These data strongly hint at a requirement for caspase activity in the activation of IMD/dJNK in vivo.
FIGURE 5.

Baculovirus p35 inhibits dJNK activation in vivo. A, agarose gel electrophoresis of RT-PCR products amplified from RNA extracted from UASp35/yolkGAL4 male flies (1st and 3rd lanes) or UASp35/yolkGAL4 female flies (2nd and 4th lanes) using p35-specific primers. RT-PCR was performed without (1st and 2nd lanes) or with (3rd and 4th lanes) reverse transcriptase (RT) enzyme. The female fat body-specific yolk-driver induced a strong expression of p35 in female flies, whereas males showed only a weak expression of p35. B, Western blot analysis of lysates from UASp35/yolkGAL4 male (1st to 3rd lanes) and UASp35/yolkGAL4 female (4th to 6th lanes) flies, infected with E. coli and recovered for the indicated times. Protein levels are visualized with total dJNK- (lower panel) and P-dJNK (upper panel)-specific antibodies. In contrast to UASp35/yolkGAL4 male flies, infection with E. coli does not induce a transient increase in P-dJNK levels in UASp35/yolkGAL4 female flies. Molecular weights are indicated to the right of each panel. C and D, quantitative real time PCR analysis of yolkGAL4 male flies (as indicated to the left of each box plot) and yolkGAL4 female flies (as indicated to the right of each box plot), infected with E. coli, and recovered for the indicated times. The relative expression levels for diptericin (C), and puckered (D) are standardized to actin levels. The expression levels for yolkGAL4 male and yolkGAL4 female flies at the indicated time points after infection are reported relative to uninfected yolkGAL4 male or yolkGAL4 female flies, respectively. The measurements for each transcript are presented as a box plot to graphically illustrate the results of three independent experiments. E–H, quantitative real time PCR analysis of UASp35/yolkGAL4 male flies (as indicated on the left of each box plot) and UASp35/yolkGAL4 female flies (as indicated on the right of each box plot) infected with E. coli and recovered for the indicated times. The relative expression levels for attacin (E), diptericin (F), puckered (G), and mmp-1 (H) are standardized to actin levels. The expression levels for UASp35/yolkGAL4 male and UASp35/yolkGAL4 female flies at the indicated time points after infection are reported relative to uninfected UASp35/yolkGAL4 male or UASp35/yolkGAL4 female flies. The measurements for each transcript are presented as a box plot to graphically illustrate the results of three independent experiments. Comparison of UASp35/yolkGAL4 male flies to UASp35/yolkGAL4 female flies show a strong reduction of attacin, diptericin, puckered, and mmp-1 expression levels in UASp35/yolkGAL4 female flies.
To explore the impact of p35 on IMD/dJNK activation further, we determined if the expression of p35 in the fly blocks the infection-mediated induction of dJNK-dependent transcripts. Initially, we confirmed that the expression of GAL4 in female fat bodies alone does not have an appreciable impact on IMD pathway responses, as yolk-GAL4 females express dipt and puc at levels comparable with yolk-GAL4 males upon bacterial challenge (Fig. 5, C and D).
We then analyzed the transcriptional profiles of yolk-GAL4,UAS-p35 male and female flies in response to immune challenge. Despite a minor variability between replicates, likely a reflection of the efficacy of our bacterial delivery between replicates, each transcript showed a clear and reproducible trend throughout the experiments (Fig. 5, E–H). For example in our control experiment, quantification of the Rel-dependent antimicrobial peptides att and dipt repeatedly demonstrated a gradual increase of peptide expression after infection of yolk-GAL4/UAS-p35 males (Fig. 5, E and F). Likewise, bacterial challenges of yolk-GAL4/UAS-p35 males always resulted in a transient induction of the dJNK-dependent transcripts puc and mmp-1 (Fig. 5, G and H). In contrast, we observed a greatly diminished induction of att and dipt in infected yolk-GAL4/UAS-p35 female flies (Fig. 5, E and F, respectively). Most importantly, we also observed a markedly impaired induction of the dJNK-dependent transcripts puc and mmp-1 in infected yolk-GAL4/UAS-p35 female flies (Fig. 5, G and H, respectively). In summary, our in vivo observations recapitulate our cell culture data and establish a requirement for caspase activity in the activation of IMD/dJNK and the attendant induction of dJNK-responsive transcripts.
Dredd Is Required for dJNK Activation in IMD Signaling in Vivo
Our cell culture analyses strongly suggest that Dredd is required for IMD/dJNK activation and the expression of IMD/dJNK-responsive transcripts, and our in vivo data demonstrate a requirement for caspase activity in the activation of the IMD/dJNK module. The simplest explanation for these findings is that Dredd is required for IMD/dJNK activation in vivo. To test this hypothesis, we analyzed dredd mutant flies for their ability to activate the IMD/dJNK pathway in response to infection with E. coli. For these studies, we used the dreddB118 fly line. dreddB118 encodes a truncated Dredd protein that replaces arginine 127 with a stop codon and is considered a null allele (44). We initially asked if bacterial challenge induces the phosphorylation of dJNK in dreddB118 flies. To do so, we performed a Western blot analysis of phospho-dJNK levels in immune-challenged control and dreddB118 flies. Compared with control flies, dreddB118 mutant flies were clearly impaired in their ability to induce dJNK phosphorylation upon infection (Fig. 6A).
FIGURE 6.

Dredd is required for dJNK activation in IMD signaling in vivo. A, Western blot analysis of lysates from w1118 (1st to 3rd lanes) and dreddB118 (4th to 6th lanes) flies infected with E. coli and recovered for the indicated times. Protein levels are visualized with total dJNK- (lower panel) and P-dJNK (upper panel)-specific antibodies. In contrast to w1118 flies, infection with E. coli does not induce a transient increase in P-dJNK levels in dreddB118 mutant flies. Molecular weights are indicated to the right of each panel. IB, immunoblot. B–E, quantitative real time PCR analysis of w1118 flies (as indicated on the left of each box plot) and dreddB118 flies (as indicated on the right of each box plot) infected with E. coli and recovered for the indicated times. The relative expression levels for attacin (B), diptericin (C), puckered (D), and mmp1 (E) were standardized to actin levels. The expression levels for w1118 and dreddB118 flies at the indicated time points after infection are reported relative to uninfected w1118 or dreddB118 flies. Measurements for each transcript are presented as a box plot to graphically illustrate the results of three independent experiments. Comparison of w1118 flies to dreddB118 flies show a strong reduction of attacin, diptericin, puckered, and mmp1 expression levels in dreddB118 mutant flies.
We next asked if dreddB118 flies induce dJNK-responsive transcripts after infection. Despite a minor variability between replicates, each transcript showed a clear and reproducible trend throughout the experiments. We repeatedly observed induction of att and dipt in immune-challenged wild-type flies. As anticipated, we did not detect an infection-dependent increase in att or dipt expression levels in dreddB118 flies (Fig. 6, B and C, respectively). We then followed the infection-mediated induction of puc and mmp-1. We consistently found that induction of both transcripts was greatly impaired in dreddB118 mutant flies compared with wild-type control flies (Fig. 6, D and E, respectively). Combined, our data demonstrate that the two hallmarks of IMD/dJNK activation, phosphorylation of dJNK and the transcriptional induction of puc/mmp-1, are completely absent in dreddB118 mutant flies. These data are in agreement with our observations in cell culture assays and strongly argue that Dredd is essential for the activation of an IMD/dJNK response to infection in Drosophila.
DISCUSSION
All multicellular organisms rely on their innate immune system to induce appropriate defenses against microbial invaders (55). A remarkable conservation between mammalian and insect signal transduction pathways establishes D. melanogaster as an instructive model to decipher the mechanisms of innate immune response pathways. Drosophila responds to Gram-negative bacterial challenges through the IMD pathway, a pathway with significant similarities to the human TNF pathway. Both pathways require NF-κB, caspase, and JNK modules to induce appropriate antimicrobial responses (56). Caspases are cysteinyl aspartate proteases with essential developmental and homeostatic roles in programmed cell death and immunity (57, 58). For example, human caspase-8 is an important pro-apoptotic mediator of the selective processes that determine the population of mature lymphocytes (59, 60). The Drosophila caspase Dredd is considered an evolutionary relative of caspase-8. It is not clear if Dredd is a true ortholog of caspase-8, as Dredd displays several structural feature that are unusual for established caspase-8 homologs. For example, Dredd lacks bona fide N-terminal death effector domains, and the active site pentamer of Dredd (QACQE) is different from that found in other caspase-8 homologs (QACQG) (42). In addition, Dredd does not appear to perform essential apoptotic roles in vivo. In contrast, Dredd is an indispensable element of the IMD pathway, and dredd mutant flies are greatly impaired in their ability to mount comprehensive immune responses to bacterial challenges (44). A considerable body of literature demonstrated a role for Dredd in the proteolytic activation of Rel (43, 44, 49). More recent data indicated a requirement for Dredd in the phosphorylation of dJNK in tissue culture assays (45, 46). However, the precise involvement of Dredd in the dJNK arm of the IMD pathway is unexplored. In particular, there are no data on the degree to which Dredd contributes to the IMD/dJNK transcriptional response; the molecular basis for Dredd-mediated activation of dJNK is unexplored; and the epistatic relationship of Dredd to additional IMD/dJNK pathway elements requires clarification. Most importantly, there is no evidence for an involvement of Dredd in IMD/dJNK activation in vivo.
In this study, we present the results of a comprehensive analysis of Dredd in the activation of IMD/dJNK in cell culture and in vivo. Our initial cell culture assays demonstrated a fundamental requirement for Dredd in the activation of dJNK, including dJNK phosphorylation and the expression of dJNK-dependent target genes. Our interaction experiments identified Dredd as a central component of a rich network of interactions among proximal IMD signal transduction molecules and placed Dredd upstream of dTAK1 in the activation of IMD/dJNK. We demonstrated a dependence of the IMD/dJNK arm on caspase activity in cell culture and in vivo, and we showed a direct requirement for Dredd in the IMD/dJNK arm in vivo. Our results establish the position of Dredd within the IMD/dJNK pathway and enhance our understanding of signal transduction events in the IMD response.
Caspase-dependent signal transduction often proceeds through multiprotein complexes formed through homotypic interactions (61). For example, human caspase-8 interacts with FADD through death effector domains (62). Dredd and dFADD lack death effector domains. However, both proteins are characterized by N-terminal “death-inducing domains,” and mis-expressed Dredd interacts with mis-expressed dFADD in the human HeLa cell line (21). This led us to ask if Dredd interacts with dFADD in a more physiologically relevant Drosophila tissue culture line. We demonstrated a strong interaction between Dredd and dFADD in S2 cells. As dFADD is a proximal signal transduction element in the IMD pathway (25), we elaborated our studies to probe interactions among dFADD, Dredd, and additional IMD pathway members. We showed for the first time that Dredd forms a molecular complex with dIAP2 and that dFADD and dIAP2 interact in S2 cells. Our previous observation that Dredd, dFADD, dIAP2, and Imd are essential for PGN-mediated phosphorylation of dJNK supports the molecular interactions described in this study (45). Somewhat surprisingly, addition of PGN did not visibly alter interactions in our assays, and we failed to detect the reported interaction between Imd and dIAP2 (27). It is possible that these observations reflect the fact that our interaction studies relied on the induced expression of tagged constructs that mask more subtle or dynamic features of protein-protein interactions during IMD pathway signal transduction.
Our interaction studies prompted us to explore the relative position of Dredd in the IMD/dJNK pathway. Given the extensive interactions among Dredd and early IMD pathway members, we speculated that Dredd acts at an early stage of IMD/dJNK activation. Our epistasis analysis confirmed that dMKK4/7 acts downstream of dTAK1 in the activation of dJNK. In contrast, we demonstrated for the first time that Dredd acts upstream of dTAK1 in the IMD/dJNK arm. As a caveat, these epistatic data require confirmation in an in vivo model. We note that our data overlap with previous studies that proposed a role for Dredd upstream of dTAK1 in the activation of the IMD/Rel pathway (46). These observations led us to propose that Dredd is an essential element of an IMD pathway phospho-relay that diverges downstream of dTAK1 and is required for the full activation of the IMD/Rel and IMD/dJNK immune responses.
As the molecular basis of Dredd-dependent dJNK activation is unclear, we asked if caspase activity is essential to induce dJNK-dependent immune responses. Specifically, we asked if the caspase inhibitor p35 blocks IMD/dJNK activation in cell culture and in vivo assays. In both cases, we found that p35 attenuates signal transduction through the IMD/Rel and IMD/dJNK cassettes. These observations agree with established requirements for Dredd in the IMD/Rel arm and support a general requirement for a caspase in the IMD/dJNK arm. p35 is a viral caspase “suicide substrate” that is proteolytically cleaved by caspases to generate 25- and 10-kDa products (52, 53). The 25-kDa product forms a stable complex with the corresponding caspase, rendering the bound caspase proteolytically inactive. We note that key features of our p35-Dredd data are not consistent with the suicide inhibitor model described above. For example, we detected interactions between p35 and a proteolytically inactive Dredd, and the molecular weights of the individual members of the p35-Dredd complex were not consistent with p35 cleavage by a mature caspase. Despite these caveats, we believe that p35 acts directly on Dredd, potentially as a competitive inhibitor, as we demonstrated a physical interaction of p35 with Dredd in S2 cells, and our RNAi experiments indicate that Dredd is the sole essential caspase in the IMD/dJNK pathway. However, we cannot currently exclude the possibility that additional caspases are required for JNK activation in the IMD pathway.
In addition, and in strong agreement with our cell culture data, we demonstrated for the first time that p35 blocks dJNK phosphorylation and the induction of dJNK-dependent genes in vivo. Given our interaction and epistasis data, we find it attractive to speculate that Dredd proteolytic activity acts on a proximal IMD pathway member upstream of dTAK1 to activate dJNK. In this context, it is particularly noteworthy that Imd is cleaved at a caspase consensus cleavage site and that Imd cleavage requires the proteolytic activity of Dredd (27). We suggest that p35 forms a covalent adduct with Dredd and in this way inhibits Dredd-dependent Imd cleavage. This scenario results in a block of the Dredd-dependent phospho-relay and consequently blocks IMD/dJNK and IMD/Rel activation. We note that we did not detect a physical interaction between Imd and Dredd. However, caspase cleavage generally occurs rapidly, and it is possible that Dredd binds and cleaves Imd and then quickly dissociates from the active Imd molecule to facilitate the activation of IMD/Rel and IMD/dJNK.
So far, there is no evidence for the requirement of Dredd in IMD/dJNK activation in vivo. The genetically tractable model system Drosophila allows us to test cell culture observations in a more physiologically relevant in vivo context. We exploited this advantage to explore a requirement for Dredd in IMD/dJNK activation in a whole animal setting. Our studies clearly demonstrated a direct requirement for Dredd in IMD/dJNK activation in vivo. We showed that loss of Dredd blocks infection-responsive phosphorylation of dJNK and prevents a bacterial challenge-dependent induction of dJNK-dependent target genes in vivo. In our Western blot analysis, we noticed a basal amount of phospho-dJNK in samples from adult flies, which probably reflects a broad requirement for dJNK activity in the adult fly. Importantly, in our studies bacterial infection always resulted in a brief increase of phospho-dJNK levels that parallels IMD/dJNK pathway activation, and we did not observe such an increase in dreddB118 mutant flies. We believe that the ability of Dredd to modify dJNK is likely specific to the IMD pathway, as dredd null mutants do not display the traditional dorsal closure phenotype of dJNK pathway mutants. In summary, our data clearly demonstrate that Dredd is an essential component in the activation of the appropriate IMD/dJNK response in vivo.
The failure of dJNK activation in dredd mutant flies is phenocopied by the observed reduction of phospho-dJNK and dJNK-dependent transcripts in our p35 experiments. Our in vivo data are entirely in agreement with our cell culture data, and both approaches demonstrate that loss of Dredd or loss of caspase activity causes a failure to activate IMD/dJNK. In summary, our work establishes Dredd as an essential proximal element of a phospho-relay in the IMD pathway that is required for full activation of IMD/dJNK and IMD/Rel.
Acknowledgments
p35 DNA was provided by Pascal Meier. Bruno Lemaitre and Jean Marc Reichhart provided the dreddB118 and yolkGAL4 fly stocks, respectively. Relish antibodies were provided by Neal Silverman. David Bond generated the pMT-HAdTAK1CA expression plasmid. Rejish Thomas assisted with the generation of expression plasmids. cDNA was obtained from Drosophila Genomics Resource Center, and flies were provided by the Bloomington Drosophila Stock Center. We are grateful to David Bond and Brendon Parsons for critical reading of the manuscript. Infrastructure funds were provided by the Canada Foundation for Innovation and the Alberta Science and Research Investment Program.
This work was supported by Natural Sciences and Engineering Research Council Grant RGPIN 371188 (to E. F.).
- IMD
- Drosophila immune deficiency
- PGN
- peptidoglycan
- dsRNA
- double-stranded RNA
- dFADD
- Drosophila Fas-associated death domain.
REFERENCES
- 1. Medzhitov R., Janeway C. A., Jr. (1997) Cell 91, 295–298 [DOI] [PubMed] [Google Scholar]
- 2. Janeway C. A., Jr. (2001) Microbes Infect. 3, 1167–1171 [DOI] [PubMed] [Google Scholar]
- 3. Janeway C. A., Jr., Medzhitov R. (2002) Annu. Rev. Immunol. 20, 197–216 [DOI] [PubMed] [Google Scholar]
- 4. Anderson K. V., Jürgens G., Nüsslein-Volhard C. (1985) Cell 42, 779–789 [DOI] [PubMed] [Google Scholar]
- 5. Ip Y. T., Reach M., Engstrom Y., Kadalayil L., Cai H., González-Crespo S., Tatei K., Levine M. (1993) Cell 75, 753–763 [DOI] [PubMed] [Google Scholar]
- 6. Lemaitre B., Meister M., Govind S., Georgel P., Steward R., Reichhart J. M., Hoffmann J. A. (1995) EMBO J. 14, 536–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lemaitre B., Nicolas E., Michaut L., Reichhart J. M., Hoffmann J. A. (1996) Cell 86, 973–983 [DOI] [PubMed] [Google Scholar]
- 8. Medzhitov R., Preston-Hurlburt P., Janeway C. A., Jr. (1997) Nature 388, 394–397 [DOI] [PubMed] [Google Scholar]
- 9. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 10. Qureshi S. T., Larivière L., Leveque G., Clermont S., Moore K. J., Gros P., Malo D. (1999) J. Exp. Med. 189, 615–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rock F. L., Hardiman G., Timans J. C., Kastelein R. A., Bazan J. F. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 588–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Akira S., Takeda K., Kaisho T. (2001) Nat. Immunol. 2, 675–680 [DOI] [PubMed] [Google Scholar]
- 13. Lemaitre B., Hoffmann J. (2007) Annu. Rev. Immunol. 25, 697–743 [DOI] [PubMed] [Google Scholar]
- 14. Choe K. M., Werner T., Stöven S., Hultmark D., Anderson K. V. (2002) Science 296, 359–362 [DOI] [PubMed] [Google Scholar]
- 15. Gottar M., Gobert V., Michel T., Belvin M., Duyk G., Hoffmann J. A., Ferrandon D., Royet J. (2002) Nature 416, 640–644 [DOI] [PubMed] [Google Scholar]
- 16. Kaneko T., Goldman W. E., Mellroth P., Steiner H., Fukase K., Kusumoto S., Harley W., Fox A., Golenbock D., Silverman N. (2004) Immunity 20, 637–649 [DOI] [PubMed] [Google Scholar]
- 17. Leulier F., Parquet C., Pili-Floury S., Ryu J. H., Caroff M., Lee W. J., Mengin-Lecreulx D., Lemaitre B. (2003) Nat. Immunol. 4, 478–484 [DOI] [PubMed] [Google Scholar]
- 18. Rämet M., Manfruelli P., Pearson A., Mathey-Prevot B., Ezekowitz R. A. (2002) Nature 416, 644–648 [DOI] [PubMed] [Google Scholar]
- 19. Georgel P., Naitza S., Kappler C., Ferrandon D., Zachary D., Swimmer C., Kopczynski C., Duyk G., Reichhart J. M., Hoffmann J. A. (2001) Dev. Cell 1, 503–514 [DOI] [PubMed] [Google Scholar]
- 20. Gesellchen V., Kuttenkeuler D., Steckel M., Pelte N., Boutros M. (2005) EMBO Rep. 6, 979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu S., Yang X. (2000) J. Biol. Chem. 275, 30761–30764 [DOI] [PubMed] [Google Scholar]
- 22. Kleino A., Valanne S., Ulvila J., Kallio J., Myllymäki H., Enwald H., Stöven S., Poidevin M., Ueda R., Hultmark D., Lemaitre B., Rämet M. (2005) EMBO J. 24, 3423–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lemaitre B., Kromer-Metzger E., Michaut L., Nicolas E., Meister M., Georgel P., Reichhart J. M., Hoffmann J. A. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 9465–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leulier F., Vidal S., Saigo K., Ueda R., Lemaitre B. (2002) Curr. Biol. 12, 996–1000 [DOI] [PubMed] [Google Scholar]
- 25. Naitza S., Rossé C., Kappler C., Georgel P., Belvin M., Gubb D., Camonis J., Hoffmann J. A., Reichhart J. M. (2002) Immunity 17, 575–581 [DOI] [PubMed] [Google Scholar]
- 26. Zhuang Z. H., Sun L., Kong L., Hu J. H., Yu M. C., Reinach P., Zang J. W., Ge B. X. (2006) Cell. Signal. 18, 964–970 [DOI] [PubMed] [Google Scholar]
- 27. Paquette N., Broemer M., Aggarwal K., Chen L., Husson M., Ertürk-Hasdemir D., Reichhart J. M., Meier P., Silverman N. (2010) Mol. Cell 37, 172–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Silverman N., Zhou R., Erlich R. L., Hunter M., Bernstein E., Schneider D., Maniatis T. (2003) J. Biol. Chem. 278, 48928–48934 [DOI] [PubMed] [Google Scholar]
- 29. Vidal S., Khush R. S., Leulier F., Tzou P., Nakamura M., Lemaitre B. (2001) Genes Dev. 15, 1900–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boutros M., Agaisse H., Perrimon N. (2002) Dev. Cell 3, 711–722 [DOI] [PubMed] [Google Scholar]
- 31. Park J. M., Brady H., Ruocco M. G., Sun H., Williams D., Lee S. J., Kato T., Jr., Richards N., Chan K., Mercurio F., Karin M., Wasserman S. A. (2004) Genes Dev. 18, 584–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen W., White M. A., Cobb M. H. (2002) J. Biol. Chem. 277, 49105–49110 [DOI] [PubMed] [Google Scholar]
- 33. Geuking P., Narasimamurthy R., Lemaitre B., Basler K., Leulier F. (2009) PLoS One 4, e7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delaney J. R., Stöven S., Uvell H., Anderson K. V., Engström Y., Mlodzik M. (2006) EMBO J. 25, 3068–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu Y., Wu L. P., Anderson K. V. (2001) Genes Dev. 15, 104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rutschmann S., Jung A. C., Zhou R., Silverman N., Hoffmann J. A., Ferrandon D. (2000) Nat. Immunol. 1, 342–347 [DOI] [PubMed] [Google Scholar]
- 37. Silverman N., Zhou R., Stöven S., Pandey N., Hultmark D., Maniatis T. (2000) Genes Dev. 14, 2461–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu L. P., Anderson K. V. (1998) Nature 392, 93–97 [DOI] [PubMed] [Google Scholar]
- 39. Dushay M. S., Asling B., Hultmark D. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 10343–10347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hedengren M., Asling B., Dushay M. S., Ando I., Ekengren S., Wihlborg M., Hultmark D. (1999) Mol. Cell 4, 827–837 [DOI] [PubMed] [Google Scholar]
- 41. Ertürk-Hasdemir D., Broemer M., Leulier F., Lane W. S., Paquette N., Hwang D., Kim C. H., Stöven S., Meier P., Silverman N. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 9779–9784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen P., Rodriguez A., Erskine R., Thach T., Abrams J. M. (1998) Dev. Biol. 201, 202–216 [DOI] [PubMed] [Google Scholar]
- 43. Stoven S., Silverman N., Junell A., Hedengren-Olcott M., Erturk D., Engstrom Y., Maniatis T., Hultmark D. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 5991–5996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leulier F., Rodriguez A., Khush R. S., Abrams J. M., Lemaitre B. (2000) EMBO Rep. 1, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bond D., Foley E. (2009) PLoS Pathog. 5, e1000655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou R., Silverman N., Hong M., Liao D. S., Chung Y., Chen Z. J., Maniatis T. (2005) J. Biol. Chem. 280, 34048–34055 [DOI] [PubMed] [Google Scholar]
- 47. Ditzel M., Wilson R., Tenev T., Zachariou A., Paul A., Deas E., Meier P. (2003) Nat. Cell Biol. 5, 467–473 [DOI] [PubMed] [Google Scholar]
- 48. Foley E., O'Farrell P. H. (2004) PLoS Biol. 2, E203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stöven S., Ando I., Kadalayil L., Engström Y., Hultmark D. (2000) EMBO Rep. 1, 347–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aggarwal K., Rus F., Vriesema-Magnuson C., Ertürk-Hasdemir D., Paquette N., Silverman N. (2008) PLoS Pathog. 4, e1000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bond D., Primrose D. A., Foley E. (2008) Biol. Proced. Online 10, 20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Riedl S. J., Renatus M., Snipas S. J., Salvesen G. S. (2001) Biochemistry 40, 13274–13280 [DOI] [PubMed] [Google Scholar]
- 53. Xu G., Cirilli M., Huang Y., Rich R. L., Myszka D. G., Wu H. (2001) Nature 410, 494–497 [DOI] [PubMed] [Google Scholar]
- 54. Brand A. H., Perrimon N. (1993) Development 118, 401–415 [DOI] [PubMed] [Google Scholar]
- 55. Hoffmann J. A., Kafatos F. C., Janeway C. A., Ezekowitz R. A. (1999) Science 284, 1313–1318 [DOI] [PubMed] [Google Scholar]
- 56. Hoffmann J. A., Reichhart J. M. (2002) Nat. Immunol. 3, 121–126 [DOI] [PubMed] [Google Scholar]
- 57. Lamkanfi M., Festjens N., Declercq W., Vanden Berghe T., Vandenabeele P. (2007) Cell Death Differ. 14, 44–55 [DOI] [PubMed] [Google Scholar]
- 58. Shi Y. (2002) Mol. Cell 9, 459–470 [DOI] [PubMed] [Google Scholar]
- 59. Fernandes-Alnemri T., Armstrong R. C., Krebs J., Srinivasula S. M., Wang L., Bullrich F., Fritz L. C., Trapani J. A., Tomaselli K. J., Litwack G., Alnemri E. S. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 7464–7469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maelfait J., Beyaert R. (2008) Biochem. Pharmacol. 76, 1365–1373 [DOI] [PubMed] [Google Scholar]
- 61. Micheau O., Tschopp J. (2003) Cell 114, 181–190 [DOI] [PubMed] [Google Scholar]
- 62. Shi Y. (2006) Curr. Opin. Cell Biol. 18, 677–684 [DOI] [PubMed] [Google Scholar]