

Abstract
Cholangiocytes, the epithelial cells lining the bile ducts in the liver, are periodically exposed to potentially injurious microbes and/or microbial products. As a result, cholangiocytes actively participate in microbe-associated, hepatic proinflammatory responses. We previously showed that infection of cultured human cholangiocytes with the protozoan parasite, Cryptosporidium parvum, or treatment with Gram-negative bacteria-derived LPS, activates NFκB in a myeloid differentiation 88 (MyD88)-dependent manner. Here, we describe a novel signaling pathway initiated by Toll-like receptors (TLRs) involving the small GTPase, Ras, that mediates cholangiocyte proinflammatory cytokine production and induction of cholangiocyte proliferation. Using cultured human cholangiocytes and a Ras activation assay, we found that agonists of plasma membrane TLRs (TLR 1, 2, 4, 5, and 6) rapidly (<10 min) activated N-Ras, but not other p21 Ras isoforms, resulting in the rapid (<15 min) phosphorylation of the downstream Ras effector, ERK1/2. RNA interference-induced depletion of TRAF6, a downstream effector of MyD88 and known activator of MAPK signaling, had no effect on N-Ras activation. Following N-Ras activation the proinflammatory cytokine, IL6, is rapidly secreted. Using a luciferase reporter, we demonstrated that LPS treatment induced IL6 promoter-driven luciferase which was suppressed using MEK/ERK pharmacologic inhibitors (PD98059 or U0126) and RNAi-induced depletion of N-Ras. Finally, we showed that LPS increased cholangiocyte proliferation (1.5-fold), which was inhibited by depletion of N-Ras; TLR agonist-induced proliferation was also inhibited following pretreatment with an IL6 receptor-blocking antibody. Together, our results support a novel signaling axis involving microbial activation of N-Ras likely involved in the cholangiocyte pathogen-induced proinflammatory response.
Keywords: Cell Proliferation, Epithelial Cell, Inflammation, Innate Immunity, Ras, Toll-like receptor (TLR), Cholangiocyte
Introduction
Intrahepatic bile ducts form a complex three-dimensional tubular network that modifies the composition of bile and serves as a conduit for delivery of bile to the duodenum. Although bile is thought to be sterile under normal physiological conditions, biliary epithelial cells lining bile ducts (cholangiocytes) are periodically exposed to potentially pathogenic organisms or products derived from these microbes (1–4). Indeed, cholangiocytes are exposed to bacteria (5–9), viruses (10–12), and parasites (13–16). Cholangiocyte pathogen recognition promotes innate and adaptive immune responses (4, 17–19) through the expression of adhesion molecules (20–22) and the secretion of cytokines/chemokines (23, 24) and antimicrobial peptides (18, 23). Thus, cholangiocytes once thought of as an exclusively secretory/absorptive epithelium with a primary role in bile modification, are now recognized as important in the hepatic proinflammatory response to microorganisms or microbially derived products. However, the molecular pathways regulating cholangiocyte recognition and response to microbial insults are obscure and are the focus of the work presented here. Toll-like receptor (TLR)2 activation and associated nuclear factor κB (NFκB) signal transduction are essential for eliciting an efficient and effective innate immune response (25, 26). Human cholangiocytes express all 10 TLRs (27). Using a cell culture model to assess cholangiocyte responses to microbes, we demonstrated that Cryptosporidium parvum, a protozoon parasite, and Gram-negative bacterial lipopolysaccharide (LPS), induced TLR-dependent activation of NFκB (27). Furthermore, cholangiocyte pathogen recognition-dependent NFκB activation induces secretion of innate immune response-associated molecules, including antimicrobial peptides and cytokines/chemokines, in a myeloid differentiation 88 (MyD88)-dependent manner (27). One of these cytokines, IL6, is central to the proinflammatory and proliferative response of cholangiocytes. Indeed, cholangiocyte IL6 expression is responsive to a variety of signal cascades, including NFκB and MAPK activation. Whether MAPK signaling is involved in pathogen recognition-induced IL6 expression in cholangiocytes is unknown.
Ras family GTPase homologs (Harvey-, H-; Kirsten-, K-; and neuroblastoma-, N-Ras) are expressed throughout mammalian tissue and transduce signals from extracellular stimulus-activated receptors, including cytokine/chemokine receptors. Activated Ras promotes a variety of signal transduction cascades including the MAPK, PI3K, and RAL-GTPase pathways that regulate cell proliferation, survival, differentiation, and proinflammatory cytokine production (28). Although the Ras homologs redundantly activate downstream signaling cascades, specificity in Ras signaling has been described; for example, low level stimulation of the T cell receptor of Jurkat T cells resulted in specific activation of N-Ras (29). This specificity is likely conferred by relative homolog abundance, differential subcellular localization, selectivity in downstream effectors, and utilization of distinct guanine nucleotide exchange factors (GEFs) to activate Ras proteins (28).
Although it is accepted that pathogen recognition promotes cholangiocyte proliferation (24), the role of TLRs and activated Ras in this process has not been thoroughly investigated. Furthermore, the normal physiological role of Ras in the cholangiocyte response to pathogen recognition and how disturbances in Ras function contribute to altered cholangiocyte proinflammatory gene expression remain unclear, as do the physiological consequences of pathogen-induced Ras activation. In the present paper, using a cell culture model of cholangiocyte response to microbial insult, we found that N-Ras is the predominant Ras isoform expressed in these epithelia. We also demonstrated that TLR agonists, including LPS, specifically and rapidly induce N-Ras activation and induce signaling through the MEK/ERK pathway. Moreover, small interfering (si)RNA-induced depletion of TLR4 inhibits N-Ras activation, whereas depletion of TRAF6, a known mediator of TLR-dependent MAPK activation, has no effect on this pathway. Finally, depletion of N-Ras diminishes IL6 expression and correlates with diminished cholangiocyte proliferation, whereas transfection with a constitutively active N-Ras construct increases LPS-induced IL6 expression. These results indicate that TLR-dependent activation of N-Ras is a central regulator of the cholangiocyte proinflammatory and proliferative response to microbial insult.
EXPERIMENTAL PROCEDURES
Cell Culture and TLR Agonist Treatment
H69 cells (a gift from Dr. D. Jefferson, Tufts University, Boston, MA) are SV40-transformed normal human cholangiocytes originally derived from normal liver harvested for transplant. These cholangiocytes continue to express biliary epithelial cell markers (CK19 and GGT) and have been extensively characterized (30). Twenty-four hours after plating, the cells were rinsed, and complete medium was replaced with medium lacking FBS and growth factors. TLR agonists were purchased from a commercial source (Invivogen, San Diego, CA) and added to the medium at the following recommended concentrations: LPS (TLR4 agonist, 200 ng/ml), Pam3CSK4 (TLR1/2 agonist, 1 μg/ml), heat-killed Listeria monocytogenes (HKLM; TLR2 agonist, 1 × 108 cells/ml), poly(I·C) (TLR3 agonist, 10 μg/ml), flagellin (TLR5 agonist, 10 μg/ml), FSL1 (TLR2/6 agonist, 1 μg/ml), imiquimod (TLR7 agonist, 10 μg/ml); ssRNA40 (TLR8 agonist, 10 μg/ml), and ODN2006 (TLR9 agonist, 10 μg/ml).
Immunoblotting
Cells were lysed using M PER protein extraction reagent (Thermo Scientific), and immunoblotting was performed on total cell lysates as previously described (22). Briefly, protein lysate was separated by SDS-PAGE and blotted onto nitrocellulose membranes. The membranes were incubated overnight in primary antibodies against N-Ras (Santa Cruz Biotechnology, Santa Cruz, CA), H-Ras (Santa Cruz Biotechnology), K-Ras (Santa Cruz Biotechnology), TRAF6 (Cell Signaling Technologies, Danvers, MA), p-ERK1/2 (Cell Signaling Technologies), ERK1/2 (Cell Signaling Technologies), STAT3 (Cell Signaling Technologies), p-STAT3 (Cell Signaling Technologies), IL6 (Santa Cruz Biotechnology) or actin (Sigma-Aldrich). The blots were washed and incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibody, and bands were revealed using the enhanced chemiluminescent plus detection system (ECL Plus; Amersham Biosciences).
Luciferase Assay
H69 cells were grown to 80% confluence in a 24-well plate. The cells were transfected with the 0.5 μg of IL6 promoter (Switch Gear Genomics, Menlo Park, CA), or the NFκB reporter luciferase construct (Promega, Madison, WI) and 30 ng of TK-Renilla luciferase plasmid (Promega) which served as a transfection control. Transfections were performed using FuGENE HD following the manufacturers' protocol (Roche Diagnostics). For microbial stimulus experiments the transfected cells were incubated for 6 h and subsequently treated with LPS, HKLM, or PAM3CSK4. For experiments involving RNAi, cells were transfected with siRNA using siPORT (Ambion, Austin, TX) according to the manufacturer's directions or shRNA using FuGENE HD for 24 h prior to analysis. The values are expressed as -fold change between the relative IL6 or NFκB-driven firefly luciferase to transfection control Renilla luciferase and normalized to pGL4 empty vector.
RAS Activation Assay
Cells were grown as above prior to treatment with TLR agonists. 500 μg of protein from lysed cells was used for each Ras activation assay (Cytoskeleton, Denver, CO). Following immunoprecipitation with the Raf1-RBD beads, the pull-down product was separated by SDS-PAGE and transferred to nitrocellulose. Total protein was stained using Ponceau Red; subsequent immunoblotting was performed using primary antibodies specific to N-Ras, K-Ras, or H-Ras.
IL6 ELISA
An IL6 ELISA was performed to quantify secreted IL6 present in the cell culture medium (Invitrogen). Briefly, H69 cells were treated with LPS (200 ng/ml) for 15 min, washed, and incubated in medium for up to 48 h. At each time point, the cell culture was harvested for ELISA, and the cells were lysed for Ras activation analysis (as above). The cell culture medium at each time point was centrifuged to remove cellular debris, and 100 μl of cleared cell medium was used in the IL6 ELISA following the manufacturer's instructions. The IL6 ELISA was read using a 450-nm wavelength against a 630-nm reference wavelength. A standard curve was generated with known quantities of IL6, and sample concentrations were calculated.
Quantitative PCR
H69 cells were grown to confluence, and total RNA was extracted using TRIzol reagent (Invitrogen) and synthesized into cDNA using SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). Quantitative PCR was performed using 1 μl of cDNA and the Light Cycler Fast Start DNA MasterPlus SYBR Green I kit (Roche Diagnostics) as described previously (39). The primers used to evaluate the RAS isoforms were: N-Ras sense (5′-CTTGCTGGTGTGAAATGACTGAG-3′) and antisense (5′-AAGCCTTCGCCTGTCCTCATGT-3′); K-Ras sense (5′-AGGAAGCAAGTAGTAATTGATGGA-3′) and antisense (5′-GCCTGTTTTGTGTCTACTGTTCT-3′); H-Ras sense (5′-TGCCTGTTGGACATCCTGGATA-3′) and antisense (5′-ATGTAGGGGATGCCGTAGCTT-3′). The samples were normalized to 18 S rRNA.
Proliferation Assay
H69 cells were transfected with N-Ras siRNA, and after 24 h 200 ng/ml LPS was added to the cells and incubated for 24 and 48 h prior to the assay. The rate of cell proliferation was determined using CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega) according to the manufacturer's instructions.
Statistical Analysis
All values are given as mean ± S.E. Means of groups were compared with Student's t test (unpaired) or the ANOVA test when appropriate. p values <0.05 were considered statistically significant.
RESULTS
N-Ras Is the Most Abundant p21 Ras Isoform in Cholangiocytes and Is Activated following LPS Treatment
Quantitative PCR was performed, using PCR primers specific to human N-Ras, K-Ras, or H-Ras, to determine the relative expression of the p21Ras isoforms in cholangiocytes. The basal expression of N-Ras in cultured cholangiocytes was significantly higher (*, p < 0.05) than K-Ras (2-fold greater) or H-Ras (5-fold greater) (Fig. 1A). We next asked whether LPS treatment induced the activation of these Ras isoforms. GTP-bound Ras interacts with Raf1 kinase, a property that can be exploited to assess the activated state of this GTPase. Using the Ras binding domain (RBD) of RAF1 kinase-bound beads, GTP-bound Ras isoforms were isolated from untreated and LPS-treated cells. Activated N-Ras was detected by Western blotting when cells were treated with LPS at a concentration of 50 ng/ml, whereas activated K-Ras and H-Ras was not detected at concentrations up to 200 ng/ml LPS (Fig. 1B). A time course of LPS-induced N-Ras activation was performed and demonstrates rapid (within 10 min after treatment) and persistent (through 60 min after treatment) activation (Fig. 1C). Total N-Ras expression was assessed by quantitative RT-PCR following LPS treatment. No increase in total N-Ras expression was observed over the course of 12 h (Fig. 1D). Together, these data suggest that N-Ras is the dominant p21 Ras isoform expressed in cultured cholangiocytes and that this small GTPase is rapidly activated following LPS treatment. Hence, sensitivity to LPS may reflect an important difference between the p21 Ras isoforms.
FIGURE 1.

N-Ras is the predominant p21 Ras isoform expressed in cultured cholangiocytes. A, RNA was extracted from confluent H69 cells, and quantitative RT-PCR was performed using p21 Ras isoform-specific PCR primers. N-Ras mRNA is expressed at a level 2-fold and 5-fold greater than K-Ras and H-Ras, respectively (p < 0.05). Data were normalized to the 18 S rRNA level and expressed as copies of RAS mRNA/106 copies of 18 S rRNA. B, LPS activates N-Ras in a dose-dependent manner. Confluent H69 cells were incubated in the presence of LPS (0, 10, 50, 100, or 200 ng/ml) for 15 min. Activated RAS was pulled down using RAF-RBD-agarose beads, and immunoblotting for N-Ras, K-Ras, and H-Ras was performed. Activated N-Ras was detected at doses as low as 50 ng/ml, whereas activated K-Ras and H-Ras were not detected after LPS treatment. Total RBD-GST was used as a loading control. C, LPS-induced N-Ras activation is rapid and persistent. Activated N-Ras was detected within 10 min following LPS treatment and remained activated through 60 min after LPS treatment. Total RBD-GST was used as a loading control. D, expression of N-Ras in cholangiocytes following treatment with LPS was quantitated by RT-PCR and normalized to 106 copies of 18 S rRNA. No significant increase was observed in N-Ras mRNA expression following LPS treatment. Quantitative PCR data are presented as mean ± S.E. (error bars) from three independent experiments; *, p < 0.05 compared with N-Ras.
Cholangiocyte N-Ras Activation Correlates with NFκB Reporter Luciferase Expression following Treatment with TLR Agonists
Having established that LPS treatment rapidly activates N-Ras (Fig. 1C), we next assessed whether other TLR ligands induce N-Ras activation. To demonstrate first that cholangiocytes are responsive to these TLR ligands, we performed a NFκB reporter luciferase assay. The NFκB luciferase reporter construct contains multiple NFκB consensus sequences that drive the expression of the luciferase reporter. The synthetic lipoprotein, FSL1, a TLR2/6 agonist derived from Mycoplasma salivarium, exhibited the greatest induction of NFκB reporter luciferase activity (Fig. 2A). Indeed, multiple agonists for plasma membrane-associated TLRs induced luciferase expression, including flagellin (TLR5 agonist), LPS (TLR4 agonist), HKLM (TLR2 agonist), and a synthetic lipopeptide representing the acylated amino terminus of bacterial lipoproteins (Pam3CSK4; TLR1/2 agonist) (Fig. 2A). Conversely, the intracellular TLR agonists poly(I·C) (TLR3 agonist), imiquimod (TLR7 agonist), ssRNA40 (TLR8 agonist), and ODN2006 (TLR9 agonist) exhibited minimal NFκB activation in the cultured cholangiocytes (Fig. 2A). Having demonstrated that a subset of TLR agonists induces NFκB activation, we addressed whether these agonists induce cholangiocyte N-Ras activation (Fig. 2B). The TLR ligands demonstrating the greatest induction of NFκB luciferase reporter activity also appeared to exhibit the most robust N-Ras activation (flagellin > LPS > HKLM > Pam3CSK4). Hence, a regression analysis was performed to assess the relationship between NFκB reporter luciferase activity and N-Ras activation. This calculation demonstrated a strong correlation (correlation coefficient = 0.87) between NFκB reporter luciferase activity and N-Ras activation (Fig. 2C). Together, these data suggest that cholangiocyte plasma membrane-localized TLRs simultaneously activate the NFκB and Ras/MAPK pathways.
FIGURE 2.
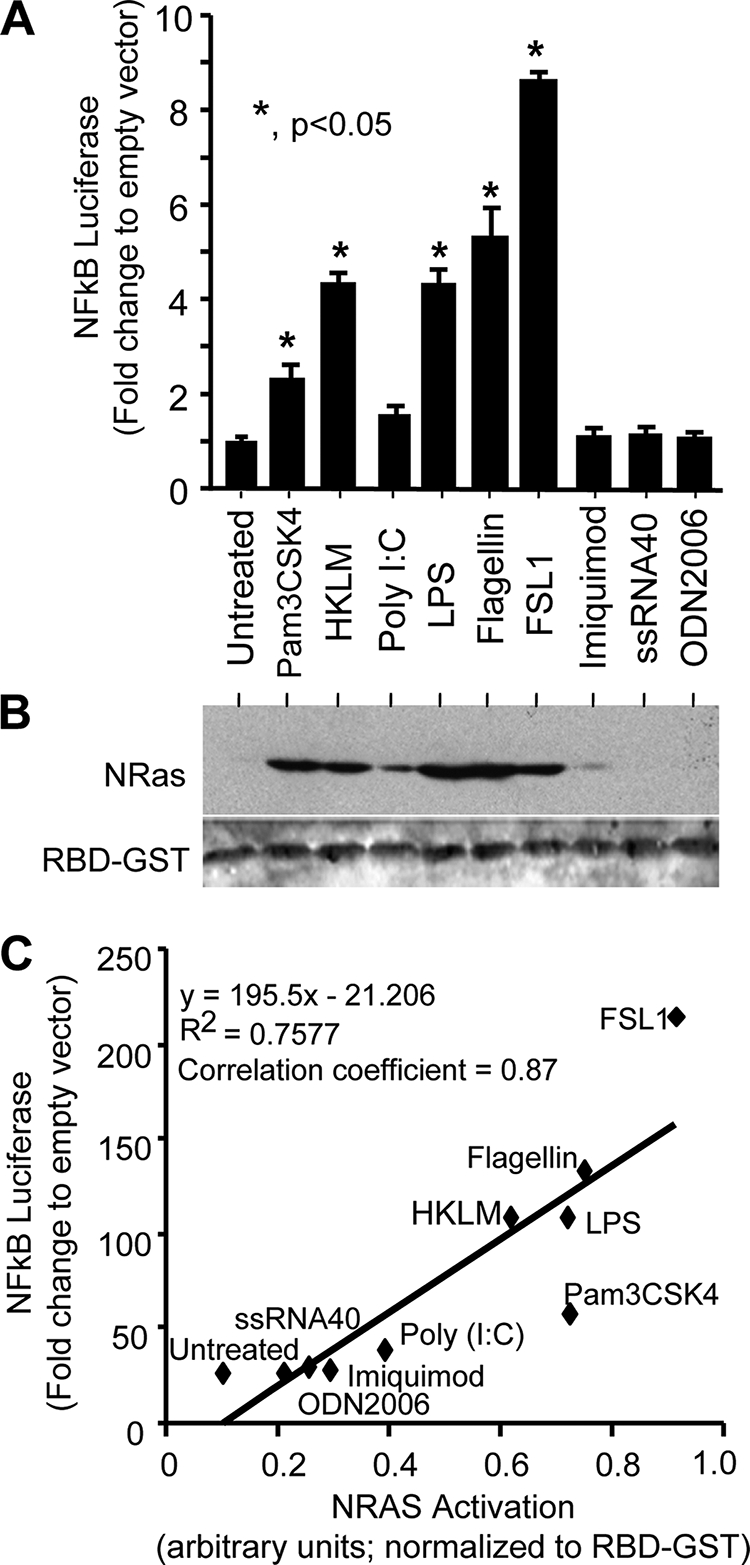
TLR agonists induce NFκB reporter luciferase and N-Ras activation. A, tandem NFκB consensus sites were cloned into the luciferase expression plasmid, pGL4.22. This plasmid was co-transfected into cholangiocytes with the transfection control plasmid TK-Renilla. Treatment with membrane-bound TLR agonists Pam3CSK4 (TLR1/2), HKLM (TLR2), LPS (TLR4), flagellin (TLR5), and FSL-1 (TLR2/6) induced NFκB-driven luciferase expression. Expression is presented as -fold change NFκB-driven firefly luciferase: Renilla luciferase compared with empty pGL4.22 vector firefly luciferase: Renilla luciferase. Data are represented as mean ± S.E. (error bars). B, an N-Ras activation assay was performed on TLR agonist-treated cultured cholangiocytes. Again, the membrane-bound TLR agonists Pam3CSK4, HKLM, LPS, flagellin, and FSL-1 exhibited robust N-Ras activation, whereas agonists for the cytoplasmic TLRs exhibited minimal N-Ras activation. Total RBD-GST was used as a normalizing control. C, a correlation coefficient was performed to assess the linear relationship between TLR agonist-induced NFκB-driven luciferase activity and N-Ras activation. A strong correlation is demonstrated with a correlation coefficient = 0.87 (y = 195.5x − 21.206; R2 = 0.7577).
LPS-induced Cholangiocyte N-Ras Activation Requires TLR4 but Not TRAF6
In an effort to determine the proximal events leading to N-Ras activation, RNA interference was used to selectively target TLR4 and TRAF6, an adaptor molecule known to transduce signals from TLRs to both NFκB and MAPKs (31). We have previously demonstrated that TLR4 depletion by siRNAs suppresses TLR4 protein expression and diminishes NFκB activation in the presence of TLR4 ligands (27). Following LPS treatment, N-Ras is activated in H69 cells and cells transfected with a scrambled control siRNA (Fig. 3A). In contrast, transfection of H69 cells with a TLR4 siRNA diminishes LPS-induced N-Ras activation (Fig. 3A). Given that Ras activates the MEK/ERK pathway, we next addressed whether LPS treatment promotes ERK phosphorylation and whether this process requires N-Ras. Following LPS treatment, ERK is rapidly phosphorylated (<15 min); however, using an siRNA that effectively depleted N-Ras (Fig. 3B), we show that LPS-induced ERK phosphorylation requires functional N-Ras (Fig. 3B). The current paradigm of MAPK signaling initiated by TLRs is through the downstream kinase, TRAF6. We therefore transfected the H69 cells with short hairpin RNAs to deplete TRAF6, validated knockdown by Western blotting (Fig. 3C), and established a cell line expressing one of these shRNAs (#80; Fig. 3C). TLR agonists failed to activate NFκB in H69 cells stably transfected with this shRNA as demonstrated with the NFκB-driven luciferase reporter (Fig. 3D) yet maintained microbial-induced N-Ras activation (Fig. 3E). The assay was performed with both the TLR4 ligand, LPS, which signals through both Myd88-dependent and -independent (Toll/Interleukin-1 receptor (TIR)-domain-containing adaptor-inducing interferon-β/TRIF-related adaptor molecule) pathways, and the TLR2 ligand, HKLM, where signal transduction occurs in a MyD88-dependent manner. Furthermore, silencing of TRAF6 had no effect on LPS-induced ERK phosphorylation (Fig. 3F). In addition to demonstrating a primary role for TRAF6 in cholangiocyte NFκB activation, these results suggest that TLRs rapidly activate MAPK signaling (ERK) through an N-Ras-dependent, yet TRAF6- and NFκB-independent mechanism.
FIGURE 3.

N-Ras activation is dependent on TLR4 but not TRAF6. A, H69 cells were transfected with TLR4-siRNA (50 nm), scrambled (Scr)-siRNA (50 nm), or FuGENE HD alone (control, Ctrl) 24 h prior to treatment. The cells were treated with LPS (200 ng/ml) for 15 min, and the RAS activation assay was performed. Ponceau Red stain was utilized to detect RBD-GST to confirm equal loading. N-Ras activation was observed following LPS treatment; however, the TLR4-siRNA diminished N-Ras activation compared with both LPS-treated control and LPS-treated cell transfected with the scrambled control (Scr siRNA). B, following a 15-min LPS treatment, an increase in phosphorylated ERK is detected in H69 cells. However, transfection of H69 cell with an N-Ras siRNA, which effectively depleted N-Ras, blocked LPS-induced ERK phosphorylation. C, stable transfections of H69 cells, using three different TRAF-6 shRNA constructs (#77, 78, and 80) or with the pGIPZ empty vector were performed. Following stable clone selection, immunoblotting was performed for TRAF6. The 60-kDa TRAF6 protein was detected in the pGIPZ-transfected cells, but TRAF6 was largely diminished in the shRNA expressing cell lines. D, the NFκB luciferase reporter system was utilized to confirm the functional depletion of TRAF6 in the cells expressing shRNA #80. Cholangiocytes stably transfected with either the pGIPZ empty vector (black bars) or TRAF6-shRNA (gray bars) were treated with LPS (200 ng/ml) or HKLM (1 × 108 cells/ml) for 5 h followed by the Dual Luciferase Reporter assay. Both LPS and HKLM induced NFκB-driven luciferase in pGIPZ-transfected cells (*, p < 0.05 compared with control untreated cells), whereas the TRAF6-shRNA-expressing cells showed no increase in NFκB-driven luciferase activity. Data are presented as mean ± S.E. (error bars). E, an N-Ras activation assay was also performed on LPS-treated TRAF6-depleted cells. The pGIPz control transfected or TRAF6-depleted cells were treated with LPS or HKLM for 15 min. N-Ras activation was not diminished in those cells depleted of TRAF6. The membrane was stained with Ponceau Red to confirm equal loading. EV, empty vector. F, depletion of TRAF6 had no effect on LPS-induced ERK phosphorylation. LPS (200 ng/ml for 15 min) treatment of both the pGIPZ empty vector control cells and TRAF6-depleted cells resulted in a similar increase in phosphorylation of ERK1/2 as demonstrated by Western blotting.
LPS-induced IL6 Expression Is Enhanced by N-Ras Activation
LPS induces robust IL6 expression in cultured cholangiocytes (24). We therefore assessed whether the N-Ras/ERK pathway influenced the cholangiocyte IL6 response. Following LPS treatment, cellular IL6 protein was increased at 6 h after treatment as assessed by immunoblotting (Fig. 4A). However, siRNA-mediated knockdown of N-Ras diminished LPS-induced IL6 expression (Fig. 4A). We further analyzed IL6 protein expression by ELISA of cell culture supernatant at various time points following brief (15 min) LPS treatment. Following LPS treatment, the cells were washed repeatedly and activated N-Ras and IL6 protein in culture supernatant was assessed immediately (15 min), 30 min, 1 h, 3 h, and 6 h after LPS treatment. Activated N-Ras was detected at 15 min and remained elevated until 1 h after LPS treatment. By 3 h after LPS treatment, activated N-Ras diminished, yet rebounded at the 6-h time point, suggesting a secondary, autocrine mechanism of N-Ras activation (Fig. 4B). IL6 expression was elevated by 3 h after LPS treatment (Fig. 4B). Immunoblotting was performed on the cellular lysates to assess the activation status of signaling molecules downstream of N-Ras. A dramatic increase of p-ERK1/2 was detected at 15 min after LPS treatment, correlating with N-Ras activation, and gradually decreased over the course of 3 h (Fig. 4C). In contrast, STAT3 activation (p-STAT3), a downstream mediator of IL6 signaling, was detected at 2 h after LPS treatment and exhibited sustained activation through 3 h after LPS treatment (Fig. 4C), which was maintained through 48 h after LPS treatment (data not shown). Therefore, a brief LPS treatment induces rapid N-Ras activation, which correlates with ERK1/2 phosphorylation. IL6 secretion is subsequently up-regulated, as is the sustained activation of STAT3. Because these data suggest biphasic N-Ras activation over the course of 6 h, we addressed whether an autocrine response to IL6 mediated the delayed activation of N-Ras. Cholangiocytes were cultured in the presence of a blocking antibody specific to IL6 receptor (1 μg/ml), treated with LPS for 15 min, washed, and assessed for N-Ras activation immediately or following a 6-h incubation. The IL6 receptor-blocking antibody had no effect on LPS-induced N-Ras activation at the 15-min time point (Fig. 4D). Conversely, the IL6 receptor-blocking antibody diminishes LPS-induced N-Ras activation at the 6-h time point (Fig. 4D), suggesting that an IL6 autocrine signaling mechanism regulates the delayed activation of N-Ras. Together, these results suggest that LPS-induced N-Ras activation contributes to cholangiocyte IL6 expression, and the induction of IL6 promotes a delayed N-Ras response in an autocrine-dependent manner. Furthermore, LPS treatment induced immediate ERK1/2 and delayed STAT3 activation.
FIGURE 4.

LPS induces IL6 expression in an N-Ras-dependent manner. A, H69 cells transfected with either the scrambled (Scr) or N-Ras siRNA were cultured in the presence or absence of LPS (200 ng/ml) for 6 h, and IL6 immunoblotting was performed. IL6 expression was induced in the cells transfected with the scrambled siRNA; however, cells depleted of N-Ras exhibited no increase in IL6 expression at this time point. Actin was blotted as a loading control. B, cholangiocytes were treated with LPS (200 ng/ml) for 15 min, washed, and cultured over the course of 6 h. An IL6 ELISA was performed on tissue culture supernatant, and an N-Ras activation assay was performed on the cellular lysate (n = 3). N-Ras activation exhibited a biphasic response to LPS-treatment. Activation of N-Ras occurred immediately following LPS exposure (15 min), decreased at 1 h, and increased again after 3 h. In contrast, an increase in IL6 secretion was first detected 1 h after LPS treatment and was consistently elevated from 3 h after LPS treatment. C, the cellular lysate from each time point was utilized for an N-Ras activation assay and immunoblotting for phosphorylated ERK1/2, total ERK1/2 (loading control), and phosphorylated STAT3. N-Ras activation was observed at the 15-min time point and correlated with the phosphorylation of ERK1/2. In contrast, STAT3 phosphorylation was delayed (1 h) and remained elevated through 6-h after LPS treatment. D, an IL6 receptor inhibitory antibody (IL6R Ab) diminishes N-Ras activation at the 6-h time point. Cells were cultured in the presence or absence of an IL6 inhibitory antibody and in the presence or absence of LPS. The IL6-inhibiting antibody did not inhibit N-Ras activation 15 min after LPS treatment, but diminished N-Ras activation at the 6-h time point.
LPS Modulates IL6 Transcription in an ERK1/2- and N-Ras-dependent Manner
An IL6 promoter luciferase reporter construct was utilized to assess the role of activated N-Ras in LPS-induced IL6 transcription in cultured cholangiocytes. In control H69 cells, LPS treatment induced IL6 reporter luciferase ∼4-fold compared with cells cultured in the absence of LPS (Fig. 5A). Forced expression of wild-type N-Ras did not induce IL6 promoter-dependent luciferase transcription; however, LPS treatment of cells expressing this construct exhibited increased luciferase expression compared with control LPS-treated cholangiocytes (Fig. 5A). In addition, forced expression of constitutively active N-Ras mutant, G12D, resulted in a modest increase in IL6 reporter luciferase in the absence of LPS compared with control untreated cells (Fig. 5A). However, in the presence of LPS, the cells expressing the constitutively active mutant exhibited IL6 reporter luciferase expression that was significantly elevated compared with control untreated cells as well as control and N-Ras wild type expressing LPS-treated cells (Fig. 5A). To confirm that N-Ras contributed to the observed increase in luciferase expression, the N-Ras siRNA (50 μm) or a dominant negative N-Ras construct was co-transfected with the IL6 promoter luciferase reporter plasmid. RNA-induced silencing of N-Ras suppressed LPS-induced IL6 promoter-dependent luciferase expression compared with control LPS-treated cells (Fig. 5A). Similarly, forced expression of the N-Ras S17N dominant negative diminished LPS-induced IL6 promoter-dependent luciferase expression. These results suggest that N-Ras is required for robust LPS-induced IL6 transcription. We next addressed whether inhibition of MEK/ERK signaling modulated LPS-induced IL6 promoter-dependent luciferase expression. When cells were preincubated for 30 min in the presence of highly specific MEK/ERK pharmacological inhibitors PD98059 (10 μm) or U0126 (10 μm), the LPS-induced increase of IL6 reporter luciferase was abolished to the levels observed in the untreated cells (Fig. 5B). Hence, the IL6 promoter is responsive to N-Ras/MEK/ERK signaling. Together, these results suggest that LPS-induced N-Ras activation and subsequent MEK/ERK signaling are key players in the cholangiocyte response to pathogens.
FIGURE 5.

LPS induces IL6 promoter-driven luciferase reporter in an N-Ras- and ERK-dependent manner. A, the H69 cells were transfected with N-Ras WT, N-Ras G12D constitutively active (C/A), N-Ras siRNA, or N-Ras S17N dominant negative plasmid and treated with LPS (200 ng/ml), and a Dual Luciferase assay was performed. In the absence of LPS, the N-Ras G12D constitutively active mutant was the only constructed exhibiting a significant increase in IL6 reporter luciferase (*, p < 0.05). Following LPS stimuli, both N-Ras WT and the constitutive active mutant (G12D C/A) showed significant increases in IL6 reporter luciferase compared with LPS-treated control cells. In contrast, the N-Ras siRNA and dominant negative mutant (S17N D/N) significantly decreased IL6 reporter luciferase compared with control LPS-treated cells. B, the IL6 promoter-driven luciferase reporter assay was performed in the presence and absence of the MEK/ERK inhibitors PD98059 and U0126. Both inhibitors significantly diminished LPS-dependent IL6 promoter-driven luciferase activity compared with cholangiocytes cultured in the absence of inhibitors (*, p < 0.05). Expression is presented as -fold change in IL6 promoter-driven firefly luciferase: Renilla luciferase compared with empty pGL4.22 vector firefly luciferase: Renilla luciferase. Data are represented as mean ± S.E. (error bars; n = 5).
LPS Induces Cholangiocyte Proliferation in an N-Ras- and IL6-dependent Manner
LPS induces proliferation in cultured cholangiocytes (24). We therefore addressed whether depletion of N-Ras altered LPS-induced cholangiocyte proliferation. H69 cells were transfected with the N-Ras siRNA or a control scrambled siRNA and treated with LPS, and cell proliferation was assessed. LPS treatment increases the proliferation rate in scramble control treated cells compared with cells cultured in the absence of LPS (Fig. 6A). However, LPS treatment of cells depleted of N-Ras exhibited a proliferation rate similar to cells cultured in the absence of LPS (Fig. 6A). Hence, N-Ras is required for LPS-induced cholangiocyte proliferation. Our initial observations demonstrated that N-Ras activation and p-ERK promote LPS-induced IL6 promoter-dependent luciferase expression (Fig. 5, A and B). Furthermore, LPS has been shown to promote cholangiocyte proliferation through IL6 and ERK activation (24). Hence, to validate our system, we addressed whether LPS-induced cholangiocyte proliferation required IL6. As expected, an IL6 inhibitory antibody blocks IL6-induced cholangiocyte proliferation (Fig. 6B). Furthermore, LPS-induced proliferation is diminished when the cells are cultured in the presence of an IL6 inhibitory antibody (Fig. 6B). Thus, LPS-induced proliferation in cultured cholangiocytes requires IL6, yet depletion of N-Ras abrogates the proliferative effect of LPS. Together, our results suggest that cholangiocyte N-Ras is central to LPS-induced proinflammatory signaling and proliferation in response to microbial insult.
FIGURE 6.

N-Ras modulates LPS-induced cholangiocyte proliferation through IL6. A, cholangiocytes were transfected with N-Ras siRNA (50 nm) for 24 h. Proliferation was assessed using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay. Cells treated with LPS (200 ng/ml) exhibited a significant (*, p < 0.05) increase in proliferation 24 h after LPS treatment compared with uninfected cells. However N-Ras depletion using the N-Ras siRNA significantly reduced LPS-induced cholangiocyte proliferation (#, p < 0.05) compared with the LPS-treated cells in the absence of N-Ras siRNA. B, to functionally validate the importance of IL6 autocrine signaling in LPS-induced proliferation the cells were treated with LPS or IL6 in the absence or presence of the IL6R-blocking antibody or a control, nonspecific isotype-matched control antibody. The proliferation assay was performed 24 h after treatment. In the absence of the IL6R-blocking antibody, LPS and IL6 significantly increased cholangiocyte proliferation (*, p < 0.05) compared with control cholangiocytes. In contrast, when the cells were pretreated with IL6-blocking antibody the proliferation rates following LPS or IL6 treatment were significantly reduced (*, p < 0.05) compared with treated cells cultured in the presence of the control Ab.
DISCUSSION
It is well known that TLR activation in epithelial cells triggers an array of epithelial defense responses, including production and release of cytokines/chemokines and antimicrobial peptides and cell proliferation (32, 33). The results of our current study provide the first direct evidence that TLR-dependent cholangiocyte pathogen recognition also results in N-Ras and associated ERK activation mediating cholangiocyte IL6 expression/secretion and proliferation. Using a human cholangiocyte cell culture model we have shown that (i) N-Ras is the predominant Ras isoform expressed in cultured cholangiocytes and multiple TLR agonists rapidly induce N-Ras activation and associated ERK phosphorylation; (ii) TLR agonist-induced N-Ras activation correlates with NFκB reporter luciferase expression; (iii) TLR4 is required for LPS-induced N-Ras/ERK activation but the canonical TLR signal transducer, TRAF6, is not; (iv) LPS-induced N-Ras/ERK activation is required for a robust increase in IL6 expression; and (v) depletion of N-Ras and/or IL6 diminishes LPS-induced cholangiocyte proliferation. Thus, our results support a novel role for N-Ras in the cholangiocyte proinflammatory response to microbial insult.
In addition to sensing and modifying the composition of bile, cholangiocytes can interact with and respond to potentially injurious insults. Upon pathogen recognition, many immune response and inflammatory genes are up-regulated, including adhesive proteins, cytokines, and chemokines (4, 18, 19). The TLRs, one class of pathogen recognition receptors, recognize distinct pathogen-associated molecular patterns from bacteria, viruses, protozoa, and fungi. Signaling from TLRs requires interactions with TIR domain-containing adaptors and all TLRs can signal through the adaptor MyD88. The “canonical” TLR4 signaling pathway requires sequential MyD88 activation of IL1 receptor-associated kinases and ultimately TRAF6-dependent NFκB nuclear translocation. Additionally, TLR4 initiates signaling through the activation of interferon response factor-3. Given that agonists for several plasma membrane-associated TLRs induced N-Ras activation, including those that do not activate interferon response factor-3, this “noncanonical” TLR4 signal transduction cascade is likely not involved in N-Ras activation. Thus, the data presented here demonstrate that in addition to the well characterized TRL4 signaling cascades, N-Ras activation is an early event downstream of TLR4 activation. Furthermore, our data demonstrate that the rapid activation of N-Ras/ERK signaling is a common signaling pathway initiated by plasma membrane-associated TLRs, and LPS-induced N-Ras and ERK activation requires TLR4, but occurs independently of TRAF6.
The mammalian p21ras gene family consists of H-ras, K-ras, and N-ras, which encode four highly related small GTPases: K-RasA and K-RasB (formed by alternative splicing), H-Ras, and N-Ras which cycle between an active GTP- and inactive GDP-bound state. All Ras homologs are highly conserved, with the exception of the C-terminal hypervariable domain, and exhibit redundancy in signal transduction. Lipid modification of the C-terminal CAAX motif localizes the protein to membranes where they interact with upstream activators (i.e. GEFs) and downstream effectors (e.g. Raf). We demonstrate that N-Ras is the predominant Ras isoform expressed in cultured cholangiocytes and is the only isoform activated following pathogen recognition in our system. It seems likely, therefore, that the relative abundance of N-Ras and, perhaps, differences in subcellular localization of the Ras homologs confer selective activation of N-Ras. Indeed, N-Ras and H-Ras co-localize to sphingolipid-enriched membrane microdomains (34), whereas LPS treatment induces the aggregation of TLR4 to these kinase-rich lipid microdomains (35). Furthermore, GTPase-activating proteins and GEFs tightly control the on/off cycle of Ras proteins. Although our studies demonstrate a functional role for TLR4 in N-Ras activation, the precise mechanism, which likely involves the localized activation of GEFs, remains an area of active investigation.
Activated Ras molecules engage multiple effector molecules, including Raf. The activation of Raf promotes signaling through the MEK/ERK pathway and ultimately alters gene expression. IL6 is an acute phase cytokine that is both secreted by cholangiocytes and, through autocrine signaling, alters cholangiocyte physiology, including increased proliferation (24). Our current study demonstrates that LPS-induced cholangiocyte IL6 expression requires N-Ras and ERK activation. Furthermore, we observed immediate phosphorylation of ERK (<15 min) and a delayed activation of the IL6-responsive transcription factor STAT3, supporting autocrine signaling mediated by IL6. Interestingly, STAT3 has been shown to sustain the expression of IL6 in cholangiocarcinoma cells (36). Given that inhibition of N-Ras or the downstream effectors of N-Ras activation (MEK/ERK) diminish LPS-induced IL6 transcription, we propose that the immediate activation of N-Ras serves as a proximal signaling event required for IL6 expression, whereas persistent expression of IL6 is N-Ras-independent.
The proliferative capacity of cholangiocytes is a typical property of this epithelium and occurs in virtually all pathological conditions of liver injury where it is associated with inflammation, regeneration, and repair (37). Our data show that LPS treatment induces proliferation in cultured cholangiocytes in an N-Ras-dependent manner. However, this increase in proliferation observed in our model of infection is prevented in the presence of a blocking antibody to the IL6 receptor. We therefore propose that in our culture system, pathogen recognition-induced N-Ras activation is required for cholangiocyte IL6 secretion, and IL6 autocrine-mediated signaling events promote cholangiocyte proliferation. Although it is accepted that pathogen recognition promotes cholangiocyte proliferation, our data suggest that N-Ras activation plays a central role in this process and is a likely contributor to the phenotypic alterations observed upon pathogen recognition.
Alterations of gene expression result from the concerted effect of both transcriptional and post-transcriptional regulatory events. In previous work, we demonstrated that let-7i, a microRNA that regulates TLR4 expression in cholangiocytes (17), is transcriptionally repressed by the NFκB p50 subunit and CCAAT/enhancer-binding protein β (C/EBPβ) following LPS treatment of cholangiocytes or infection with the protozoan parasite, C. parvum (39). The C/EBP family of transcription factors, together with AP-1 and ATF/CREB, belong to the b-ZIP class of DNA-binding proteins (40). C/EBPβ is an acute phase transcription factor regulated by Ras/MAPK signaling and is a known regulator of IL6 (41). Hence, this Ras-responsive, acute phase transcription factor contributes to the diminished expression of a miRNA, let-7i, which others have demonstrated targets the Ras oncogenes (42). This sequence of events suggests that the p21ras gene family may be integrally involved in the cholangiocyte response to microbes through both the activation of Ras-responsive transcription factor-dependent gene expression and the regulation of Ras-induced alterations of microRNA expression.
In summary, using a cell culture model of cholangiocyte response to microbial insult, we have identified a novel signaling axis involving TLR-dependent N-Ras activation and the regulated expression of the proinflammatory cytokine, IL6. The precise mechanism of N-Ras activation remains obscure but may involve homolog abundance, subcellular compartmentalization of this Ras homolog, and/or interactions of N-Ras-specific GEFs. Furthermore, we speculate that in addition to promoting proinflammatory cytokine expression and proliferation, Ras activation likely influences other aspects of the cellular phenotype. Indeed, the cellular response to Ras activation is complex and may contribute to the plasticity of biliary epithelial cells. Investigations of the events induced as a result of pathogen recognition may provide insight into the reactive cholangiocyte phenotype, characterized by increased proliferation, altered expression of numerous proinflammatory cytokines, cell adhesion molecules, and autocrine/paracrine signaling mediators. Although heterogeneity of cholangiocytes exists within the bile duct, and the reactive cholangiocyte phenotype is attributed to those epithelial cells within the finest branches of the biliary tree, phenotypic alterations likely occur in both small and large cholangiocytes upon pathogen recognition. We propose that upon pathogen recognition, cholangiocytes exhibit a proinflammatory cellular phenotype, including proliferation, at least in part regulated by Ras activation and the regulated expression of microRNAs. Investigations of the pleiotropic effects of N-Ras activation in the cholangiocyte response to pathogen recognition are ongoing and will likely provide insight into the plasticity of biliary epithelial cells in health and disease.
Acknowledgment
We thank Deb Hintz for secretarial assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants AI089713 (to S. P. O.) and DK57993 (to N. F. L.). This work was also supported by an American Gastroenterological Association summer research fellowship (to P. N. L.) and the Genetics Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology Grant P30DK084567.
- TLR
- Toll-like receptor
- C/EBPβ
- CCAAT/enhancer-binding protein β
- GEF
- guanine nucleotide exchange factor
- HKLM
- heat-killed Listeria monocytogenes
- MyD88
- myeloid differentiation 88
- NFκB
- nuclear factor κB
- RBD
- Ras binding domain
- TRAF
- tumor necrosis factor receptor-associated factor.
REFERENCES
- 1. Jafri M., Donnelly B., Bondoc A., Allen S., Tiao G. (2009) J. Pediatr. Surg. 44, 500–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harada K., Shimoda S., Sato Y., Isse K., Ikeda H., Nakanuma Y. (2009) Clin. Exp. Immunol. 157, 261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karrar A., Broomé U., Södergren T., Jaksch M., Bergquist A., Björnstedt M., Sumitran-Holgersson S. (2007) Gastroenterology 132, 1504–1514 [DOI] [PubMed] [Google Scholar]
- 4. Chen X. M., O'Hara S. P., LaRusso N. F. (2008) Immunol. Cell Biol. 86, 497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harada K., Ozaki S., Kono N., Tsuneyama K., Katayanagi K., Hiramatsu K., Nakanuma Y. (2001) J. Pathol. 193, 218–223 [DOI] [PubMed] [Google Scholar]
- 6. Silva C. P., Pereira-Lima J. C., Oliveira A. G., Guerra J. B., Marques D. L., Sarmanho L., Cabral M. M., Queiroz D. M. (2003) J. Clin. Microbiol. 41, 5615–5618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kosaka T., Tajima Y., Kuroki T., Mishima T., Adachi T., Tsuneoka N., Fukuda K., Kanematsu T. (2010) Br. J. Surg. 97, 544–549 [DOI] [PubMed] [Google Scholar]
- 8. Hamada T., Yokota K., Ayada K., Hirai K., Kamada T., Haruma K., Chayama K., Oguma K. (2009) Helicobacter 14, 545–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takayama S., Takahashi H., Matsuo Y., Okada Y., Takeyama H. (2010) Dig. Dis. Sci. 55, 1905–1910 [DOI] [PubMed] [Google Scholar]
- 10. Rauschenfels S., Krassmann M., Al-Masri A. N., Verhagen W., Leonhardt J., Kuebler J. F., Petersen C. (2009) Eur. J. Pediatr. 168, 469–476 [DOI] [PubMed] [Google Scholar]
- 11. Domiati-Saad R., Dawson D. B., Margraf L. R., Finegold M. J., Weinberg A. G., Rogers B. B. (2000) Pediatr. Dev. Pathol. 3, 367–373 [DOI] [PubMed] [Google Scholar]
- 12. Fischler B., Svensson J. F., Nemeth A. (2009) Acta Paediatr. 98, 1600–1602 [DOI] [PubMed] [Google Scholar]
- 13. O'Hara S. P., Small A. J., Gajdos G. B., Badley A. D., Chen X. M., Larusso N. F. (2009) J. Infect. Dis. 199, 1195–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen X. M., LaRusso N. F. (2002) Semin. Liver Dis. 22, 277–289 [DOI] [PubMed] [Google Scholar]
- 15. Sripa B., Bethony J. M., Sithithaworn P., Kaewkes S., Mairiang E., Loukas A., Mulvenna J., Laha T., Hotez P. J., Brindley P. J. (July 23, 2010) Acta Trop. 10.1016/j.actatropica.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sripa B., Kaewkes S. (2000) Int. J. Parasitol. 30, 735–740 [DOI] [PubMed] [Google Scholar]
- 17. Chen X. M., Splinter P. L., O'Hara S. P., LaRusso N. F. (2007) J. Biol. Chem. 282, 28929–28938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harada K., Ohba K., Ozaki S., Isse K., Hirayama T., Wada A., Nakanuma Y. (2004) Hepatology 40, 925–932 [DOI] [PubMed] [Google Scholar]
- 19. Yokoyama T., Komori A., Nakamura M., Takii Y., Kamihira T., Shimoda S., Mori T., Fujiwara S., Koyabu M., Taniguchi K., Fujioka H., Migita K., Yatsuhashi H., Ishibashi H. (2006) Liver Int. 26, 467–476 [DOI] [PubMed] [Google Scholar]
- 20. Cruickshank S. M., Southgate J., Selby P. J., Trejdosiewicz L. K. (1998) J. Hepatol. 29, 550–558 [DOI] [PubMed] [Google Scholar]
- 21. Morita M., Watanabe Y., Akaike T. (1994) Hepatology 19, 426–431 [PubMed] [Google Scholar]
- 22. Scholz M., Cinatl J., Blaheta R. A., Kornhuber B., Markus B. H., Doerr H. W. (1997) Tissue Antigens 49, 640–643 [DOI] [PubMed] [Google Scholar]
- 23. Chen X. M., Levine S. A., Splinter P. L., Tietz P. S., Ganong A. L., Jobin C., Gores G. J., Paya C. V., LaRusso N. F. (2001) Gastroenterology 120, 1774–1783 [DOI] [PubMed] [Google Scholar]
- 24. Park J., Gores G. J., Patel T. (1999) Hepatology 29, 1037–1043 [DOI] [PubMed] [Google Scholar]
- 25. Wu M., McClellan S. A., Barrett R. P., Zhang Y., Hazlett L. D. (2009) J. Immunol. 183, 8054–8060 [DOI] [PubMed] [Google Scholar]
- 26. Romano Carratelli C., Mazzola N., Paolillo R., Sorrentino S., Rizzo A. (2009) FEMS Immunol. Med. Microbiol. 57, 116–124 [DOI] [PubMed] [Google Scholar]
- 27. Chen X. M., O'Hara S. P., Nelson J. B., Splinter P. L., Small A. J., Tietz P. S., Limper A. H., LaRusso N. F. (2005) J. Immunol. 175, 7447–7456 [DOI] [PubMed] [Google Scholar]
- 28. Hancock J. F. (2003) Nat. Rev. Mol. Cell Biol. 4, 373–384 [DOI] [PubMed] [Google Scholar]
- 29. Perez de Castro I., Bivona T. G., Philips M. R., Pellicer A. (2004) Mol. Cell. Biol. 24, 3485–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grubman S. A., Perrone R. D., Lee D. W., Murray S. L., Rogers L. C., Wolkoff L. I., Mulberg A. E., Cherington V., Jefferson D. M. (1994) Am. J. Physiol. 266, G1060–1070 [DOI] [PubMed] [Google Scholar]
- 31. Lee H. Y., Takeshita T., Shimada J., Akopyan A., Woo J. I., Pan H., Moon S. K., Andalibi A., Park R. K., Kang S. H., Kang S. S., Gellibolian R., Lim D. J. (2008) BMC. Infect. Dis. 8, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akira S., Takeda K. (2004) Nat. Rev. Immunol. 4, 499–511 [DOI] [PubMed] [Google Scholar]
- 33. Takeda K., Kaisho T., Akira S. (2003) Annu. Rev. Immunol. 21, 335–376 [DOI] [PubMed] [Google Scholar]
- 34. Prior I. A., Hancock J. F. (2001) J. Cell Sci. 114, 1603–1608 [DOI] [PubMed] [Google Scholar]
- 35. Triantafilou M., Miyake K., Golenbock D. T., Triantafilou K. (2002) J. Cell Sci. 115, 2603–2611 [DOI] [PubMed] [Google Scholar]
- 36. Isomoto H., Mott J. L., Kobayashi S., Werneburg N. W., Bronk S. F., Haan S., Gores G. J. (2007) Gastroenterology 132, 384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alvaro D., Mancino M. G., Glaser S., Gaudio E., Marzioni M., Francis H., Alpini G. (2007) Gastroenterology 132, 415–431 [DOI] [PubMed] [Google Scholar]
- 38. Deleted in proof.
- 39. O'Hara S. P., Splinter P. L., Gajdos G. B., Trussoni C. E., Fernandez-Zapico M. E., Chen X. M., LaRusso N. F. (2010) J. Biol. Chem. 285, 216–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vinson C. R., Sigler P. B., McKnight S. L. (1989) Science 246, 911–916 [DOI] [PubMed] [Google Scholar]
- 41. Akira S., Isshiki H., Sugita T., Tanabe O., Kinoshita S., Nishio Y., Nakajima T., Hirano T., Kishimoto T. (1990) EMBO J. 9, 1897–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnson S. M., Grosshans H., Shingara J., Byrom M., Jarvis R., Cheng A., Labourier E., Reinert K. L., Brown D., Slack F. J. (2005) Cell 120, 635–647 [DOI] [PubMed] [Google Scholar]