

Abstract
Helix V in LacY, which abuts and crosses helix I in the N-terminal helix bundle of LacY, contains Arg144 and Trp151, two residues that play direct roles in sugar recognition and binding, as well as Cys154, which is important for conformational flexibility. In this study, paired Cys replacement mutants in helices V and I were strategically constructed with tandem factor Xa protease cleavage sites in the loop between the two helices to test cross-linking. None of the mutants form disulfides spontaneously; however, three mutants (Pro28 → Cys/Cys154, Pro28 → Cys/Val158 → Cys, and Phe29 → Cys/Val158 → Cys) exhibit cross-linking after treatment with copper/1,10-phenanthroline (Cu/Ph) or 1,1-methanediyl bismethanethiosulfonate ((MTS)2-1), 3–4 Å), and cross-linking is quantitative in the presence of ligand. Remarkably, with one mutant, complete cross-linking with (MTS)2-1 has no effect on lactose transport, whereas quantitative disulfide cross-linking catalyzed by Cu/Ph markedly inhibits transport activity. The findings are consistant with a number of previous conclusions suggesting that sugar binding to LacY causes a localized scissors-like movement between helices V and I near the point where the two helices cross in the middle of the membrane. This ligand-induced movement may act to initiate the global conformational change resulting from sugar binding.
Keywords: Lactose, Membrane, Membrane Proteins, Membrane Transport, Transport, Transporters, Electrochemical Proton Gradient, Membrane Proteins, Symport, Thiol Cross-linking
Introduction
Typical of many transport proteins from organisms as disparate as Archaea and Homo sapiens, the lactose permease of Escherichia coli (LacY), a member of the major facilitator superfamily (1), catalyzes the coupled, stoichiometric translocation of an H+ and a galactopyranoside (galactoside/H+ symport). Because transport is obligatorily coupled, active transport of sugar against a concentration gradient is achieved by transduction of free energy released from the downhill movement of H+ with the electrochemical H+ gradient (Δμ̃H+; interior negative and/or alkaline). Conversely, downhill sugar translocation by LacY drives uphill H+ translocation with the generation of Δμ̃H+, the polarity of which depends on the direction of the sugar concentration gradient (reviewed in Refs. 2–4).
X-ray structures of wild type LacY (5) and a conformationally restricted mutant (C154G) (6, 7) reveal 12 mostly irregular transmembrane helices arranged in 2 pseudo-symmetrical 6-helix bundles surrounding a large central water-filled cavity open to the cytoplasm only (an inward-facing conformation). The inward-open conformation is the predominant conformation of wild-type LacY in the native membrane and in solution (8, 9). Both the N and C termini are on the cytoplasmic side of the membrane, and several lines of evidence indicate that LacY is structurally (6, 10–13), as well as functionally (see Ref. 14), a monomer. Although an outward-facing conformation of LacY is yet to be observed crystallographically, FucP a distantly related protein, has been crystallized in an outward-facing conformation (15) that agrees well with the model proposed for the outward-facing structure of LacY (8, 16). Moreover, multiple independent biochemical and biophysical studies with LacY, which include thiol cross-linking (17–24), site-directed alkylation (reviewed in Refs. 9 and 24–30), single molecule fluorescence resonance energy transfer (31), double electron-electron resonance (8), and Trp fluorescence quenching (32), not only demonstrate that an outward-facing conformation exists, but that a large periplasmic cavity must open and close for transport to occur (24). By this means, the cytoplasmic cavity closes with opening of a wide hydrophilic cavity on the periplasmic side of LacY, which allows exposure of sugar- and H+-binding sites to either side of the membrane (the alternating access mechanism) (reviewed in Ref. 33).
LacY appears to be highly dynamic. As shown by attenuated total reflectance Fourier transform infrared spectroscopy (34), the average helix tilt in LacY is ∼51° at a low lipid:protein ratio and decreases to ∼33° at high lipid:protein ratios. In addition, although ∼70% of the side chains in LacY are hydrophobic and buried (3), ∼85% of the backbone amide protons exchange with deuterium in 10 min at room temperature (35, 36) with 100% exchange at elevated, but non-denaturing temperatures (37).
Early site-directed mutagenesis experiments on the native Cys residues in LacY led to the isolation and characterization of a mutant with Gly in place of native Cys154 (Helix V) (38, 39). Remarkably, this single mutation completely changes the functional properties and physical characteristics of LacY (40). C154G LacY binds ligand as well or better than wild-type LacY, but catalyzes very little translocation across the membrane (7). Moreover, whereas sugar binding is mostly entropic with wild-type LacY, it is enthalpic with the C154G mutant (41). Indeed, the mutant does not exhibit the long-range conformational changes normally observed upon ligand binding, and it is also thermostable with respect to ligand binding and aggregation (40) and is arrested in a partially open-outward conformation(s) (27). However, the x-ray structure of the mutant exhibits an open-inward conformation (6, 7), whereas it is clearly open on the periplasmic side in the membrane (27).
X-ray crystal structures yield a clue as to why the C154G mutant is conformationally restricted. As shown (Fig. 1), helices V and I cross in the approximate middle of the membrane where Cys154 (helix V) and Gly24 (helix I) are in close proximity (6). Two Gly residues close to each other in adjacent helices can lead to significantly tighter helix packing (42–45), which may partly explain the lack of conformational flexibility of the C154G mutant. Therefore, Gly24 (helix I) was replaced with Cys in the C154G mutant. The G24C/C154G double mutant exhibits a marked increase in transport activity with sugar binding and thermostability similar to wild-type LacY. Replacement of Gly24 with Val or Asp in mutant C154G yields a low degree of rescue, but surprisingly, replacement with Ala, Ile, Met, Thr, Ser, or Glu has no significant effect. Interestingly, replacement of Cys154 with Ser or Thr yields active mutants, whereas the same replacements for Gly24 in the C154G mutant yield little or no activity. Moreover, a mutant with Cys at both positions 24 and 154 transports like the wild type. Taken together, the results indicate that it is not merely the bulk of the side chains at positions 24 and 154 that accounts for the properties of the C154G mutant. The finding also supports the conclusion that mutations disrupting interactions between helix IV and loop 6–7 or between helices II and IV in the intracellular face of LacY also rescue mutant C154G activity (46). All of these positions are located at a distance from positions 14 or 154. Thus, highly precise, dynamic interactions between helices V and I are likely required for proper structural rearrangements during turnover. Further evidence for this proposal is provided by the thiol cross-linking studies between helices V and I presented here.
FIGURE 1.

Side view of the overall structure of lactose permease (PDB code 2V8N). A, LacY is shown as a ribbon model; helix I is colored blue and helix V is colored orange, cytoplasmic and periplasmic sides are indicated with IN and OUT, respectively. The proteolytic cleavage sites (fXa) are colored in magenta. B, enlarged section from A (green rectangle) in a view turned by 90°. Cys positions for cross-linking are denoted by sticks, based on the structure of wild-type LacY (PDB code 2V8N), and cross-linked pairs are indicated by broken lines: red, cross-linking with native Cys154; blue, cross-linking with the Cys replacement for Val158.
EXPERIMENTAL PROCEDURES
Materials
Restriction endonucleases, T4 DNA ligase, and factor Xa (fXa) protease were purchased from New England Biolabs (Beverly, MA). QuikChange II kits were from Stratagene (La Jolla, CA). DNA plasmid purification and DNA fragment gel extraction kits were purchased from Qiagen (Valencia, CA). 1,1-Methanediyl bismethanethiosulfonate ((MTS)2-1))4 was obtained from Toronto Research Chemicals, Inc. (Toronto, Canada). Site-directed rabbit polyclonal antiserum against a dodecapeptide corresponding to the C terminus of LacY was prepared as described (47). Micro-BCA protein determination and SuperSignal West Pico Chemiluminescent substrate kits were from Pierce (Rockford, IL). All other materials were of reagent grade and obtained from commercial sources.
Construction of Mutants
Plasmid pT7-5/Cys-less/fXa IV-V encoding Cys-less LacY with two tandem fXa sites (Ile-Glu-Gly-Arg)2 between Ser136 and Asn137 in cytoplasmic loop IV-V (Fig. 1A) was constructed as described previously (48, 49). Double Cys mutants were constructed by QuikChange and cutting and pasting the BamHI/SptI fragment containing helix I or the BssHII/KpnI fragment containing helix V from plasmid pT7-5 containing given single Cys mutants (50) into pT7-5/Cys-less/fXa IV-V. DNA sequencing of the entire lacY gene and the restriction digestion profile confirmed all constructs.
Protein Expression
E. coli T184 (lacI+O+Z−Y− (A) rpsL met− thr− recA hsdM hsdR/F′ lacIq O+ZD118 (Y+ A+) was transformed with a plasmid encoding a given double Cys mutation by electroporation using 0.2-cm cuvettes, and cells were grown at 37 °C in Luria-Bertani broth with 100 mg/liter of ampicillin. Overnight cultures were diluted 10-fold and allowed to grow for 2 h at 37 °C before induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside. After additional growth for 2–3 h at 37 °C, cells were harvested by centrifugation.
Preparation of Right Side-out (RSO) Membrane Vesicles
RSO membrane vesicles were prepared in 0.4 to 1-liter cultures of E. coli T184 expressing a given mutant by lysozyme/EDTA and osmotic lysis (51, 52). The vesicles were washed and resuspended to a protein concentration of 10–20 mg/ml in 100 mm potassium phosphate (KPi; pH 7.5) and 10 mm MgSO4, frozen in liquid N2, and stored at 80 °C until use.
Thiol Cross-linking
Cross-linking reactions were carried out with 1 ml of suspension of RSO vesicles (1 mg/ml of protein) containing a given paired Cys mutant on ice or at room temperature for 5 min, as indicated. Oxidative cross-linking was catalyzed by adding 0.5 mm (Cu/Ph) freshly prepared by mixing 0.4 ml of a stock solution of 1.25 m 1,10-phenanthroline in 50% ethanol and 0.6 ml of 250 mm CuSO4 (29), and the reaction was terminated by addition of 50 mm EDTA. Alternatively, (MTS)2-1 was used, and the final concentration of the reagent was 0.05 mm. Reactions were quenched by addition of 5 mm N-ethylmaleimide. Following 10-fold dilution with cold 100 mm KPi (pH 7.5) and immediate centrifugation (14,000 × g for 10 min) at 4 °C, the samples were washed 3 times with cold 100 mm KPi (pH 7.5) and resuspended in 100 mm KPi (pH 7.5), 10 mm MgSO4 at 3–5 mg of protein/ml. A portion of the preparation was used to assay lactose transport, and the remainder for analysis of cross-linking and protein determination.
Transport Assays
RSO membrane vesicles containing LacY mutants were adjusted to an A600 of 3.0 (3 mg of protein/ml). Transport was assayed by incubation of samples with 20 mm ascorbate, 0.2 mm phenazine methosulfate, and 0.4 mm [1-14C]lactose (10 mCi/mmol) under oxygen, followed by dilution and rapid filtration at the times given, as described (53, 54).
Analysis of Cross-linking
Cross-linking was analyzed as follows: aliquots (10 μg of protein) were digested overnight with fXa protease as described (48), and samples (2 μg of protein) were subjected to SDS-12% polyacrylamide gel electrophoresis and Western blotting with site-directed polyclonal antibody against the C terminus of LacY followed by an horseradish peroxidase-coupled anti-rabbit antibody (Amersham Biosciences, NA-934) as described (47).
Protein Assays
Protein was assayed with the Micro-BCA protein determination kit (Pierce).
RESULTS
Mutant Construction and Functional Analyses
To investigate dynamic interactions between the periplasmic halves of helices I and V, paired Cys mutants (F21C/C154, G24C/C154, A25C/C154, P28C/C154, F29C/C154, A25C/V158C, F27C/V158C, P28C/V158C, F29C/V158C, and P31C/F162C) at the interface between these helices (Fig. 1) were constructed as described under “Experimental Procedures.” As shown, in addition to a given pair of Cys replacements, each construct contains two tandem fXa protease cleavage sites in the loop between helices IV and V (loop IV/V).
Lactose accumulation by RSO vesicles containing given mutants was assayed at 5 min and normalized to Cys-less LacY (Fig. 2). Most double Cys mutants exhibit significant activity, except for mutants F21C/C154 and P28C/C154, which accumulate only about 40 and 20%, respectively, of the lactose accumulated by the Cys-less LacY. Mutant P31C/F162C accumulates less than 5% of the lactose transported by Cys-less LacY in 5 min and therefore was excluded from this study (data not shown).
FIGURE 2.
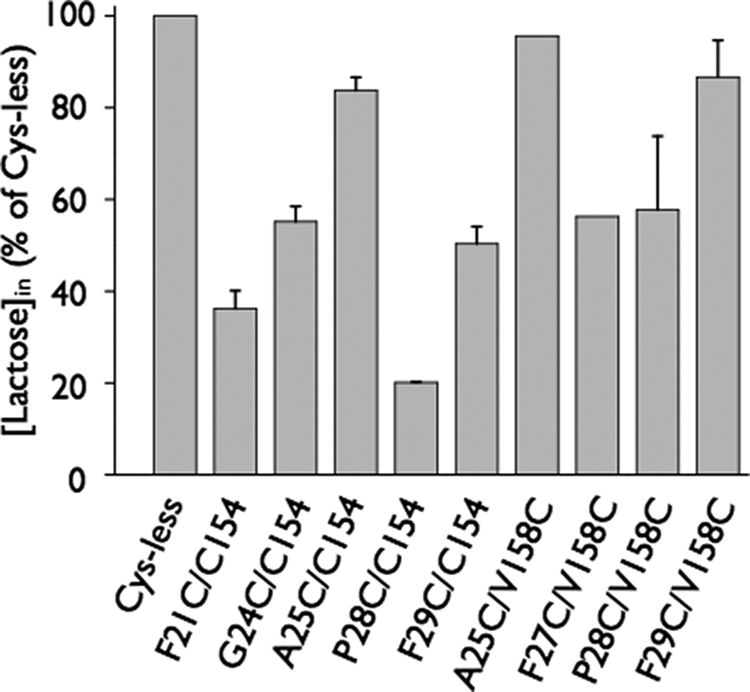
Transport activity of double Cys mutants in RSO membrane vesicles. The double Cys mutants in the “Cys-less” background were expressed in E. coli T184 as described under “Experimental Procedures.” Active transport was assayed in the presence of 20 mm ascorbate and 0.2 mm phenazine methosulfate under oxygen with [1-14C]lactose (2.5 mCi/mmol) at a final concentration of 0.4 mm at room temperature for 5 min, measurements were carried out in triplicate. Results are expressed as a percentage of activity in the Cys-less mutant.
Cross-linking
To investigate interactions between helices V and helix I, cross-linking of native Cys154 (helix V) or a mutant containing Cys in place of Val158 (helix V) with Cys replacements in helix I (Fig. 1) was tested in RSO membrane vesicles after incubation with either Cu/Ph as an oxidant or the homobifunctional cross-linking reagent (MTS)2-1 (3–4 Å). Cross-linking was also performed in the absence or presence of the lactose homologue β-d-galactopyranosyl 1-thio-β-d-galactopyranoside (TDG), and the effect on transport activity was tested as well.
G24C/C154
Although Cys154 is in close proximity to Gly24 (Fig. 3C), mutant G24C/C154 untreated with cross-linking agent but digested with fXa protease exhibits a single band migrating at an molecular mass of ∼25 kDa after Western blotting with anti-C-terminal antibody (Fig. 3A, lane 1). This band corresponds to a fragment containing the C-terminal eight helices and the central loop (C8). Because digestion with the fXa protease is complete and no anti-C-reactive band was present at ∼35 kDa (the position of full-length LacY), it is apparent that this pair does not form a disulfide bond spontaneously. After incubation with Cu/Ph alone, no effect was observed (lane 2), but when TDG was also added, a small amount of protein migrating at about 35 kDa was observed (lane 3). With (MTS)2-1 alone, a small amount of the 35-kDa band was observed (lane 4), and a slight increase is seen when TDG was also present (lane 5). Finally, lactose transport is essentially unchanged after the treatments described (Fig. 3B).
FIGURE 3.

Correlation between cross-linking and transport activity of G24C/C154 in RSO membrane vesicles. A, Western blots. RSO vesicles containing the G24C/C154 mutant with tandem fXa sites at 1 mg/ml were washed 3 times with 0.1 m KPi (pH 7.5) after treatment with 0.5 mm Cu/Ph or 0.05 mm (MTS)2-1 at room temperature for 5 min; and aliquots (2 μg of protein) were loaded after cleavage with fXa protease, and detected with anti-C-terminal antibody as described under “Experimental Procedures.” B, active transport. Transport assays with RSO vesicles containing the G24C/C154 mutant were performed in the presence of 20 mm ascorbate and 0.2 mm phenazine methosulfate under oxygen with [1-14C]lactose (2.5 mCi/mmol) at a final concentration of 0.4 mm for the times given. ■, control; ●, treated with 0.5 mm Cu/Ph; ▴, treated with 10 mm TDG and 0.5 mm Cu/Ph; ▾, treated with 0.05 mm (MTS)2-1; ♦, treated with 10 mm TDG and 0.05 mm (MTS)2-1; ◀, vector pT7-5. C, native Cys154 and a Cys replacement for Gly24 are denoted by sticks based on the structure of LacY (PDB code 2V8N).
P28C/C154
Mutant P28C/C154 (Fig. 4A) also exhibits no spontaneous cross-linking, as the only anti-C-reactive band observed migrates at 25 kDa (Fig. 4A, lane 1). However, about one-third to one-half of the protein is cross-linked in the presence of Cu/Ph without or with TDG, as evidenced by the presence of a relatively large amount of the 35-kDa band after incubation with oxidant in the absence or presence of ligand (lanes 2 and 3, respectively). A slightly greater amount of cross-linking was observed with (MTS)2-1 alone (lane 4), and remarkably, when TDG was added in addition, cross-linking goes to completion (lane 5). Because transport activity by this mutant is relatively low (Fig. 2), it appears that activity is almost abolished after cross-linking under each of the conditions tested (Fig. 4B).
FIGURE 4.

Correlation between cross-linking and transport activity of P28C/C154 in RSO membrane vesicles. A, Western blots of RSO vesicles containing the P28C/C154 mutant with tandem fXa sites. B, active transport. Cross-linking, fXa digestion, and [1-14C]lactose transport assays were performed as described in the legend to Fig. 3. ■, control; ●, treated with 0.5 mm Cu/Ph; ▴, treated with 10 mm TDG and 0.5 mm Cu/Ph; ▾, treated with 0.05 mm (MTS)2-1; ♦, treated with 10 mm TDG and 0.05 mm (MTS)2-1; ◀, vector pT7-5. C, native Cys154 and a Cys replacement for Pro28 are denoted by sticks based on the structure of LacY (PDB code 2V8N).
To explore the effect of temperature on movement between helices V and I, rates of Cu/Ph-induced cross-linking between P28C and Cys154 were studied at 0 or 23 °C (Fig. 5, A and B). At 0 °C, cross-linking by Cu/Ph reaches ∼23% after 5 min and stays relatively constant thereafter, whereas at room temperature more than 75% was cross-linked after 5 min, and cross-linking is virtually complete after 10 min (Fig. 5C).
FIGURE 5.

Time courses of Cu/Ph-induced cross-linking of mutant P28C/C154 at different temperatures. A and B, Western blots. RSO vesicles containing P28C/C154 LacY with tandem fXa sites were cross-linked on ice (A) or at room temperature (B) at the times given, and subsequently cleaved with fXa protease. LacY was detected with anti-C-terminal antibody as described under “Experimental Procedures.” C, time courses of cross-linking. Cross-linking is expressed as the density a band at a given time point relative to that of the undigested band. Solid line with a filled circle, room temperature; broken line with filled square, 0 °C.
A25C/V158C
Mutant A25C/V158C (Fig. 6A) also does not undergo disulfide formation spontaneously (Fig. 6A, lane 1) nor is disulfide formation observed in the presence of Cu/Ph (lane 2), and addition of TDG has no effect (lane 3). Furthermore, no cross-linking was observed with (MTS)2-1 (lane 4); however, when TDG is added in the presence of (MTS)2-1, more than half of the anti-C-reactive protein migrates at 35 kDa, signifying relatively effective cross-linking (lane 5). However, no effect of cross-linking is observed on the transport activity of this paired mutant (Fig. 6B).
FIGURE 6.

Correlation between cross-linking and transport activity of A25C/V158C in RSO membrane vesicles. Cross-linking experiments and [1-14C]lactose transport assays were performed as described in the legend to Fig. 3. A, Western blots. B, active lactose transport. ■, control; ●, treated with 0.5 mm Cu/Ph; ▴, treated with 10 mm TDG and 0.5 mm Cu/Ph; ▾, treated with 0.05 mm (MTS)2-1; ♦, treated with 10 mm TDG and 0.05 mm (MTS)2-1; ◀, vector pT7-5. C, mutation V158C and a Cys replacement for Ala25 are denoted by sticks based on the structure of LacY (PDB code 2V8N).
P28C/V158C
With mutant P28C/V158C (Fig. 7A), spontaneous disulfide formation is not observed (Fig. 7A, lane 1). But strikingly, complete cross-linking is observed after treatment with either Cu/Ph or (MTS)2-1 (lanes 2 and 3). Furthermore, treatment with either reagent in the presence of TDG has no effect whatsoever (data not shown). Unremarkably, quantitative cross-linking with both reagents largely inhibits the already low level of lactose transport in the mutant (Fig. 6B).
FIGURE 7.

Correlation between cross-linking and transport activity of P28C/V158C in RSO. Cross-linking experiments and [1-14C]lactose transport assays were performed as described in the legend to Fig. 3. A, Western blots. B, active transport. ■, control; ●, in presence of 0.5 mm Cu/Ph; ▴, in presence of 0.05 mm (MTS)2-1; ▾, in presence of 0.5 mm o-phenylenedimaleimide (o-PDM); ♦, vector pT7-5. C, V158C mutation and a Cys replacement for Pro28 are denoted by sticks based on the structure of LacY (PDB code 2V8N).
F29C/V158C
Oxidation with Cu/Ph for the times indicated yield little disulfide formation with this double Cys mutant (Fig. 8A, lane 2), but oxidation in the presence of TDG causes almost quantitative cross-linking (Fig. 8A, lane 3). Correspondingly, Cu/Ph treatment alone has little effect on lactose transport activity, but Cu/Ph treatment in the presence of TDG causes marked inhibition (Fig. 8B). Similarly, (MTS)2-1 treatment alone results in significantly less cross-linking than treatment with the reagent in the presence of TDG, which is quantitative (Fig. 8A, lanes 4 and 5). However, it is remarkable that (MTS)2-1 treatment without or with TDG has no significant effect on transport activity (Fig. 8C). Thus, it is apparent that the activity is critically dependent upon the distance and/or flexibility between helices V and I at this level.
FIGURE 8.

Correlation between cross-linking and transport activity of F29C/V158C in RSO. Cross-linking experiments and [1-14C]lactose transport assays were performed as described in the legend to Fig. 3. A, Western blots. B, active transport. ■, control; ●, in the presence of 0.5 mm Cu/Ph; ▴, in presence of 0.5 mm Cu/Ph and 10 mm TDG; ▾, vector pT7-5. C, active transport of F29C/V158C after cross-linking with (MTS)2-1 as assayed identically as in B: ■, control; ●, in the presence of 0.05 mm (MTS)2-1; ▴, in the presence of 0.05 mm (MTS)2-1 and 10 mm TDG; ▾, vector pT7-5. D, mutation V158C and a Cys replacement for Phe29 are denoted by sticks based on the structure of LacY (PDB code 2V8N).
DISCUSSION
Site-directed thiol cross-linking in situ with mutants containing an engineered protease site is a powerful approach for estimating helix packing and probing ligand-induced conformational changes, as cross-linking reactions can be performed before and after interaction with substrate. Availability of a plethora of cross-linking reagents with different arm lengths can also provide valuable information about the distances between interacting residues (see Ref. 24). Additionally, functional analyses can be performed after intra-molecular cross-linking.
Here we describe the use of two reagents, the oxidizing agent Cu/Ph and the homo-bifunctional cross-linking agent (MTS)2-1 (3–4 Å) to detect dynamic movements in the periplasmic domains of helices V and I of LacY. In the x-ray crystal structures of LacY (5–7), helices V and I in the N-terminal helix bundle are in close contact with helices VII and VIII, respectively, in the C-terminal bundle and play an important role in sealing the inward-facing hydrophilic cavity from the outside (8, 24, 32). There are four Gly residues in helix V, as well as two Gly and two Pro residues in helix I, and both amino-acyl residues are often associated with conformational changes (55–58). In addition, helix V contains Arg144 and Trp151, which are directly involved in sugar binding and Cys148, which either interacts directly with the galactopyranoside moiety of LacY ligands or is very close to the binding site (see Ref. 3). Finally, it has been shown that the C154G mutation, which is on the same face of helix V, results in a more compact structure with decreased conformational flexibility, an alteration that specifically blocks the structural changes necessary for substrate translocation with little or no effect on ligand binding (40). Indeed, isothermal titration calorimetry shows that the C154G mutant binds ligand with slightly better affinity than wild-type LacY, but is restricted conformationally (41).
The x-ray structures of C154G LacY demonstrate that helix V crosses helix I about halfway through the thickness of the membrane in such a manner that Cys154 lies close to Gly24 (helix I) (5–7). Moreover, replacement of Gly24 with Cys with the C154G mutant results in rescue of ∼80% of wild-type transport activity (46). But cross-linking induced by Cu/Ph suggests that interaction between a Cys replacement at position 28 and native Cys154 (mutant P28C/C154) is minimal at 0 °C in the presence of TDG, but changing to room temperature increases cross-linking to ∼75% after 5 min and essentially 100% in 10 min (Fig. 5). Pro28, an absolutely conserved residue in the major facilitator superfamily, seems essential for activity because replacement with Ala, Gly, or Leu almost completely abolishes lactose transport (55). However, because mutant P28S retains the ability to accumulate TDG to ∼50% of the wild-type level, it is unlikely that a Pro residue per se at this position is irreplaceable with respect to transport (59). Mutant P28C/V158C retains about 60% of Cys-less transport activity and is inactivated by quantitative cross-linking with either Cu/Ph or (MTS)2-1 (Fig. 7). The data are consistent with the interpretation that Val158 (helix V) is proximal to Pro28 (helix I).
Although mutant F29C/V158C cross-links to completion with either Cu/Ph or (MTS)2-1 in the presence of TDG (Fig. 8A), significant inactivation of transport activity is observed only after disulfide formation between these Cys residues (Fig. 8B). This finding indicates that ligand binding induces movement between helices V and I so that positions 29 and 158 come into closer proximity. Furthermore, the dramatic difference between the effect of Cu/Ph-catalyzed disulfide formation (-S-S-) versus cross-linking with (MTS)2-1, which introduces a distance of 3–4 Å between the two S atoms of the cross-linked Cys residues (–S-S-CH2-S-S-), on transport activity suggests that a critical distance and/or flexibility is required. This interpretation is supported by the finding that mutant A25C/V158C is cross-linked significantly by (MTS)2-1 in the presence of TDG with no significant effect on activity (Fig. 6A). However, with this mutant, no cross-linking was observed in the presence of Cu/Ph.
Site-directed cross-linking depends on the distance of the cross-linking sites, their geometry with respect to each other, and their accessibility to the cross-linking agents. As mentioned, it has been shown by multiple independent biochemical and biophysical methods that the opening probability of the periplasmic cavity depends on the presence of substrate sugar, if sugar is present, LacY preferably adopts an outward-facing conformation. Mapping the cross-linking sites on the solvent-accessible surface of LacY calculated from the inward-open x-ray structure (CAST-p web server, probe size 1.4 Å) in the inward-facing conformation reveals that only P28C/V158C is accessible to the solvent (Fig. 9A). However, in the outward-facing conformation, mutants A25C, P28C, F29C, and V158C are accessible to solvent (Fig. 9B) as calculated from the outward-open model of LacY (16). A similar TDG-dependent accessibility pattern for single Cys mutants A25C, P28C, and F29C were also observed with site-directed alkyation (60). The cross-linking data for mutants A25C/V158C, P28C/V158C, and F29C/V158C correlate very well with sugar-dependent opening of the periplasmic cavity. In the absence of TDG, only mutants P28C and V158C are cross-linked because these positions are the only sites in contact with the aqueous phase. Upon ligand binding, Cys replacements at positions 25, 28, and 29 become exposed to the solvent and can be cross-linked with a Cys at position 158.
FIGURE 9.

Comparison of solvent accessibility between inward-facing and outward-facing conformations of LacY. Residues A25C (green), P28C (red), F29C (yellow), and V158C (magenta) are highlighted in space-filling models of LacY. The N-terminal 6-helix bundle is shown, viewed as indicated from the C-terminal bundle with the exposed plane shown as gray surface. A, inward-facing conformation, constructed from the wild-type LacY structure (PDB code 2V8N). Only residues V158C and P28C are accessible to the aqueous phase. B, model for outward-facing conformation (14). In this conformation, all of the colored residues (A25C, P28C, F29C, and V158C) are accessible to the aqueous phase.
The level of cross-linking by Cu/Ph correlates reasonably well with transport activity. When residues are cross-linked by Cu/Ph, transport is inhibited, but this correlation is not observed with (MTS)2-1, except with mutants P28C/C154 and P28C/V158C. One disulfide bond (-S-S-) between double Cys pairs is formed when an oxidizing agent is present, as long as they are in close proximity. However, when (MTS)2-1 is present, two disulfide bonds are formed (–S-S-CH2-S-S-), and movement is consequently less restricted due to the parallel arrangement of the chemical bonds. Therefore, the functional effects of cross-linking by different reagents at the periplasmic ends of helices V and I argues against rigid-body movement of the two 6-helix bundles during turnover of LacY. It is likely that precise rearrangement between helices V and I within the N-terminal six-helix bundle is critical for completing the transport cycle.
In the case of P28C/C154, the level of cross-linking by Cu/Ph is time- and temperature-dependent (Fig. 5). Comparison of cross-linking at 23 and 0 °C provides a qualitative indication of the contribution of backbone dynamics to the reactivity and accessibility of a given Cys residue. The α-C atom distance between Pro28 and Cys154 is ∼11 Å, whereas it is ∼9 Å between Pro28 and Val158. The shorter distance between Pro28 and Val158 results in increased rates of cross-linking induced by Cu/Ph with the P28C/V158C mutant relative to the P28C/C154 mutant (compare Fig. 4C with Fig. 7C). It is not surprising that higher temperatures increase protein backbone movement and the rate of cross-linking. However, it should be emphasized that cross-linking reflects dynamic collisions and chemical reactions between residues, not simply their proximities (61). For example, Cys pairs that frequently undergo collisions and are highly reactive chemically could form cross-links at relatively rapid rates even though they may be distant in the average structure. However, a strong correlation is expected between collision rates and proximity (61).
Cross-linking between paired Cys residues in helices V and I clearly reveals a ligand-induced conformational change at the interface between helices V and I, and a scissors-like movement (46) probably best explains the available data. Based on the structural observations, an ordered mechanism for the initial step in galactopyranoside/H+ symport has been postulated (7). In the ligand-free conformation, the irreplaceable residues for substrate binding ((Arg144 (helix V), Glu126 (helix IV), and Glu269 (helix VIII)) are not in the correct configuration to bind substrate. Sugar first recognizes Trp151 (helix V) through nonspecific hydrophobic stacking between the galactopyranosyl and indole rings (62, 63). This interaction orients the galactopyranosyl moiety for recognition by Arg144, Glu126, and Glu269. Moreover, interaction of sugar with Trp151 is thought to disrupt the salt bridge between Arg144 and Glu126 when strong H-bonding occurs between Arg144 and OH groups on the galactopyranosyl ring (64). The preponderance of the available findings (reviewed in Ref. 3) indicates that sugar binding and/or dissociation is the primary driving force for the global conformational change that is central to the alternating access model (reviewed in Ref. 33). Therefore, it seems reasonable to suggest that an initiating event involves sugar-induced conformational change(s) in helix V that is transmitted to helix I and then propagated through the remaining helices in LacY in a cooperative manner.
Acknowledgments
We are particularly indebted to Salete Newton for editorial help with the manuscript and Irina Smirnova and Vladimir Kasho for advice and discussions.
This work was supported by National Institutes of Health Grants DK51131, DK069463, GM073210, and GM074929 and National Science Foundation Grant 0450970 (to H. R. K.).
- (MTS)2-1
- 1,1-methanediyl bismethanethiosulfonate
- RSO
- right-side-out
- TDG
- β-d-galactopyranosyl 1-thio-β-d-galactopyranoside
- Cu/Ph
- copper/1,10-phenanthroline.
REFERENCES
- 1. Saier M. H., Jr. (2000) Mol. Microbiol. 35, 699–710 [DOI] [PubMed] [Google Scholar]
- 2. Kaback H. R., Sahin-Tóth M., Weinglass A. B. (2001) Nat. Rev. Mol. Cell Biol. 2, 610–620 [DOI] [PubMed] [Google Scholar]
- 3. Guan L., Kaback H. R. (2006) Annu. Rev. Biophys. Biomol. Struct. 35, 67–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garcia-Celma J. J., Smirnova I. N., Kaback H. R., Fendler K. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 7373–7378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guan L., Mirza O., Verner G., Iwata S., Kaback H. R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 15294–15298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abramson J., Smirnova I., Kasho V., Verner G., Kaback H. R., Iwata S. (2003) Science 301, 610–615 [DOI] [PubMed] [Google Scholar]
- 7. Mirza O., Guan L., Verner G., Iwata S., Kaback H. R. (2006) EMBO J. 25, 1177–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smirnova I., Kasho V., Choe J. Y., Altenbach C., Hubbell W. L., Kaback H. R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 16504–16509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nie Y., Kaback H. R. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 9903–9908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Costello M. J., Escaig J., Matsushita K., Viitanen P. V., Menick D. R., Kaback H. R. (1987) J. Biol. Chem. 262, 17072–17082 [PubMed] [Google Scholar]
- 11. Sun J., Kaback H. R. (1997) Biochemistry 36, 11959–11965 [DOI] [PubMed] [Google Scholar]
- 12. Guan L., Murphy F. D., Kaback H. R. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 3475–3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ermolova N., Guan L., Kaback H. R. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 10187–10192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sahin-Tóth M., Lawrence M. C., Kaback H. R. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 5421–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dang S., Sun L., Huang Y., Lu F., Liu Y., Gong H., Wang J., Yan N. (2010) Nature 467, 734–738 [DOI] [PubMed] [Google Scholar]
- 16. Radestock S., Forrest L. R. (2011) J. Mol. Biol. 407, 698–715 [DOI] [PubMed] [Google Scholar]
- 17. Wu J., Kaback H. R. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 14498–14502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu J., Kaback H. R. (1997) J. Mol. Biol. 270, 285–293 [DOI] [PubMed] [Google Scholar]
- 19. Wu J., Hardy D., Kaback H. R. (1998) Biochemistry 37, 15785–15790 [DOI] [PubMed] [Google Scholar]
- 20. Wu J., Hardy D., Kaback H. R. (1998) J. Mol. Biol. 282, 959–967 [DOI] [PubMed] [Google Scholar]
- 21. Wu J., Hardy D., Kaback H. R. (1999) Biochemistry 38, 1715–1720 [DOI] [PubMed] [Google Scholar]
- 22. Wu J., Hardy D., Kaback H. R. (1999) Biochemistry 38, 2320–2325 [DOI] [PubMed] [Google Scholar]
- 23. Sorgen P. L., Hu Y., Guan L., Kaback H. R., Girvin M. E. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 14037–14040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Y., Guan L., Freites J. A., Kaback H. R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 3774–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaback H. R., Dunten R., Frillingos S., Venkatesan P., Kwaw I., Zhang W., Ermolova N. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 491–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nie Y., Ermolova N., Kaback H. R. (2007) J. Mol. Biol. 374, 356–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nie Y., Sabetfard F. E., Kaback H. R. (2008) J. Mol. Biol. 379, 695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nie Y., Zhou Y., Kaback H. R. (2009) Biochemistry 48, 738–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Y., Nie Y., Kaback H. R. (2009) J. Mol. Biol. 394, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang X., Nie Y., Kaback H. R. (2011) Biochemistry 50, 1634–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Majumdar D. S., Smirnova I., Kasho V., Nir E., Kong X., Weiss S., Kaback H. R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 12640–12645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smirnova I., Kasho V., Sugihara J., Kaback H. R. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 21561–21566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaback H. R., Smirnova I., Kasho V., Nie Y., Zhou Y. (2011) J. Membr. Biol. 239, 85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. le Coutre J., Narasimhan L. R., Patel C. K., Kaback H. R. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 10167–10171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. le Coutre J., Kaback H. R., Patel C. K., Heginbotham L., Miller C. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 6114–6117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patzlaff J. S., Moeller J. A., Barry B. A., Brooker R. J. (1998) Biochemistry 37, 15363–15375 [DOI] [PubMed] [Google Scholar]
- 37. Sayeed W. M., Baenziger J. E. (2009) Biochim. Biophys. Acta 1788, 1108–1115 [DOI] [PubMed] [Google Scholar]
- 38. van Iwaarden P. R., Driessen A. J., Lolkema J. S., Kaback H. R., Konings W. N. (1993) Biochemistry 32, 5419–5424 [DOI] [PubMed] [Google Scholar]
- 39. Menick D. R., Sarkar H. K., Poonian M. S., Kaback H. R. (1985) Biochem. Biophys. Res. Commun. 132, 162–170 [DOI] [PubMed] [Google Scholar]
- 40. Smirnova I. N., Kaback H. R. (2003) Biochemistry 42, 3025–3031 [DOI] [PubMed] [Google Scholar]
- 41. Nie Y., Smirnova I., Kasho V., Kaback H. R. (2006) J. Biol. Chem. 281, 35779–35784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cosson P., Bonifacino J. S. (1992) Science 258, 659–662 [DOI] [PubMed] [Google Scholar]
- 43. Lemmon M. A., Flanagan J. M., Treutlein H. R., Zhang J., Engelman D. M. (1992) Biochemistry 31, 12719–12725 [DOI] [PubMed] [Google Scholar]
- 44. Smith S. O., Bormann B. J. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 488–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Javadpour M. M., Eilers M., Groesbeek M., Smith S. O. (1999) Biophys. J. 77, 1609–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ermolova N. V., Smirnova I. N., Kasho V. N., Kaback H. R. (2005) Biochemistry 44, 7669–7677 [DOI] [PubMed] [Google Scholar]
- 47. Carrasco N., Herzlinger D., Mitchell R., DeChiara S., Danho W., Gabriel T. F., Kaback H. R. (1984) Proc. Natl. Acad. Sci. U.S.A. 81, 4672–4676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wolin C. D., Kaback H. R. (2000) Biochemistry 39, 6130–6135 [DOI] [PubMed] [Google Scholar]
- 49. Sahin-Tóth M., Dunten R. L., Kaback H. R. (1995) Biochemistry 34, 1107–1112 [DOI] [PubMed] [Google Scholar]
- 50. Frillingos S., Sahin-Tóth M., Wu J., Kaback H. R. (1998) FASEB J. 12, 1281–1299 [DOI] [PubMed] [Google Scholar]
- 51. Kaback H. R. (1971) in Methods in Enzymolology (Kaplan N. P., Jakoby W. B., Colowick N. P. eds) pp. 99–120, Elsevier Science Publishing Co., Inc., New York [Google Scholar]
- 52. Short S. A., Kaback H. R., Kohn L. D. (1975) J. Biol. Chem. 250, 4291–4296 [PubMed] [Google Scholar]
- 53. Konings W. N., Barnes E. M., Jr., Kaback H. R. (1971) J. Biol. Chem. 246, 5857–5861 [PubMed] [Google Scholar]
- 54. Kaback H. R. (1974) Methods Enzymol. 31, 698–709 [DOI] [PubMed] [Google Scholar]
- 55. Consler T. G., Tsolas O., Kaback H. R. (1991) Biochemistry 30, 1291–1298 [DOI] [PubMed] [Google Scholar]
- 56. Jung K., Jung H., Colacurcio P., Kaback H. R. (1995) Biochemistry 34, 1030–1039 [DOI] [PubMed] [Google Scholar]
- 57. Weinglass A. B., Smirnova I. N., Kaback H. R. (2001) Biochemistry 40, 769–776 [DOI] [PubMed] [Google Scholar]
- 58. Zhou Y., Kanner B. I. (2005) J. Biol. Chem. 280, 20316–20324 [DOI] [PubMed] [Google Scholar]
- 59. Overath P., Weigel U., Neuhaus J. M., Soppa J., Seckler R., Riede I., Bocklage H., Müller-Hill B., Aichele G., Wright J. K. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 5535–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ermolova N., Madhvani R. V., Kaback H. R. (2006) Biochemistry 45, 4182–4189 [DOI] [PubMed] [Google Scholar]
- 61. Chervitz S. A., Falke J. J. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 2545–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guan L., Hu Y., Kaback H. R. (2003) Biochemistry 42, 1377–1382 [DOI] [PubMed] [Google Scholar]
- 63. Vázquez-Ibar J. L., Guan L., Svrakic M., Kaback H. R. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 12706–12711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sahin-Tóth M., Lawrence M. C., Nishio T., Kaback H. R. (2001) Biochemistry 40, 13015–13019 [DOI] [PubMed] [Google Scholar]