

Abstract
Nisin is a posttranslationally modified antimicrobial peptide containing the cyclic thioether amino acids lanthionine and methyllanthionine. Although much is known about its antimicrobial activity and mode of action, knowledge about the nisin modification process is still rather limited. The dehydratase NisB is believed to be the initial interaction partner in modification. NisB dehydrates specific serine and threonine residues in prenisin, whereas the cyclase NisC catalyzes the (methyl)lanthionine formation. The fully modified prenisin is exported and the leader peptide is cleaved off by the extracellular protease NisP. Light scattering analysis demonstrated that purified NisB is a dimer in solution. Using size exclusion chromatography and surface plasmon resonance, the interaction of NisB and prenisin, including several of its modified derivatives, was studied. Unmodified prenisin binds to NisB with an affinity of 1.05 ± 0.25 μm, whereas the dehydrated and the fully modified derivatives bind with respective affinities of 0.31 ± 0.07 and 10.5 ± 1.7 μm. The much lower affinity for the fully modified prenisin was related to a >20-fold higher off-rate. For all three peptides the stoichiometry of binding was 1:1. Active nisin, which is the equivalent of fully modified prenisin lacking the leader peptide did not bind to NisB, nor did prenisin in which the highly conserved FNLD box within the leader peptide was mutated to AAAA. Taken together our data indicate that the leader peptide is essential for initial recognition and binding of prenisin to NisB.
Keywords: Antimicrobial Peptides, Enzyme Purification, HPLC, Peptide Interactions, Surface Plasmon Resonance (SPR), Nisin, Binding Affinity, Dehydratase NisB
Introduction
The development of bacterial resistance against clinically relevant antibiotics is on the rise and represents a major scientific challenge (1). There is an ever growing demand for novel and improved antimicrobial agents. As potential antibiotics, bactericidal peptides that are secreted by many bacteria, mainly for self-defense purposes, have gained special interest (2). Among these are the so-called lantibiotics, which are ribosomally synthesized peptides that are posttranslationally modified (3) and produced by a large number of Gram-positive bacteria.
Nisin, probably the most well known lantibiotic, is produced by the Gram-positive bacterium Lactococcus lactis and its antimicrobial activity is directed against microorganisms of similar type. Nisin has been used for more than 40 years as a food preservative, but so far, no significant bacterial resistance has arisen. Recently, L. lactis subspecies have been identified that are capable to inactivate nisin via proteolytic cleavage (4).
Nisin exerts its bactericidal mode of action in a dual fashion. First, nisin inhibits bacterial cell-wall synthesis by binding to lipid II, an essential cell-wall precursor molecule (5). Second, binding to lipid II leads to the formation of lipid II-nisin hybrid pores, which depolarizes and permeabilizes the cytoplasmic membrane of the target cell, leading to starvation and cell death (6–8).
The nisin biosynthesis operon comprises the nisin structural gene nisA and genes involved in modification (nisB and nisC), transport (nisT), and activation via processing (nisP) (9). Nisin biosynthesis is autoregulated via the two-component system nisRK (10, 11), whereas the autoimmunity factors of L. lactis are encoded by genes nisFEG and nisI (12). For the maturation of nisin, the 57-amino acid prenisin peptide (NisA) requires modification by several enzymes. The dehydratase NisB catalyzes the dehydration of specific serine and threonine residues to didehydroalanine and didehydrobutyrine residues, respectively (13, 14). These, in turn, are stereospecifically coupled to cysteines by the cyclase NisC (14, 15) yielding five thioether rings (13–15). Finally, the fully modified prenisin is secreted by the ATP-binding cassette transporter NisT (16, 17) and extracellularly processed by the cell-wall anchored protease NisP to liberate active nisin. These modifications are schematically depicted in Fig. 1 (18).
FIGURE 1.

Posttranslational modification of nisin. For nisin maturation, the ribosomally synthesized precursor peptide undergoes a series of modifications. A, the leader peptide (in gray) directs the prenisin to the nisin modification and transport machinery. The conserved FNLD box is highlighted in blue. B, specific serine and threonine residues (highlighted in yellow) are converted by the dehydratase NisB into dehydroalanines (dha) and dehydrobutyrines (dhb), respectively. C, the dehydrated residues, in turn, are specifically coupled to cysteine residues (highlighted in orange) by the cyclase NisC yielding five thioether rings comprising one lanthionine (a) and four methyllanthionines (b-e). D, fully modified prenisin is subsequently exported by the ABC-type transporter NisT and processed by the extracellular protease NisP that cleaves off the leader peptide to liberate active nisin. Note that Ser-29 is never dehydrated in nisin.
The dehydratase NisB has a central role in nisin biosynthesis. An increase of NisB expression leads to an enhanced dehydration efficiency of prenisin (19). NisB can, however, function in the absence of the cyclase NisC, producing dehydrated prenisin devoid of thioether rings, whereas deletion of NisB results in a substantially reduced production of (unmodified) prenisin (17). Remarkably, NisB can also function in the absence of both the cyclase NisC and the transporter NisT. L. lactis cells lacking NisCT secreted dehydrated peptide when the nisin leader peptide is preceded by a Sec signal sequence (22). Yeast two-hybrid and co-immunoprecipitation studies signified interactions between NisB, NisC, and prenisin (9).
Recent studies revealed that the modification of prenisin is a progressive and directional process requiring NisB and NisC to function cooperatively in an alternating fashion (20, 21). However, isolation of the proposed nisin modification complex NisBC has so far been unsuccessful (9, 19).
NisB is capable of dehydrating therapeutic peptides (unrelated to nisin) that are fused C-terminal to the nisin leader peptide (13, 22). Combined in vivo and in vitro data indicate that the leader peptide is essential for NisB and NisC modification (13, 15, 23) as well as for targeting the substrate peptide to the dedicated transporter NisT (17, 24). In addition, it was demonstrated that the leader peptide attached to the fully modified lantibiotic abolishes its antimicrobial activity, suggesting also a role in self-protection (15, 25).
The introduction of thioether rings in therapeutic peptides, either via enzymatic activity (NisBC) or chemical synthesis, has been shown to increase the resistance to proteolytic degradation (26, 27). Thus, a detailed understanding of the molecular mechanisms involved in nisin modification might be very important in the development of novel and improved peptide antibiotics as well as clinically relevant peptides that may be augmented by enzymatic posttranslational modification. However, whereas NisC cyclase activity has been successfully reconstituted in vitro and was shown to be independent of NisB and NisT (15), thus far, an in vitro activity of NisB has not been reported (2, 28).
Here we show a detailed biochemical and biophysical analysis of NisB and its interaction with prenisin and its modified derivatives, i.e. dehydrated and fully modified prenisin. Our results provide strong experimental evidence that the leader peptide and especially the FNLD box herein is a key determinant in substrate recognition and specificity of NisB.
EXPERIMENTAL PROCEDURES
Expression and Purification of His-tagged NisB
L. lactis NZ9000 containing the plasmid pNGnisBhis was grown overnight in 100 ml of M17 medium containing 0.5% (w/v) glucose (GM17) and 5 μg/ml of chloramphenicol at 30 °C. Cells were transferred to 2 liters of fresh GM17 medium and growth was continued to an A600 of 0.8, whereupon NisB expression was induced by the addition of nisin (Sigma) to a final concentration of 25 ng/ml. Three hours after induction cells were harvested by centrifugation at 8000 × g for 20 min at 4 °C. The cell pellet was suspended in 14 ml of buffer composed of 50 mm HEPES-NaOH, pH 8.0, and 150 mm NaCl and stored at −20 °C until use.
For purification, cells were thawed at 4 °C, supplemented with protease inhibitor mixture (Roche Applied Science) and DNase I (Sigma), and lysed with a cell disruptor (IUL Instruments), generally 4–5 cycles at a pressure of 2.5 kbar. The remaining cells were removed at 18,000 × g for 30 min at 4 °C, and the resulting supernatant was cleared from membranes by centrifugation at 130,000 × g for 75 min at 4 °C. The supernatant containing NisB was supplemented with imidazole, pH 8.0, to a final concentration of 5 mm and incubated at 4 °C for 10 min. The solution was then loaded on a 5-ml HiTrap chelating column (GE Healthcare) saturated with Ni2+ ions, using a flow rate of 2 ml/min at 4 °C. The column was washed with buffer containing 50 mm HEPES-NaOH, pH 8.0, 500 mm NaCl, 10 mm imidazole, and 10% (v/v) glycerol followed by a wash step in which the imidazole concentration was increased to 50 mm imidazole to remove unspecifically bound proteins. Finally, NisB was step-eluted by increasing the imidazole concentration to 250 mm using a flow rate of 1 ml/min. Eluted protein was monitored by measuring the absorbance at 280 nm and analyzed by SDS-PAGE. NisB containing fractions were pooled and applied to a HiLoad 16/60 Superdex 200 (GE Healthcare) size exclusion column using 50 mm HEPES-NaOH, pH 8.0, 500 mm NaCl, and 10% (v/v) glycerol as elution buffer. NisB containing fractions were pooled and concentrated with an Amicon Ultracentrifugal filter Ultracel (100 kDa cut-off). Protein concentration was determined by measuring the absorbance at 280 nm with a NanoDrop ND-1000 spectrophotometer (peqlab) using the theoretical extinction coefficient of NisB of 128,400 liters mol−1 cm−1, as calculated for the His-tagged NisB using ProtParam webserver (EXPASY).
Expression and Purification of Prenisin and Its Derivatives
Production of prenisin (NisA) and its derivatives was performed with L. lactis strain NZ9000 containing pNZnisA-E3 (17) together with pIL3BTC (for fully modified prenisin) (29), pIL3hpBT (for dehydrated prenisin) (30), or pIL3hpT (for unmodified prenisin) (30) as described in Ref. 21. The FNLD/AAAA mutant was produced as described in Ref. 24. Purification of the various prenisin peptides was performed as described in Ref. 21 with modifications. Cell-free medium containing the peptide was diluted 1:1 with 50 mm lactic acid, pH 3, and subjected to SP-Sepharose chromatography. After peptide binding, the lactic acid buffer, pH 3, was gradually changed to 50 mm HEPES-NaOH, pH 7, by applying a gradient (0–100% 50 mm HEPES-NaOH, pH 7) for 4 column volumes at a flow rate of 2 ml/min followed by a wash step for 8 column volumes with 50 mm HEPES-NaOH, pH 7. At this stage, the eluent showed a pH of 7. Finally, bound prenisin was eluted with 50 mm HEPES-NaOH, pH 7, 1 m NaCl, and 10% (v/v) glycerol. Peptide elution was monitored at 215 nm and fractions were analyzed by SDS-PAGE. Prenisin containing fractions were pooled and filtered through an Amicon Ultracentrifugal filter (30 kDa cut-off) to remove high molecular mass contaminants. The flow-through containing the prenisin was concentrated with an Amicon Ultracentrifugal filter (3 kDa cut-off). Peptide concentrations were determined with a Pierce BCA Protein Assay Kit (Thermo Scientific) at 584 nm.
Purification of Nisin
Nisin was obtained as a lyophilized powder from a commercial source (Sigma), which contains ∼2.5% (w/w) nisin. The active nisin was purified as described elsewhere (41). In brief, about 1.3 g of powder (corresponding to ∼32 mg of nisin) was diluted in 100 ml of 50 mm lactic acid, pH 3, and filtered through a 0.45-μm membrane filter (Pall Corporation). The nisin solution was then applied to a 5-ml HiTrap SP HP ion exchange column (GE Healthcare) using a flow of 2 ml/min. After binding, the column was washed with 50 mm lactic acid, pH 3, until a stable base line was reached. The elution was performed by block elution in which the NaCl concentration was increased from 0 to 1 m in 200 mm steps. Protein elution was monitored at 215 nm and fractions were analyzed by SDS-PAGE. Active nisin eluted at 400 mm NaCl. Nisin containing fractions were pooled, and protein was precipitated with 25% (v/v) trichloroacetic acid overnight at 4 °C. Precipitated protein was washed two times with ice-cold acetone to remove residual NaCl and TCA and then suspended in 50 mm lactic acid, pH 3. Peptide concentrations were determined with a Pierce BCA Protein Assay Kit (Thermo Scientific) at 584 nm.
HPLC Analysis of Prenisin and Its Derivatives
Analytical RP-HPLC was performed with a LiChrospher WP 300 RP-18 end capped column (Merck) at room temperature. Purified prenisin or nisin were injected at a concentration of 50 μm and eluted by mixing the aqueous buffer A (10% acetonitrile, 0.1% (v/v) trifluoroacetic acid) with the organic solvent buffer B (90% acetonitrile, 0.1% (v/v) trifluoroacetic acid). Elution was performed by applying a gradient of 0–100% of buffer B over the course of 60 min at a flow rate of 1 ml/min. The eluent was monitored by measuring the absorbance at 220 nm.
Static Light Scattering
Size exclusion chromatography (SEC)2 and multiangle light scattering were performed in line on an Äkta purifier (GE Healthcare) connected to a triple-angle light scattering detector (miniDawnTM TREOS, Wyatt Technology) and a differential refractive index detector (Optilab® rEX, Wyatt Technology). SEC was performed using an analytical Superdex 200 10/300 column (GE Healthcare) equilibrated with 50 mm HEPES-NaOH, pH 7.4, and 250 mm NaCl. The protein concentration of the purified NisB in elution buffer was 2 mg/ml, whereas the sample volume was 100 μl. Data were analyzed with the ASTRA software package (Wyatt Technology).
Interaction Studies Using SEC
For complex formation, 10 μl of a 300 μm solution of purified prenisin or nisin (in 50 mm HEPES-NaOH, pH 7, 1 m NaCl, and 10% (v/v) glycerol) was mixed with 100 μl of 15 μm purified NisB (in 50 mm HEPES-NaOH, pH 8, 500 mm NaCl, and 10% (v/v) glycerol), giving a molar ratio of 2:1. Complex formation was allowed to proceed for 1 h at room temperature. Next, the samples were applied to a Superdex 200 pc 3.2/30 size exclusion column on an ÄKTA micro (GE Healthcare) system at 4 °C using 50 mm HEPES-NaOH, pH 7.4, and 500 mm NaCl as elution buffer. Protein elution was monitored at 215 and 280 nm and the co-elution of NisB and prenisin was analyzed by SDS-PAGE. Protein was visualized by silver staining.
Surface Plasmon Resonance (SPR) Measurements
All measurements were performed with a Biacore X (GE Healthcare) at 25 °C and at a constant flow rate of 30 μl/min using a nitrilotriacetic acid sensor chip (GE Healthcare). The SPR buffer was composed of 50 mm HEPES-NaOH, pH 7.4, 250 mm NaCl, and 50 μm EDTA. Prior to immobilization of His-tagged NisB, both flow cells were saturated with Ni2+ by injecting 5 μl of a 10 mm Ni2+ solution at a flow rate of 10 μl/min. For immobilization, purified NisB was diluted in SPR buffer to a final concentration of 170 nm, whereupon 5 μl was injected on flow cell 2 with a flow rate of 10 μl/min. The first flow cell was left with just Ni2+ as reference. Prenisin and its derivatives (including active nisin and the FNLD/AAAA mutant) were diluted from stock solutions in SPR buffer to final concentrations ranging from 46 nm to 2.9 μm. Due to the low binding responses of the fully modified prenisin, higher concentrations were tested in addition, with no apparent effect on the kinetic constants and the affinity (data not shown). For each peptide concentration, 50 μl of the prenisin samples were injected at a flow rate of 30 μl/min. Sensorgrams were recorded for 250 s. After every analyte injection, the chip surface was regenerated by injecting successively 10 μl of 350 mm EDTA, 10 μl of 100 mm NaOH, and 10 μl of 0.5% (w/v) SDS at a flow rate of 10 μl/min. Subsequently NisB was immobilized again as described above. All sensorgrams presented are corrected for background and bulk flow effects. Each measurement was repeated at least three times. Data were analyzed using BIAevaluation 4.1 software (GE Healthcare) according to the Equation 1, which describes the association, Equation 2, which describes the dissociation, and Equation 3, which describes the dissociation constant, KD, as reported in Ref. 32. Where dR/dt is the binding rate, kon is the association rate, C is the concentration of injected analyte, Rmax is proportional to the ligand concentration, R is proportional to the formed ligand-analyte complex, and koff is the dissociation rate.
 |
 |
As the binding responses from SPR measurements correlates to the mass of molecules bound to the surface, the stoichiometry of the interaction can be evaluated by Equation 4 (33, 34).
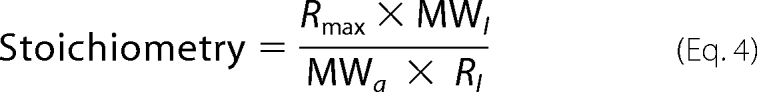 |
Where the analyte binding capacity Rmax can be extrapolated from experimental data and the immobilized ligand response Rl is obtained directly from a sensorgram recorded during ligand immobilization.
RESULTS
Purification of NisB and Prenisin Peptides
Nisin biosynthesis requires the dehydratase NisB to interact intimately with the nisin precursor peptide. To investigate the interaction of NisB and the nisin precursor peptide in vitro, NisB and unmodified prenisin and several modified derivatives thereof, were purified to homogeneity. NisB carrying a carboxyl-terminal His6 tag was expressed in L. lactis, and purified from the cytosol using immobilized metal ion affinity chromatography (IMAC) followed by SEC. After IMAC, NisB (∼120 kDa) and two contaminants (∼90 and ∼30 kDa) were present (Fig. 2, lane 4). These contaminants turned out to be degradation products of NisB, as determined by immunoblotting and mass spectrometry (data not shown), and could be removed by subsequent SEC (Fig. 2, lane 5). Thus, NisB could be purified to homogeneity from the cytosol with a typical yield of 1.5 mg of NisB/liter of cell culture. Notably, in buffer containing 50 mm HEPES-NaOH, pH 8, 500 mm NaCl, and 10% (v/v) glycerol, NisB was stable for at least 1 week at 4 °C as demonstrated by SEC, where no change in the elution profile occurred, and SDS-PAGE analysis (data not shown).
FIGURE 2.
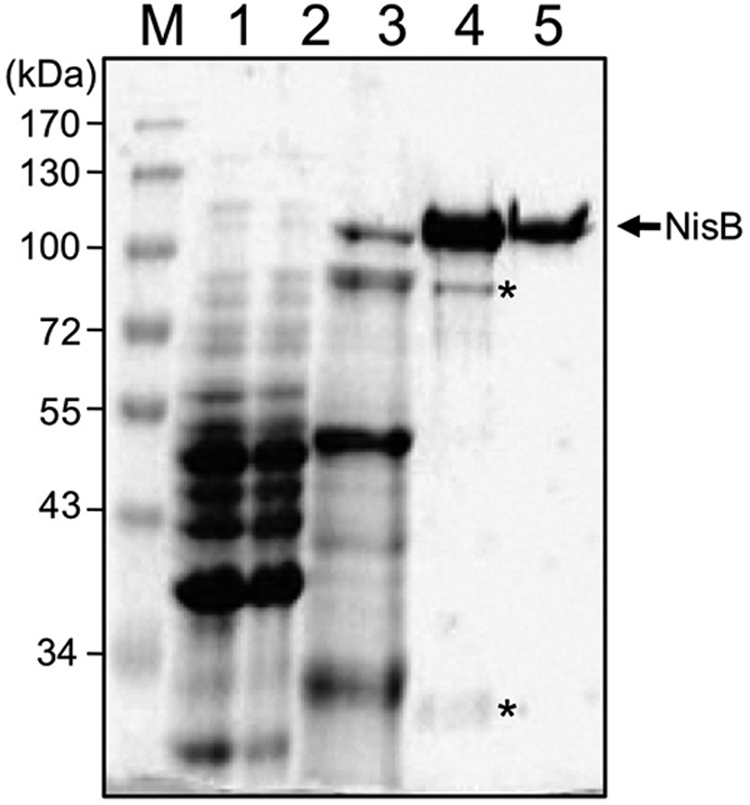
Purification of NisB. SDS-PAGE analysis of the purification of NisB. M, molecular mass marker proteins (kDa); lane 1, supernatant after high-speed centrifugation of L. lactis cell lysate; lane 2, flow-through of IMAC; lane 3, 50 mm imidazole IMAC wash; lane 4, 250 mm imidazole IMAC step elution; and lane 5, NisB after size exclusion chromatography. The arrow indicates NisB, whereas the asterisks mark the NisB degradation products.
Next, the unmodified prenisin and several of its modified derivatives including the dehydrated and the fully modified prenisin and the FNLD/AAAA mutant (see also Fig. 1) were purified from the culture medium by ion exchange chromatography (see “Experimental Procedures”). Nisin on the other hand was purified from a commercial powder (Sigma). SDS-PAGE analysis of SP-Sepharose fractions and subsequent silver staining revealed that the various peptide preparations were pure (Fig. 3A). Subsequent mass spectrometry confirmed the expected masses for the purified peptides (data not shown). Mass spectrometry can, however, not directly distinguish between dehydrated prenisin and the fully modified prenisin as these peptides have identical masses: 5688 Da (peptide without initiating methionine, see also Ref. 30). We therefore sought a simple and direct method to discriminate between these peptides. For this, the different prenisin peptides were analyzed by RP-HPLC (Fig. 3B). Nisin and the different prenisin peptides were injected separately at a concentration of 50 μm. Increasing the acetonitrile concentration eluted the peptides. Interestingly, the peptides showed markedly different elution profiles, which relates to significant differences in their hydrophobicity (Fig. 3B). The unmodified prenisin eluted between 19.0 and 20.5 min as a broad peak (Fig. 3B, black curve), whereas the dehydrated prenisin eluted in a similar broad peak but at a substantially later retention time of 19.5–22.5 min (Fig. 3B, red curve). Compared with the unmodified prenisin, the dehydrated prenisin showed reproducibly a higher absorbance at 220 nm likely due to the presence of an increased amount of double bonds as a result of the dehydration of the serine and threonine residues, which contribute to the absorbance at 220 nm (35). The fully modified prenisin eluted at 22.5 min and showed a characteristic double peak (Fig. 3B, blue curve). Nisin also showed a characteristic double peak but eluted much later from the column as compared with the fully modified prenisin, i.e. at 26.3 min (Fig. 3B, green curve). The double peak of nisin has been observed before and has been attributed to a small number of nisin molecules in which Ser-33 has escaped NisB-mediated dehydration (19). Taken together, these data demonstrate that RP-HPLC can be used to assess the identity of the different prenisin peptides in a qualitative manner. Importantly, RP-HPLC is able to distinguish directly between the dehydrated prenisin and the fully modified prenisin, which is in clear contrast to mass spectrometry and important for subsequent analysis.
FIGURE 3.

Purification and RP-HPLC analysis of prenisin and prenisin-derived peptides. A, SDS-PAGE analysis of purification of indicated peptides. M, molecular mass marker proteins (kDa); lane 1, unmodified prenisin; lane 2, dehydrated prenisin; lane 3, fully modified prenisin; lane 4, FNLD/AAAA prenisin mutant; lane 5, active nisin. Proteins were visualized by silver staining. B, RP-HPLC elution profiles of the purified peptides. Elution profiles as a function of time are shown for unmodified prenisin (black), dehydrated prenisin (red), fully modified prenisin (blue), and nisin (green). Left y axis shows the absorbance at 220 nm for unmodified and dehydrated prenisin. Right y axis shows the absorbance at 220 nm for fully modified prenisin and active nisin.
NisB Is a Dimer in Solution
The purified components in hand allow the study of the interaction between NisB and prenisin in vitro. However, whereas NisB analyzed by SDS-PAGE exhibits the expected molecular mass of ∼120 kDa (the calculated mass of His-tagged NisB is 118.3 kDa), we noticed that during preparative SEC NisB eluted as a protein with a molecular mass of ∼173 kDa (elution volume 59.6 ml). We therefore determined the molecular mass of NisB in solution using multiangle static light scattering. For this, NisB was loaded on a Superdex 200 10/300 column and the eluted protein was analyzed using a triple-angle light scattering detector. SEC combined with multiangle static light scattering revealed that the purified NisB eluted as a homogenous species with a molecular mass of 238.0 ± 1.2 kDa (Fig. 4, top panel). Analysis by SDS-PAGE confirmed that only NisB was present in this peak (Fig. 4, bottom panel). Thus, under the tested conditions, NisB is present as a dimer (calculated molecular mass for the NisB-His6 dimer is 236.6 kDa).
FIGURE 4.
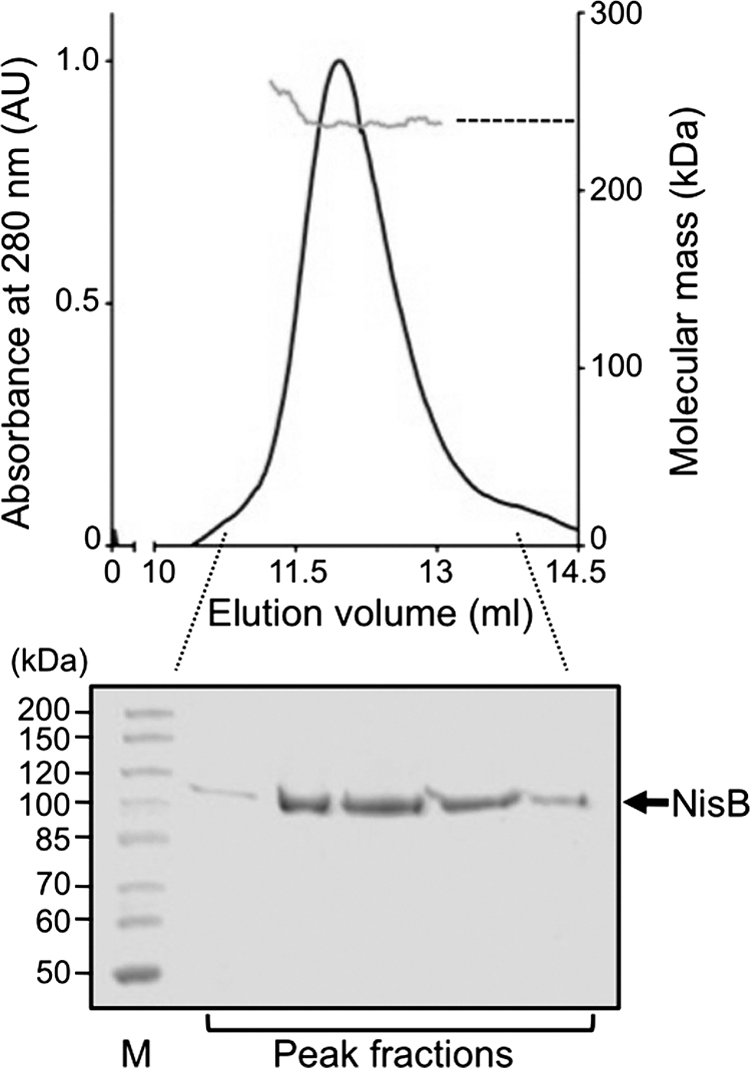
Size exclusion chromatography and static light scattering analysis of NisB. Top panel, NisB elution profile (black line) with determined molecular mass (gray line); bottom panel, SDS-PAGE analysis of the NisB peak fractions.
NisB-Prenisin Interaction Studied with SEC
To investigate the interaction of the various prenisins with NisB, analytical SEC studies were performed. For this, NisB was incubated with the unmodified, dehydrated, or the fully modified prenisin for 1 h to allow complex formation. The protein mixtures were then subjected to SEC analysis. In addition, the interaction of NisB with the FNLD/AAAA prenisin mutant as well as active nisin was investigated. Whereas NisB is readily detected at 280 nm, the prenisin peptides are not, due to a lack of Trp and Tyr residues. The peptides could, however, be detected at 215 nm. Incubation of NisB with the various peptides and subsequent SEC analysis did not reveal significant changes in the elution behavior of NisB (data not shown). However, when NisB was incubated with the dehydrated prenisins, subsequent SEC analysis showed a small decrease in absorbance at 215 nm for the free dehydrated prenisin suggesting that some of the dehydrated prenisin molecules may be bound to NisB. To verify if NisB is able to bind prenisin, the NisB peak fraction was analyzed by SDS-PAGE for co-elution of the different peptides (Fig. 5). SDS-PAGE analysis demonstrated that the unmodified, dehydrated, and the fully modified prenisin co-eluted with NisB (Fig. 5, left panels, lanes 3, 5, and 7, respectively), indicating that these peptides form a complex with NisB. The amount of the various prenisin forms co-eluting with NisB differed substantially. The dehydrated prenisin was reproducibly present in the highest amount, whereas a somewhat lower amount of unmodified prenisin was observed. In contrast, only very low amounts of fully modified prenisin were observed to co-elute with NisB. Active nisin, which lacks the leader peptide and is therefore not expected to bind to NisB, indeed did not co-elute with NisB (Fig. 5, right panels, lane 2). To further address the importance of the leader peptide in prenisin binding to NisB, a prenisin mutant, in which the conserved FNLD box within the leader peptide was mutated to AAAA, was also analyzed. This FNLD/AAAA prenisin is secreted in low levels into the culture medium by L. lactis cells harboring nisBTC, but is apparently not modified (24). Interestingly, the FNLD/AAAA prenisin did not co-elute with NisB, suggesting that it did not to bind to NisB (Fig. 5, right panels, lane 4). Taken together these data indicate that NisB shows functional binding of its native substrate in vitro and that this activity strictly depends on the presence of an intact leader peptide.
FIGURE 5.

Complex formation between NisB and prenisin. SDS-PAGE analysis of the NisB peak fractions after size exclusion chromatography showing interaction between NisB and the different prenisins. Left panels, M, molecular mass marker proteins (kDa); lane 1, purified NisB (2.5 μm); lane 2, purified unmodified prenisin (7 μm); lane 3, co-elution of unmodified prenisin and NisB; lane 4, purified dehydrated NisA (7 μm); lane 5, co-elution of dehydrated prenisin and NisB; lane 6, purified fully modified prenisin (7 μm); lane 7, co-elution of fully modified prenisin and NisB. Note, due to the presence of free cysteines in unmodified and dehydrated prenisin, oxidative products (bottom panels, upper prenisin band) are often observed. Right panels; lane 1, purified active nisin (7 μm); lane 2, NisB incubated with nisin; lane 3, purified FNLD/AAAA prenisin (7 μm); lane 4, NisB incubated with FNLD/AAAA prenisin.
NisB-Prenisin Interaction Studied with SPR
To characterize the interaction of NisB with the various prenisin peptides in more detail, the binding was quantitatively assessed by SPR. Measurements were performed by immobilizing NisB carrying a C-terminal His tag onto a Ni2+-bound nitrilotriacetic acid surface, after which the different peptides were injected at various concentrations. The peptide was injected for 100 s and sensorgrams were recorded for 250 s (Fig. 6). The real-time binding responses showed an exponential association and an exponential dissociation phase for the unmodified, dehydrated, and the fully modified prenisin (Fig. 6, A–C, respectively). The data were fitted by a 1:1 binding model with drifting baseline and local fitted Rmax. The obtained association and dissociation rate constants as well as the calculated equilibrium binding constants are summarized in Table 1. The unmodified prenisin exhibits an association rate (kon) of 1.2 ± 0.4 × 104 m−1 s−1 and a dissociation rate (koff) of 0.0117 ± 0.0014 s−1. Dehydrated prenisin on the other hand showed a ∼4-fold higher kon (5.1 ± 1.4 × 104 m−1 s−1), but similar koff (0.0149 ± 0.0025 s−1). The fully modified prenisin displayed a kon of 3.1 ± 0.4 × 104 m−1 s−1 and a more than 20-fold higher koff of 0.323 ± 0.035 s−1, when compared with the dehydrated and unmodified prenisin. The equilibrium constant or binding affinity (KD) for the different peptides were all found to be in the low micromolar range. The KD for the unmodified, dehydrated, and the fully modified prenisin peptides were 1.05 ± 0.25, 0.31 ± 0.07, and 10.5 ± 1.7 μm, respectively. Thus, NisB binds the dehydrated prenisin with highest affinity, whereas the affinity for fully modified prenisin is ∼30-fold lower. These results are in line with the SEC experiments where the amount of the different prenisin peptides that co-eluted with NisB followed the order: dehydrated > unmodified > fully modified (Fig. 5). Consistent with SEC analysis, mature nisin essentially did not bind to NisB. Only at the highest concentration tested was a very weak binding response observed (Fig. 6D). Similarly, the unmodified prenisin carrying the FNLD/AAAA mutation did not bind to NisB (Fig. 6E). Thus the data indicate that the leader peptide, and especially the FNLD box herein, is essential for binding of prenisin to NisB. Moreover, NisB shows substrate specificity as it binds its natural substrate, i.e. the unmodified prenisin and its modified derivatives, i.e. the dehydrated and the fully modified prenisin with different affinity.
FIGURE 6.

SPR analysis of the interaction of NisB with prenisin. Sensorgrams showing the interaction of immobilized NisB with unmodified prenisin (A), dehydrated prenisin (B), fully modified prenisin (C), nisin (D), and the FNLD/AAAA prenisin mutant (E). Injected peptide concentrations, from bottom to top, were 46 nm, 183 nm, 731 nm, 1.5 μm, and 2.9 μm.
TABLE 1.
Kinetic constants for the NisB-prenisin interaction
| Peptide | kon | koff | KD |
|---|---|---|---|
| m−1 s−1 | s−1 | μm | |
| Unmodified prenisin | 1.2 ± 0.4 ·104 | 0.0117 ± 0.0014 | 1.05 ± 0.25 |
| Dehydrated prenisin | 5.1 ± 1.4 ·104 | 0.0149 ± 0.0025 | 0.31 ± 0.07 |
| Fully modified prenisin | 3.1 ± 0.3 ·104 | 0.323 ± 0.035 | 10.5 ± 1.7 |
| Active nisin | NBa | ||
| FNLD/AAAA prenisin | NB | ||
a NB, no binding observed under tested experimental conditions.
SPR binding responses are generally proportional to mass and therefore one can assess the stoichiometry of an interaction by comparing the amount of ligand immobilized to the amount of analyte that it can bind. The stoichiometry of the interaction of NisB with the different prenisin peptides was determined using Equation 4 (33, 34). The Rmax (maximum capacity for analyte binding) is a local fitted parameter and depends on the amount of immobilized NisB. Because NisB was freshly immobilized for each measurement and its immobilized levels differed somewhat for each measurement, the average values of Rmax and Rl were used. Assuming that the immobilized NisB is dimeric (236.6 kDa), the calculated binding stoichiometry for the unmodified, dehydrated, and the fully modified prenisin were found to be 0.9 ± 0.1, 0.8 ± 0.1, and 0.9 ± 0.1, respectively.
DISCUSSION
The antibiotic potency of nisin seems to relate to its structure and dual mode of action. Nisin contains structural elements called (methyl)lanthionine rings that are important for binding to lipid II, an essential precursor molecule of the bacterial cell wall (6–8). Upon binding to lipid II, cell wall synthesis is blocked and nisin pores are formed that permeabilize the cytoplasmic membrane (6–8). The (methyl)lanthionine rings in nisin are installed posttranslationally by the cooperative action of the serine/threonine-specific dehydratase NisB and the cyclase NisC (21). Recently such lantibiotic modification enzymes have gained special interest as they can be successfully exploited to enhance the stability and activity of therapeutic peptides (26, 27). Moreover, a molecular understanding of the reactions catalyzed by these enzymes may further aid the development of novel and improved antibiotics.
From combined in vivo and in vitro studies it is known that the proteins involved in nisin biosynthesis, i.e. the dehydratase NisB, cyclase NisC, transporter NisT, and the leader peptidase NisP can act independent of each other (17–19, 22). The dehydration of the nisin precursor peptide catalyzed by NisB presents an early step in nisin maturation and is thus critical for nisin biosynthesis (Fig. 1). In vivo studies have demonstrated that NisB is promiscuous as it is able to dehydrate a multitude of nisin derivatives and even therapeutic peptides non-related to nisin (13, 22). However, for targeting such peptides to NisB the nisin leader peptide is required (13, 22). Unfortunately detailed information about the actual dehydration reaction is lacking. Moreover, no structure of NisB is available and reconstitution of in vitro activity of NisB has, thus far, been unsuccessful. In contrast, NisC cyclase activity has been successfully reconstituted in vitro and its crystal structure is available (15).
Here, we developed an in vitro binding assay to investigate the interaction between the lantibiotic dehydratase NisB and its native substrate the unmodified prenisin, as well as its dehydrated and fully modified derivatives. For this, NisB was expressed in its natural host L. lactis and purified to homogeneity (1.5 mg/liter of cell culture), whereas the substrate peptides were purified directly from minimal medium (Fig. 2). Interestingly, NisB purified from the cytosol proved to be dimeric as determined by static light scattering. The dimer was shown to be very stable as even after a week of storage no dissociation or aggregation were observed. To our knowledge, this is the first report that shows a NisB self-interaction. A study that combined a yeast two-hybrid screen with co-immunoprecipitations revealed interactions between members of the putative nisin synthase complex (NisA, NisB, NisC, and NisT) (9). In this study, a NisB self-interaction was, however, not observed. For the yeast two-hybrid screen fragments of NisB were used rather than full-length NisB. It is therefore intriguing to speculate that the self-interaction occurs only when full-length NisB is present. For SpaB, a NisB homolog from Bacillus subtilis that catalyzes the dehydration of the lantibiotic subtilin, a self-interaction was demonstrated by yeast two-hybrid analysis and the in vitro association of His6-SpaB and Myc-SpaB (36, 37). The proposed subtilin synthase complex consists of two molecules each of SpaB, SpaC, and SpaT, whereas the assumed nisin synthase complex consists of only one NisB molecule and two molecules each of NisC and NisT (9, 36, 37). Our data raises the possibility that in the nisin synthase complex NisB is present as a dimer. Although a body of evidence exists that supports the existence of such multimeric lanthionine synthase complexes, direct isolation of these complexes, and thereby the determination of the stoichiometry of the involved proteins has so far been unsuccessful.
The isolated dimeric NisB exhibited biological activity in vitro as it binds its native substrate, the unmodified prenisin, as evidenced by SEC and SPR analysis (Figs. 5 and 6). The interaction between NisB and the unmodified prenisin occurred with an affinity of 1.05 ± 0.25 μm (Table 1). Notably, binding to NisB did not require special additives and occurred in the absence of a cellular membrane. These results are consistent with yeast two-hybrid analysis and co-immunoprecipitation studies that showed an interaction between NisB and unmodified prenisin (9). The modified versions of prenisin, i.e. the dehydrated and the fully modified form, were also bound by NisB, although with different affinity (Figs. 5 and 6). Dehydrated prenisin, carrying eight dehydrated residues, showed a 3-fold higher affinity as compared with the unmodified prenisin, which relates to a substantially increased association rate (Fig. 6 and Table 1). This can be explained by an overall increase in hydrophobic interactions due to the presence of the dehydrated residues. In addition it is possible that NisB interacts in a specific manner with the non-leader part of prenisin. Support for this view comes from binding experiments using the fully modified prenisin (Figs. 5 and 6). Whereas the association rate of the fully modified prenisin was comparable with that of the unmodified and dehydrated prenisin, this peptide dissociated >20 times faster (koff 0.323 ± 0.035 s−1). As a consequence NisB exhibits a much lower affinity for this prenisin peptide, which harbors five thioether rings (KD of 10.5 ± 1.7 μm). In line with this, dehydration of serine and threonine residues was shown to be favored when they are flanked by hydrophobic residues, whereas a hydrophilic environment disfavored dehydration. This indicates a specific interaction of NisB with the propeptide (non-leader part) (29, 38). However, nisin, which is the equivalent of fully modified prenisin minus the leader peptide, did not bind to NisB (Figs. 5 and 6). This demonstrates the requirement of the leader peptide for binding to NisB in vitro. The importance of the leader peptide for NisB recognition has been firmly established in vivo as L. lactis cells harboring NisB can modify nonlantibiotic therapeutic peptides fused to the nisin leader peptide (13, 22, 23). The FNLD box within the leader peptide (Fig. 1) is highly conserved in the precursor peptides of class I lantibiotics (39). We show that the simultaneous substitution of the FNLD residues in the leader peptide by alanines abolished the interaction of prenisin with NisB in vitro (Figs. 5 and 6). These results may explain the in vivo observation that this mutant is secreted only in an unmodified form despite the presence of functional NisB and NisC (24). Thus the FNLD box is essential for prenisin interaction with NisB. However, because the FNLD/AAAA prenisin is still secreted, this suggests that leader peptide recognition by NisT is less stringent or determined by different part(s) of the leader peptide.
Taken together the data indicate that the nisin leader peptide is important for recognition and initial binding to NisB, with an essential role for the FNLD box. However, the interaction between NisB and prenisin is not only determined by the nisin leader peptide, but also by the nature of the propeptide (non-leader part). Thus the dehydratase NisB shows substrate specificity in vitro, as it is able to discriminate between the unmodified prenisin and its modified derivatives. Similarly, NisB may act as the specificity determinant within the nisin synthase complex in vivo. Following modification by NisB and NisC, the fully modified prenisin containing the thioether rings is rapidly released from the modification complex and subsequently exported by NisT.
Recent characterization of a number of prenisin mutants affected in ring formation revealed that NisB-mediated dehydration and NisC cyclase activity are strongly coordinated events. Herein, NisB and NisC alternate in function to install the modifications in a processive and directional manner (20, 21, 30). Our in vitro observations that thioether rings reduce the affinity of prenisin for NisB, whereas dehydrated residues appear to increase the affinity, would be compatible with such an alternating mechanism. It remains, however, to be determined whether the NisB dimer is required for biological activity.
The successful in vitro reconstitution of the enzymatic activity of several bifunctional LanM enzymes, which harbor both dehydratase and cyclase activity, has provided us with insight into the mechanistic aspects of lantibiotic modification (31, 40). The dehydratases of the LanB family, i.e. the lantibiotic synthases, that require separate enzymes for dehydration and cyclization remain on the other hand enigmatic. Although for many of these LanB enzymes a role in dehydration has been established unequivocally in vivo, this still has to be demonstrated in the test tube (2). Therefore, future work will focus on the aim to reconstitute the dehydratase activity of NisB in vitro.
Acknowledgments
We thank BiOMaDe for providing us with L. lactis NZ9000 containing the plasmid pNGnisBhis and Britta Tschapek, Jan Stindt, Sabine Metzger, Christian Schwarz, and Miroslav Kirov for technical support and helpful discussions.
This work was supported by Deutsche Forschungsgemeinschaft SCHM 1279/10-1 and a grant from the Heinrich Heine University of Duesseldorf (to L. S.).
- SEC
- size exclusion chromatography
- SPR
- surface plasmon resonance
- IMAC
- immobilized metal ion affinity chromatography.
REFERENCES
- 1. Projan S. J., Bradford P. A. (2007) Curr. Opin. Microbiol. 10, 441–446 [DOI] [PubMed] [Google Scholar]
- 2. Lubelski J., Rink R., Khusainov R., Moll G. N., Kuipers O. P. (2008) Cell Mol. Life Sci. 65, 455–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hansen J. N. (1993) Annu. Rev. Microbiol. 47, 535–564 [DOI] [PubMed] [Google Scholar]
- 4. Sun Z., Zhong J., Liang X., Liu J., Chen X., Huan L. (2009) Antimicrob. Agents Chemother. 53, 1964–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hasper H. E., Kramer N. E., Smith J. L., Hillman J. D., Zachariah C., Kuipers O. P., de Kruijff B., Breukink E. (2006) Science 313, 1636–1637 [DOI] [PubMed] [Google Scholar]
- 6. Breukink E., van Heusden H. E., Vollmerhaus P. J., Swiezewska E., Brunner L., Walker S., Heck A. J., de Kruijff B. (2003) J. Biol. Chem. 278, 19898–19903 [DOI] [PubMed] [Google Scholar]
- 7. Brötz H., Josten M., Wiedemann I., Schneider U., Götz F., Bierbaum G., Sahl H. G. (1998) Mol. Microbiol. 30, 317–327 [DOI] [PubMed] [Google Scholar]
- 8. Breukink E., Wiedemann I., van Kraaij C., Kuipers O. P., Sahl H., de Kruijff B. (1999) Science 286, 2361–2364 [DOI] [PubMed] [Google Scholar]
- 9. Siegers K., Heinzmann S., Entian K. D. (1996) J. Biol. Chem. 271, 12294–12301 [DOI] [PubMed] [Google Scholar]
- 10. Qiao M., Ye S., Koponen O., Ra R., Usabiaga M., Immonen T., Saris P. E. (1996) J. Appl. Bacteriol. 80, 626–634 [DOI] [PubMed] [Google Scholar]
- 11. Ra S. R., Qiao M., Immonen T., Pujana I., Saris E. J. (1996) Microbiology 142, 1281–1288 [DOI] [PubMed] [Google Scholar]
- 12. Stein T., Heinzmann S., Solovieva I., Entian K. D. (2003) J. Biol. Chem. 278, 89–94 [DOI] [PubMed] [Google Scholar]
- 13. Kluskens L. D., Kuipers A., Rink R., de Boef E., Fekken S., Driessen A. J., Kuipers O. P., Moll G. N. (2005) Biochemistry 44, 12827–12834 [DOI] [PubMed] [Google Scholar]
- 14. Koponen O., Tolonen M., Qiao M., Wahlström G., Helin J., Saris P. E. (2002) Microbiology 148, 3561–3568 [DOI] [PubMed] [Google Scholar]
- 15. Li B., Yu J. P., Brunzelle J. S., Moll G. N., van der Donk W. A., Nair S. K. (2006) Science 311, 1464–1467 [DOI] [PubMed] [Google Scholar]
- 16. Qiao M., Saris P. E. (1996) FEMS Microbiol. Lett. 144, 89–93 [DOI] [PubMed] [Google Scholar]
- 17. Kuipers A., de Boef E., Rink R., Fekken S., Kluskens L. D., Driessen A. J., Leenhouts K., Kuipers O. P., Moll G. N. (2004) J. Biol. Chem. 279, 22176–22182 [DOI] [PubMed] [Google Scholar]
- 18. Siezen R. J., Rollema H. S., Kuipers O. P., de Vos W. M. (1995) Protein Eng. 8, 117–125 [DOI] [PubMed] [Google Scholar]
- 19. Karakas Sen A., Narbad A., Horn N., Dodd H. M., Parr A. J., Colquhoun I., Gasson M. J. (1999) Eur. J. Biochem. 261, 524–532 [DOI] [PubMed] [Google Scholar]
- 20. Kuipers A., Meijer-Wierenga J., Rink R., Kluskens L. D., Moll G. N. (2008) Appl. Environ. Microbiol. 74, 6591–6597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lubelski J., Khusainov R., Kuipers O. P. (2009) J. Biol. Chem. 284, 25962–25972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuipers A., Wierenga J., Rink R., Kluskens L. D., Driessen A. J., Kuipers O. P., Moll G. N. (2006) Appl. Environ. Microbiol. 72, 7626–7633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rink R., Kluskens L. D., Kuipers A., Driessen A. J., Kuipers O. P., Moll G. N. (2007) Biochemistry 46, 13179–13189 [DOI] [PubMed] [Google Scholar]
- 24. Plat A., Kluskens L. D., Kuipers A., Rink R., Moll G. N. (2011) Appl. Environ. Microbiol. 77, 604–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van der Meer J. R., Rollema H. S., Siezen R. J., Beerthuyzen M. M., Kuipers O. P., de Vos W. M. (1994) J. Biol. Chem. 269, 3555–3562 [PubMed] [Google Scholar]
- 26. Rew Y., Malkmus S., Svensson C., Yaksh T. L., Chung N. N., Schiller P. W., Cassel J. A., DeHaven R. N., Taulane J. P., Goodman M. (2002) J. Med. Chem. 45, 3746–3754 [DOI] [PubMed] [Google Scholar]
- 27. Rink R., Arkema-Meter A., Baudoin I., Post E., Kuipers A., Nelemans S. A., Akanbi M. H., Moll G. N. (2010) J. Pharmacol. Toxicol. Methods 61, 210–218 [DOI] [PubMed] [Google Scholar]
- 28. Chatterjee C., Paul M., Xie L., van der Donk W. A. (2005) Chem. Rev. 105, 633–684 [DOI] [PubMed] [Google Scholar]
- 29. Rink R., Kuipers A., de Boef E., Leenhouts K. J., Driessen A. J., Moll G. N., Kuipers O. P. (2005) Biochemistry 44, 8873–8882 [DOI] [PubMed] [Google Scholar]
- 30. van den Berg van Saparoea H. B., Bakkes P. J., Moll G. N., Driessen A. J. (2008) Appl. Environ. Microbiol. 74, 5541–5548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shioya K., Harada Y., Nagao J., Nakayama J., Sonomoto K. (2010) Appl. Microbiol. Biotechnol. 86, 891–899 [DOI] [PubMed] [Google Scholar]
- 32. Karlsson R. (1994) Anal. Biochem. 221, 142–151 [DOI] [PubMed] [Google Scholar]
- 33. Mistrík P., Moreau F., Allen J. M. (2004) Anal. Biochem. 327, 271–277 [DOI] [PubMed] [Google Scholar]
- 34. Morton T. A., Myszka D. G. (1998) Methods Enzymol. 295, 268–294 [DOI] [PubMed] [Google Scholar]
- 35. Hesse M., Meier H., Zeeh B. (1987) Spektroskopische Methoden in der Organischen Chemie, 3rd Ed., Georg Thieme, Verlag Stuttgart [Google Scholar]
- 36. Xie L., Chatterjee C., Balsara R., Okeley N. M., van der Donk W. A. (2002) Biochem. Biophys. Res. Commun. 295, 952–957 [DOI] [PubMed] [Google Scholar]
- 37. Kiesau P., Eikmanns U., Gutowski-Eckel Z., Weber S., Hammelmann M., Entian K. D. (1997) J. Bacteriol. 179, 1475–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rink R., Wierenga J., Kuipers A., Kluskens L. D., Driessen A. J., Kuipers O. P., Moll G. N. (2007) Appl. Environ. Microbiol. 73, 1792–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oman T. J., van der Donk W. A. (2010) Nat. Chem. Biol. 6, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chatterjee C., Miller L. M., Leung Y. L., Xie L., Yi M., Kelleher N. L., van der Donk W. A. (2005) J. Am. Chem. Soc. 127, 15332–15333 [DOI] [PubMed] [Google Scholar]
- 41. Abts A., Mavaro A., Stindt J., Bakkes P. J., Metzger S., Driessen A. J. M., Smits S. H., Schmitt L. (2011) Int. J. Peptide, in press [DOI] [PMC free article] [PubMed] [Google Scholar]