

Abstract
Genetic alterations of α-actinin-4 can cause podocyte injury through multiple mechanisms. Although a mechanism involving gain-of-α-actinin-4 function was well described and is responsible for a dominantly inherited form of human focal segmental glomerulosclerosis (FSGS), evidence supporting mechanisms involving loss-of-α-actinin-4 function in human glomerular diseases remains elusive. Here we show that α-actinin-4 deficiency occurs in multiple human primary glomerulopathies including sporadic FSGS, minimal change disease, and IgA nephropathy. Furthermore, we identify a close correlation between the levels of α-actinin-4 and CLP36, which form a complex in normal podocytes, in human glomerular diseases. siRNA-mediated depletion of α-actinin-4 in human podocytes resulted in a marked reduction of the CLP36 level. Additionally, two FSGS-associated α-actinin-4 mutations (R310Q and Q348R) inhibited the complex formation between α-actinin-4 and CLP36. Inhibition of the α-actinin-4-CLP36 complex, like loss of α-actinin-4, markedly reduced the level of CLP36 in podocytes. Finally, reduction of the CLP36 level or disruption of the α-actinin-4-CLP36 complex significantly inhibited RhoA activity and generation of traction force in podocytes. Our studies reveal a critical role of the α-actinin-4-CLP36 complex in podocytes and provide an explanation as to how α-actinin-4 deficiency or mutations found in human patients could contribute to podocyte defects and glomerular failure through a loss-of-function mechanism.
Keywords: Cytoskeleton, Kidney, Mutant, Protein-Protein Interactions, Site-directed Mutagenesis, Contractility, Focal Segmental Glomerulosclerosis, Glomerular Diseases, Podocytes, Proteinuria
Introduction
Chronic and end stage kidney diseases, which are frequently caused by defects in glomerular filtration barrier function, have become a major global health problem. Although many factors can contribute to the development and progression of human glomerulopathies, genetic defects play important roles in the disease processes (1–6). Elucidation of how genetic alterations contribute to glomerular defects is therefore critical for understanding the molecular mechanisms underlying the development and progression of human glomerulopathies.
The glomerular filtration barrier is composed of an endothelium, a glomerular basement membrane, and a layer of podocytes building the slit diaphragm for filtration with their interdigitating foot processes. Although genetic alterations can cause defects in any of the three layers of the glomerular filtration barrier, podocytes appear to be a frequent target of genetic alterations in glomerulopathies (3, 4, 7–14). α-Actinin-4 is a member of the actinin protein family that consists of an actin-binding domain in the N terminus, four spectrin-like repeats in the central region, and two EF-hand motifs in the C terminus (15). Although both α-actinin-1 and -4 are expressed in mouse podocytes, α-actinin-4 is the sole member of the actinin family expressed in human podocytes (16). α-Actinin-4 is widely expressed in mammalian tissues and organs. However, despite the widespread expression, podocytes appear to be the primary site of manifestations of diseases induced by genetic alterations of ACTN4 (16, 17), the gene that encodes α-actinin-4. How do alterations of ACTN4 cause podocyte damage? A subset of FSGS2-associated α-actinin-4 mutations (e.g. K255E) were found in the actin-binding domain (16, 18). These mutations increase actin binding and aggregation of α-actinin-4 and consequently cause podocyte damages (16, 18–25). Although this gain-of-function mechanism is likely responsible for development of FSGS in patients with mutations in the actin-binding region of α-actinin-4, two lines of evidence suggesting that there are probably other mechanisms. First, genetic analyses of human patients with FSGS have identified a number of α-actinin-4 mutations outside of the actin-binding region (18), suggesting that alterations of other α-actinin-4 interactions could also be involved in the development of human glomerulopathies. Second, loss of α-actinin-4 in a mouse model causes defects in podocytes, proteinuria, and glomerular failure (17, 26), suggesting that, at least in the mouse model, alterations of α-actinin-4 can induce glomerular podocyte damage through loss-of-function mechanisms. It was not known, however, whether loss of α-actinin-4 protein or activity occurs in human glomerulopathies.
In this study, we report that deficiencies of α-actinin-4 and CLP36, an α-actinin-4-binding protein (27, 28), occur in multiple human glomerulopathies. Furthermore, we show that the formation of an α-actinin-4-CLP36 complex is crucial for maintenance of a proper level of CLP36 in podocytes. Additionally, we demonstrate that two mutations in α-actinin-4 (R310Q and Q348R) found in human FSGS patients inhibit the complex formation between α-actinin-4 and CLP36. Finally, using molecular approaches, we show that depletion of CLP36 or disruption of the α-actinin-4-CLP36 complex impedes RhoA signaling and traction force generation in podocytes. Collectively, these studies suggest an important role of the α-actinin-4-CLP36 complex in podocytes and provide an explanation as to how α-actinin-4 deficiency or mutations that inhibit CLP36-binding could contribute to podocyte defects in human glomerular diseases.
EXPERIMENTAL PROCEDURES
Antibodies and Other Reagents
Mouse monoclonal antibody (mAb) recognizing α-actinin was prepared using a GST fusion protein containing the central rod domain of α-actinin (residues 311–911) as an antigen based on a previously described method (29). Goat polyclonal anti-human CLP36 antibody (Ab) (Imegenex) was used for immunoprecipitation, immunofluorescent staining, and Western blotting. For immunohistochemical studies, mouse mAb and rabbit polyclonal Ab recognizing α-actinin were from Sigma. A rabbit polyclonal anti-CLP36 Ab was a gift from Professor Wolfgang Siess, Institute for Prophylaxis and Epidemiology of Cardiovascular Diseases, IPEK, Munich, Germany) (27). The proper control immunoglobulins for primary Abs, secondary biotinylated Abs, and the peroxidase-labeled streptavidin used for immunohistochemical staining were from Zymed Laboratories Inc. (Milan, Italy). Mouse anti-FLAG mAb (M2) was purchased from Sigma or Santa Cruz Biotechnology. Cy2-conjugated AffiniPure donkey anti-goat IgG (H+L) (minimal cross-reaction with chicken, guinea pig, Syrian hamster, horse, human, mouse, rabbit, and rat serum proteins) Ab, Rhodamine Red-X-conjugated AffiniPure donkey anti-mouse IgG(H+L) (minimal cross-reaction with bovine, chicken, goat, guinea pig, Syrian hamster, horse, human, rabbit, and sheep serum proteins) Ab, Rhodamine Red-conjugated goat anti-mouse IgG Ab, Rhodamine Red-conjugated mouse anti-goat IgG Ab, and horseradish peroxidase-conjugated secondary Ab were from Jackson ImmunoResearch Laboratories (West Grove, PA). FITC-phalloidin was from Sigma. All other chemicals were from Fisher or Sigma unless otherwise indicated. Cell culture media were from Mediatech/Cellgro (Herndon, VA).
Kidney Tissues and Immunohistochemical Studies
Kidney tissue was obtained from 38 renal biopsies and 10 control kidneys taken from the healthy pole of tumor nephrectomies. Parameters of renal function are given in Table 1. Histopathology was performed following standard methods, and 9 ± 2 (mean ± S.D.) glomeruli were examined for each biopsy. Only patients with no family history of renal disease were investigated in this study. Tissue handling for immunohistochemical analyses was based on a previously described protocol (30). Briefly, unfixed renal tissue was embedded in OCT compound (Miles Scientific, Naperville, IL), snap-frozen, and stored at −80 °C. Subsequently, 5-μm sections were placed on slides and stored at −20 °C until immunohistochemical staining. Sections were incubated with primary Abs, biotinylated secondary Abs, and peroxidase-labeled streptavidin. Peroxidase activity was detected with 3,5-diaminobenzidine, and sections were counterstained with Harry's hematoxylin, dehydrated, and mounted in Permount. Specificity of labeling was demonstrated by the lack of staining after substituting phosphate-buffered saline (PBS) and proper control immunoglobulins for the primary Abs. All peroxidase-stained sections were evaluated by an electronic image analysis system (ETC3000, Graftek, Villanterio, Pavia, Italy) after digitalization of the images. Glomerular staining for α-actinin-4 and CLP36 was evaluated using a color threshold procedure and calculated as percentage of labeling per glomerular tuft area.
TABLE 1.
Clinical parameters of the patients whose biopsies were used for immunohistochemical study (means ± S.D.)
| Diagnosisa | No. of cases | Serum creatinine | Urine proteins |
|---|---|---|---|
| mg/dl | g/24 h | ||
| Con | 10 | 0.8 ± 0.1 | 0.1 ± 0.1 |
| LN | 2 | 0.8 ± 0.1 | 2.8 ± 1.7 |
| MC | 5 | 0.9 ± 0.2 | 7.3 ± 2.6 |
| IgA | 10 | 1.2 ± 0.4 | 1.0 ± 0.5 |
| FSGS | 16 | 1.8 ± 0.8 | 7.4 ± 4.7 |
| MG | 5 | 1.1 + 0.4 | 5.3 + 2.4 |
a Con, Control; LN, lupus nephritis (World Health Organization IV); MC, minimal change disease; IgA, IgA nephropathy; FSGS, focal segmental glomerulosclerosis; MG, membranous glomerulopathy.
Podocyte Culture
Conditionally immortalized human glomerular podocytes were propagated under permissive condition at 33 °C as described (31). To induce differentiation, the cells were switched to 37 °C (nonpermissive condition) for 9–14 days. The cells were analyzed as specified in each experiment.
Immunofluorescent Staining
Podocytes were cultured under nonpermissive condition for 13 days and then replated on fibronectin (10 μg/ml)-coated coverslips. Twenty-four hours after replating, the cells were fixed with 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100 in 50 mm Tris-HCl (pH 7.4) containing 150 mm NaCl and 1 mg/ml BSA. For double staining of α-actinin-4 and CLP36, the cells were stained with primary mouse anti-α-actinin-4 mAb and goat anti-CLP36 Ab and Rhodamine Red-conjugated anti-mouse and Cy2-conjugated anti-goat IgG secondary Abs. In some experiments, cells were stained with FITC-conjugated phalloidin, goat anti-CLP36 primary Ab and Rhodamine Red-conjugated anti-goat IgG secondary Abs.
RNA Interference
Two different sets of α-actinin-4 or CLP36 siRNAs were used to knock down α-actinin-4 or CLP36 in podocytes. The sequences used for CLP36 in this study were 5′-GCAAGGCGGCTCTAGCTAA-3′ (C-KD1) and 5′-GTGGCTGCGTCGATTGGAA-3′ (C-KD2) (32). The sequences used for α-actinin-4 were 5′-CATCGCTTCCTTCAAGGTCTT-3′ (A-KD1) and 5′-GCCACACTATCGGACATCAAA-3′ (A-KD2) (33). For siRNA transfection, podocytes were cultured under nonpermissive condition for 7 days and were then transfected with α-actinin-4 or CLP36 siRNAs or an irrelevant small RNA (as a negative control) twice using Lipofectamine 2000 (Invitrogen). Two days after the second transfection, the cells were harvested and analyzed by Western blotting or other assays as specified in each experiment.
Adenoviral Expression Vectors and Infection
Adenoviral vectors encoding FLAG-tagged CLP36 ΔLIM fragment, in which residues 251–329 were deleted, was generated using the AdEasy system following a previously described protocol (34). Briefly, cDNA encoding FLAG-tagged CLP36 ΔLIM fragment was cloned into the KpnI/XhoI sites of the pAdTrack-CMV shuttle vector. The resultant plasmids were linearized with PmeI, purified, and mixed with supercoiled pAdEasy-1. The vectors were transferred into Escherichia coli BJ5183 by electroporation using a Bio-Rad Gene Pulser electroporator. Recombinants that were resistant to kanamycin were selected, and recombination was confirmed by PacI digestion. The positive plasmids were transformed into DH10B cells by electroporation for large scale amplification. The plasmid DNA was digested with PacI, ethanol-precipitated, and used to transfect 293 cells with Lipofectamine PLUS. The transfected cells were harvested 10 days after transfection. The cells were lysed by three cycles of freezing in a methanol/dry ice bath and rapid thawing at 37 °C, and the lysates containing the recombinant adenovirus were collected. Control adenoviral expression vector encoding β-galactosidase was kindly provided by Drs. Tong-Chuan He and Bert Vogelstein (Howard Hughes Medical Institute, The Johns Hopkins Oncology Center, Baltimore, MD). Podocytes that were cultured under nonpermissive condition for 7 days were infected with adenoviruses. The infection efficiency was monitored by the expression of green fluorescent protein encoded by the adenoviral vectors, which typically reached 100% within 2 days. Two days after adenoviral infection, the cells were harvested and analyzed by Western blotting and other assays as specified in each experiment.
Mutagenesis, DNA Constructs, and Transfection
Point substitution mutations (K255E, R310Q, or Q348R) were introduced into cDNA encoding FLAG-α-actinin-4 using a QuikChange site-directed mutagenesis system (Stratagene). All mutations were confirmed by DNA sequencing. Podocytes were transfected with vectors encoding mutant forms of α-actinin-4 or CLP36 using Lipofectamine 2000 (Invitrogen). Two days after transfection, the cells were harvested and analyzed as specified in each experiment.
Preparation of Triton X-100-soluble and -insoluble Fractions
Total cell lysates were prepared by extraction of the cells with 1% SDS in PBS buffer (pH 7.4) containing 2 mm Na3VO4 and protease inhibitors. Triton X-100-soluble and -insoluble fractions were prepared as we described previously (35). Briefly, cells (as specified in each experiment) were rinsed once with PBS buffer, extracted with 1% Triton X-100 in PBS buffer (pH 7.4) containing 2 mm Na3VO4 and protease inhibitors, and centrifuged at 20,800 × g at 4 °C for 15 min. The supernatants (soluble fractions) and pellets were collected. The pellets were extracted with 1% SDS in the PBS buffer containing protease inhibitors (Triton X-100-insoluble fractions). The Triton X-100-soluble and -insoluble fractions were mixed with SDS-PAGE sample buffer and analyzed by Western blotting.
Immunoprecipitation
Cells (as specified in each experiment) were lysed with 1% Triton X-100 in PBS buffer (pH 7.4) containing 2 mm Na3VO4 and protease inhibitors (the lysis buffer). The lysates were either directly mixed with agarose beads conjugated with anti-FLAG mAb M2 (Sigma) or first mixed with an anti-CLP36 Ab or control Ab and then incubated with UltraLink Immobilized protein G (Pierce) for 2 h at 4 °C. M2 beads or protein G beads were washed five times with the lysis buffer, and the immunoprecipitates were analyzed by Western blotting.
RhoA Activity
RhoA activity was determined using a luminescence-based RhoA activity assay (G-LISA; Cytoskeleton, Denver, CO) following the manufacturer's protocol. Briefly, cell lysates were prepared from podocytes in which CLP36 was knocked down or the α-actinin4-CLP36 complex was disrupted and control podocytes according to the G-LISA protocol. The lysates were incubated in wells of 96-well plates (25 μg of proteins/well) that were precoated with Rhotekin-binding domain peptide. Active RhoA was detected with a RhoA specific Ab.
Cell Traction Force Measurement
Traction, the cellular force that an individual adherent cell exerts on its substrate, was determined using traction force microscopy as described previously (36). Briefly, a polyacrylamide gel disk (120 mm thick, 10 mm in diameter) embedded with 0.2-mm red fluorescent microbeads (Molecular Probes) was made and attached to the bottom of a 35-mm glass dish (with a 14-mm circular inner glass area) (MatTek, Ashland, MA) that was consecutively pretreated with 0.1 m sodium hydroxide, 3-aminopropyltrimethoxysilane, and 0.5% glutaraldehyde. The gel surface was then pretreated with Sulfo-Sanpah (Pierce) and coated with 200 μl of 100 μg/ml collagen type I.
Podocytes were transfected with 5′-fluorescein-labeled CLP36 siRNAs (Invitrogen) or adenoviral vectors encoding FLAG-ΔLIM or β-galactosidase. Forty-eight hours after transfection, podocytes were trypsinized off the tissue culture dishes and were replated on the above-described collagen-coated polyacrylamide gel disks at 1000 cells/disk and allowed to spread on the collagen surface gel for 6 h at 37 °C. Cells expressing green fluorescence were selected and phase contrast images of individual cells as well as green fluorescent images were taken, which was followed by imaging the underlying red fluorescent beads. Cells on the gel disk were trypsinized (removing the applied contractile force) and images of the fluorescent beads at the same view and the same z plane were taken.
An image correlation method was used to compute the bead displacement field by comparing the bead image under the contracting cells with the cell-free bead image. At the beginning of the image processing, the pair of images was corrected for relative translational shift. Then the images were divided into small windows (16 × 16 pixels), and the normalized cross-correlation function was implemented for each window to find the relative displacement for each window between these two images. A hierarchical algorithm, polynomial fitting, and low-pass filtering were used to minimize the noise level. The traction field was calculated from the displacement field by using Fourier transform traction cytometry (37). The calculation of traction forces is based on the Boussinesq solution for the displacement field on a surface of a semi-infinite solid. At least 7 cells in each experimental group were used to determine their collective average traction forces.
Statistic Analysis
All statistical analyses were performed with Excel (Microsoft). Two-tailed Student's t test was used to calculate statistical significance. p values <0.05 were considered statistically significant.
RESULTS
α-Actinin-4 and CLP36 Deficiencies Occur in Multiple Human Primary Glomerulopathies
To test whether α-actinin-4 deficiency occurs in human glomerulopathies, we performed immunohistochemical staining using kidney tissues from control kidneys as well as human patients with glomerulopathies. Control kidney glomeruli showed intense and global positivity for α-actinin-4 (Fig. 1A, CON). A reduced but still almost global staining was present in lupus nephritis World Health Organization (WHO) class IV and membranous glomerulopathy (Fig. 1A, LN and MG, respectively). Glomeruli from patients with minimal change glomerulopathy, IgA nephropathy, and FSGS showed a segmental staining of α-actinin-4 (Fig. 1A, MC, IgA, and FSGS, respectively), with the most marked reduction observed in FSGS cases.
FIGURE 1.

Deficiencies of α-actinin-4 and CLP36 in human glomerulopathies. A and B, immunohistochemical staining of α-actinin-4 (A) and CLP36 (B) in human glomeruli. Arrows in CON, LN, and MG indicate some of the representative areas of α-actinin-4 and CLP36 staining (brown color). Scale bars, 50 μm. C, correlation of proteinuria and the reduction of the α-actinin-4 level in FSGS. Proteinuria determined in 24-h urine samples correlated negatively with α-actinin-4 staining (n = 16) in human FSGS patients. D, close correlation of the levels of α-actinin-4 and CLP36 in human glomerulopathies. The levels of α-actinin-4 and CLP36 were quantified as described under “Experimental Procedures.” Note that the extent of the reduction of CLP36 correlated closely with that of α-actinin-4 in glomerulopathies. CON, control kidney; LN, lupus nephritis; MG, membranous glomerulopathy; MC, minimal change disease; IgA, IgA nephropathy. Error bars, S.D.
These changes in expression were homogeneous among glomeruli and not limited to areas of damage or of segmental sclerosis, and in FSGS they closely correlated with the severity of proteinuria (Fig. 1C). Analyses of CLP36, which binds to α-actinin-4 (27, 28), revealed that its level was also reduced in glomerulopathies (Fig. 1B). Furthermore, the extent of the reduction strictly paralleled that of α-actinin-4 (Fig. 1D).
Loss of α-Actinin-4 Diminishes the Level of CLP36 in Podocytes
Consistent with immunohistochemical studies in normal human kidney glomeruli (Fig. 1B, CON), abundant CLP36 was detected in podocytes in culture, in particular under differentiation condition (Fig. 2A, lane 2). CLP36 and α-actinin-4 were co-localized along actin stress fibers (Fig. 2, C and D). Furthermore, they formed a stable complex in these cells that could be readily detected by co-immunoprecipitation (Fig. 2B, lane 2).
FIGURE 2.

CLP36 and α-actinin-4 co-localize and form a complex in human podocytes. A, human podocytes were cultured under permissive (lane 1) or nonpermissive conditions (lane 2) for 14 days. The cell lysates (4 μg of lysates/lane) were analyzed by Western blotting with Abs recognizing CLP36 or tubulin (as a loading control). B, cell lysates were mixed with an anti-CLP36 Ab or a control Ab that does not recognize CLP36. CLP36 and control immunoprecipitates were analyzed by Western blotting with anti-CLP36 and anti-α-actinin-4 Abs. C–H, human podocytes were dually stained with mouse anti-α-actinin-4 Ab and goat anti-CLP36 Ab (C and D) or FITC-conjugated phalloidin and goat anti-CLP36 Ab (F and G). The primary mouse and goat Abs were detected with secondary Rhodamine Red-conjugated anti-mouse IgG Ab and Cy2-conjugated anti-goat IgG Ab (C and D). Goat anti-CLP36 Ab in G was detected with a Rhodamine Red-conjugated anti-goat IgG Ab. Scale bar, 10 μm. The immunofluorescent images were pseudo-colored and merged using the National Institutes of Health ImageJ program (E and H).
The close correlation between the levels of CLP36 and α-actinin-4 proteins in glomerulopathies (Fig. 1D) prompted us to test whether CLP36 and α-actinin-4 depend on each other to maintain their protein levels in podocytes. To do this, we suppressed the expression of α-actinin-4 in podocytes with α-actinin-4 siRNAs (Fig. 3A, lanes 2 and 3). Analysis of the level of CLP36 in podocytes showed that it was also reduced in response to α-actinin-4 knockdown (Fig. 3A, compare lanes 2 and 3 with lane 1). The reduction of the CLP36 level was particularly obvious in Triton X-100-insoluble cytoskeleton fractions (Fig. 3B, compare lanes 5 and 6 with lane 4). Depletion of CLP36 with CLP36 siRNAs, on the other hand, did not alter the level of α-actinin-4 in podocytes (Fig. 3, C and D). These results suggest that CLP36 is dependent on α-actinin-4 for maintenance of its level in podocytes, whereas the level of α-actinin-4 is independent of that of CLP36.
FIGURE 3.

Loss of α-actinin-4 diminishes the level of CLP36 in podocytes. Human podocytes were transfected with α-actinin-4 siRNA A-KD1 or A-KD2 (A and B), CLP36 siRNA C-KD1 or C-KD2 (C and D), or a control RNA as indicated. The densities of the α-actinin-4 band in the A-KD1 and A-KD2 transfectants and those of the CLP36 band in the C-KD1 and C-KD2 transfectants were quantified using National Institutes of Health ImageJ program and compared with those of control transfectants. Under the conditions used, the levels of α-actinin-4 in the A-KD1 and A-KD2 transfectants were 23 and 34% of that of the control transfectants, and the levels of CLP36 in the C-KD1 and C-KD2 transfectants were 12 and 25% of that of the control transfectants. Total cell lysates (A and C), Triton X-100-soluble and -insoluble fractions (B and D) were prepared as described under “Experimental Procedures.” The samples were analyzed by Western blotting with Abs for α-actinin-4, CLP36,, or tubulin (as a loading control) or Coomassie Blue staining (to show equal loading).
Formation of the α-Actinin-4-CLP36 Complex Is Crucial for Maintenance of a Proper Level of CLP36
To test whether α-actinin-4 regulates the level of CLP36 through complex formation, we disrupted the α-actinin-4-CLP36 complex in podocytes. To do this, we expressed in human podocytes a FLAG-tagged CLP36 N-terminal fragment (ΔLIM), which contains an α-actinin-4-binding site (27, 28). FLAG-ΔLIM bound α-actinin-4 (Fig. 4A, lane 4). Expression of FLAG-ΔLIM in podocytes effectively prevented the complex formation between endogenous CLP-36 and α-actinin-4 (Fig. 3B, lane 4). Similar to the effect of depletion of α-actinin-4 (Fig. 3), disruption of the α-actinin-4-CLP36 complex reduced the level of CLP36 (Fig. 4C, lane 2), in particular in Triton X-100-insoluble cytoskeleton fractions (Fig. 4C, lane 6). These results suggest that the formation of the α-actinin-4-CLP36 complex is crucial for maintenance of a proper level of CLP36 in podocytes.
FIGURE 4.

Disruption of the α-actinin-4-CLP36 complex reduces the level of CLP36 in podocytes. A and B, human podocytes were infected with adenoviral vectors encoding FLAG-ΔLIM or β-galactosidase (as a control) as described under “Experimental Procedures.” FLAG-ΔLIM and CLP36 were immunoprecipitated from the cell lysates with anti-FLAG (A) or anti-CLP36 (B) Abs. The cell lysates (lanes 1 and 2) and immunoprecipitates (lanes 3 and 4) were analyzed by Western blotting with Abs for α-actinin-4, FLAG, or CLP36 or Coomassie Blue staining as indicated. A band that migrated faster than that of FLAG-ΔLIM was detected in anti-FLAG immunoprecipitates prepared from both the control β-galactosidase and FLAG-ΔLIM cells (lanes 3 and 4). This band most likely represents the light chain of the anti-FLAG Ab used in the immunoprecipitation. C, total cell lysates, Triton X-100-soluble and -insoluble fractions were prepared as described under “Experimental Procedures.” The samples were analyzed by Western blotting with Abs for α-actinin-4, CLP36, or FLAG or Coomassie Blue staining as indicated.
FSGS-associated R310Q or Q348R Substitution Mutation in α-Actinin-4 Impairs Its Interaction with CLP36
Previous studies in human patients with FSGS have shown that whereas several FSGS-associated mutations (e.g. K255E) are within the actin-binding region, some other mutations are located outside of the actin-binding region (18). We were particularly interested in R310Q and Q348R mutations for several reasons. First, R310Q substitution mutation occurs in probands of families with FSGS and individuals with sporadic FSGS at frequencies that are significantly higher than that of normal control population (18). Although the Q348R mutation was found in only one patient with sporadic FSGS, this mutation was not identified in 1092 control samples that were assayed (18). Thus, both R310Q and Q348R likely contribute to susceptibility of FSGS. Second, neither R310Q nor Q348R affects actin binding or subcellular localization of α-actinin-4 (18), suggesting that they likely impact podocytes through a mechanism that is different from that involving increased actin binding (i.e. gain-of-function). Finally, Arg-310 and Gln-348 are located within the spectrin-like repeats region, which is known to mediate CLP36 binding (27, 28). Thus, R310Q and Q348R could potentially affect CLP36 binding. To test this experimentally, we expressed FLAG-tagged α-actinin-4 bearing either R310Q or Q348R mutation, or wild type α-actinin-4 as a control, in podocytes. CLP36 was immunoprecipitated from the cells expressing wild-type or mutant forms of α-actinin-4 (Fig. 5, A and B, lanes 3 and 4). Analyses of CLP36 immunoprecipitates by Western blotting showed that, as expected, FLAG-tagged wild-type α-actinin-4 was readily co-immunoprecipitated with CLP36 (Fig. 5, A and B, lane 3). The amounts of FLAG-tagged α-actinin-4 proteins bearing R310Q or Q348R mutation that was co-immunoprecipitated with CLP36, however, were much less than that of FLAG-α-actinin-4 (Fig. 5, A and B, compare lane 4 with lane 3), suggesting that R310Q or Q348R mutation significantly inhibits the ability of α-actinin-4 to interact with CLP36. As a control, we expressed a FLAG-tagged α-actinin-4 bearing a mutation in the actin-binding region (K255E). Unlike R310Q or Q348R mutant, the binding of K255E mutant to CLP36 was not reduced but instead was increased (Fig. 5C, compare lanes 3 and 4). These results suggest that although FSGS-associated mutations were identified in both the actin-binding region (e.g. K255E) and the spectrin-like repeats region (e.g. R310Q or Q348R) of α-actinin-4, the effect of the mutations in the spectrin-like repeats region differs from that induced by the mutations in the actin-binding region, in that they cause loss-of-function (i.e. reduced CLP3 -binding) rather than gain-of-function (i.e. increased actin binding) (16).
FIGURE 5.
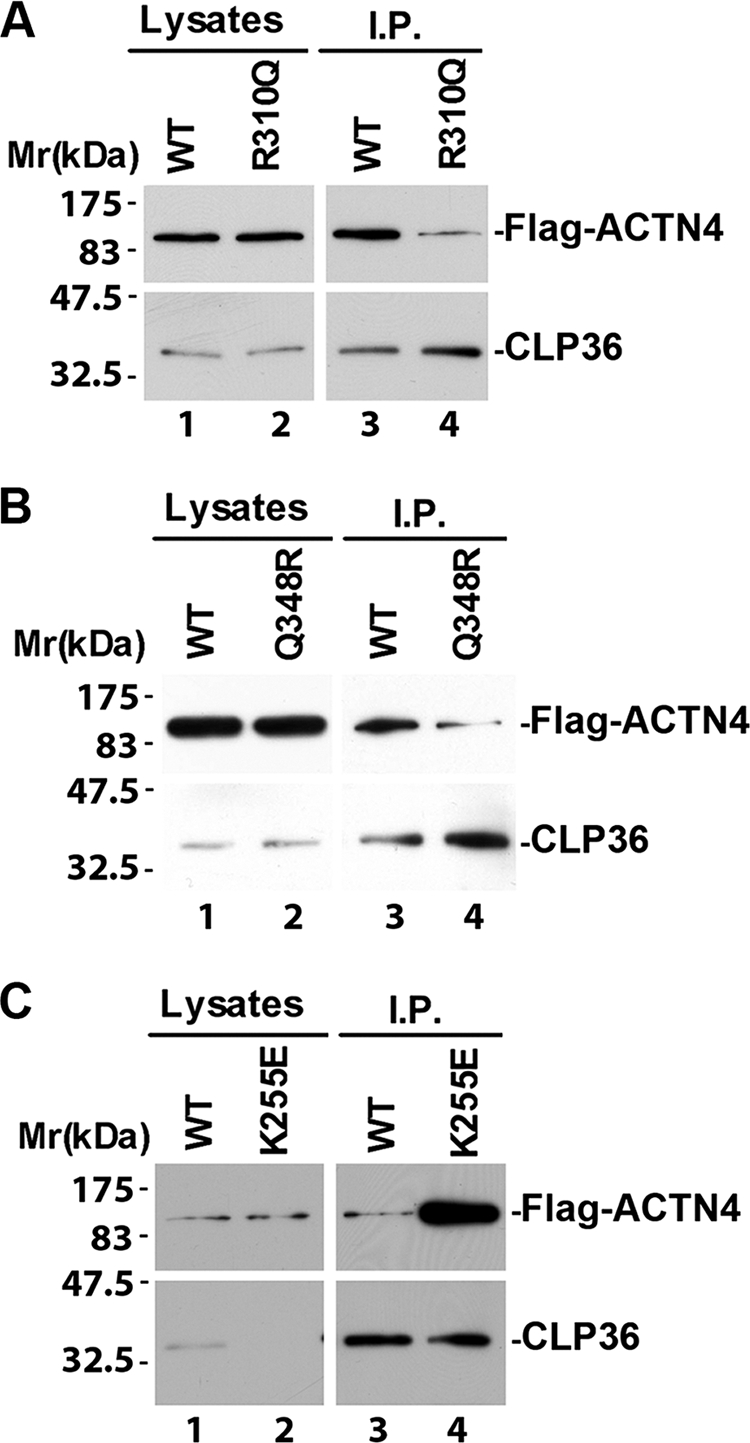
FSGS-associated R310Q or Q348R mutation inhibits α-actinin-4 interaction with CLP36. Human podocytes were transfected with expression vectors encoding FLAG-tagged wild-type or α-actinin-4 mutants bearing R310Q (A), Q348R (B), or K255E (C) mutation. CLP36 was immunoprecipitated from cell lysates with an anti-CLP36 Ab. The lysates (lanes 1 and 2) and immunoprecipitates (lanes 3 and 4) were analyzed by Western blotting with anti-FLAG and anti-CLP36 Abs as indicated. We have performed two independent immunoprecipitation experiments and obtained similar results from both experiments.
Depletion of CLP36 Reduces RhoA Activity and Cell Traction Force
α-Actinin is known to play an important role in regulation of cell contractility (38–40). Because CLP36 interacts and co-localizes with α-actinin-4, we sought to test whether CLP36 functions in regulation of podocyte contractility. To do this, we depleted CLP36 from podocytes by RNA interference (Fig. 6A). Traction force microscopic analyses showed that depletion of CLP36 significantly reduced cell traction force (Fig. 6, C and D). Furthermore, the activity of RhoA (Fig. 6B), which is known to play an essential role in control of cell traction force (41–43), was significantly reduced in response to depletion of CLP36. These results identify CLP36 as an important regulator of RhoA activity and cell traction force in podocytes.
FIGURE 6.

Depletion of CLP36 reduces RhoA activity and traction force in podocytes. A, human podocytes were transfected with CLP36 siRNA C-KD1 (lane 2), C-KD2 (lane 3), or a control RNA (lane 1). The cell lysates (4 μg of proteins/lane) were analyzed by Western blotting with Abs for CLP36 or tubulin (as a loading control). B, active RhoA was measured as described under “Experimental Procedures.” Bars represent means ± S.D. (error bars) from three independent experiments. *, p < 0.05 versus the control. C and D, cell traction force (CTF) of the control and CLP36 knockdown cells was measured as described under “Experimental Procedures.” C shows representative images of traction force microscopy of the control and CLP36 knockdown podocytes. Bars in D represent relative cell traction force (means ± S.D.) from three independent experiments. *, p < 0.05 versus the control.
Disruption of the α-Actinin-4-CLP36 Complex Impairs RhoA Signaling and Cell Traction Force Generation
We next tested whether the formation of the α-actinin-4-CLP36 complex, which is readily detectable in podocytes (Figs. 2B and 4), is required for RhoA signaling and generation of cell traction force. To do this, we expressed in podocytes FLAG-ΔLIM, which effectively disrupted the α-actinin-4-CLP36 complex (Fig. 4), and analyzed the effects on RhoA activity and cell traction force. The results showed that disruption of the α-actinin-4-CLP36 complex markedly reduced RhoA activity (Fig. 7A). Furthermore, consistent with a critical role of RhoA in regulation of cell traction force, disruption of the α-actinin-4-CLP36 complex significantly reduced traction force in podocytes (Fig. 7, B and C).
FIGURE 7.

Disruption of the α-actinin-4-CLP36 complex reduces RhoA activity and traction force in podocytes. Human podocytes were infected with adenoviral vectors encoding FLAG-ΔLIM or β-galactosidase (as a control) as in Fig. 4. A, active RhoA measured as described under “Experimental Procedures.” Bars represent means ± S.D. (error bars) from three independent experiments. *, p < 0.05 versus the control. B, representative images of traction force microscopy of podocytes infected with the control or FLAG-ΔLIM adenoviral vectors. C, bars represent relative cell traction force (CTF) (means ± S.D.) from three independent experiments. *, p < 0.05 versus the control.
DISCUSSION
Recent human genetic studies have identified a number of genetic alterations that are associated with glomerular diseases. Elucidating the molecular mechanisms by which genetic alterations cause defects in glomeruli is important for understanding the pathogenesis and progression of glomerular failure, the leading cause of end stage kidney disease. Of note, many genetic alterations associated with glomerular failure appear to impede the integrity of podocyte cytoskeleton, manifesting important roles of podocyte cytoskeleton for glomerular filtration barrier function. One of the well recognized targets of genetic mutations that impede glomerular podocyte functions is ACTN4. Although a subset of the disease-associated α-actinin-4 mutations (e.g. K255E) can cause podocyte damage through a gain-of-function mechanism (16, 18–25), there is evidence from studies in mouse models suggesting that loss of α-actinin-4 can also impair podocyte structure and consequently cause failure of glomerular filtration barrier (17). Whether or not α-actinin-4 deficiency actually occurs in human patients was not clear. The findings obtained from this study demonstrate that the level of α-actinin-4 is reduced in several (albeit not all) primary glomerulopathies, including minimal change glomerulopathy, IgA nephropathy, and FSGS (Fig. 1A). Furthermore, in FSGS the extent of the reduction in α-actinin-4 closely correlates with the severity of proteinuria (Fig. 1C). Thus, in at least a subset of human glomerulopathies, α-actinin-4 deficiency likely contributes to the failure of glomerular filtration barrier.
A second major finding of our studies is that concomitant to the reduction of α-actinin-4 in human glomerulopathies, the level of CLP36 is also reduced (Fig. 1, B and D). This finding raised two important questions. First, is there a mechanistic link between the level of CLP36 and that of α-actinin-4? Second, what is the functional significance of the reduction of the CLP36 level? To address these questions, we have performed a series of experiments using multiple approaches including RNA interference and dominant negative inhibition of the α-actinin-4-CLP36 complex. The results show that there is indeed a mechanistic link between the level of CLP36 and that of α-actinin-4. Specifically, we have found that the level of CLP36 is controlled by the complex formation between α-actinin-4 and CLP36 because depletion of α-actinin-4 (Fig. 3) or disruption of the α-actinin-4-CLP36 complex (Fig. 4) substantially reduced the level of CLP36 in podocytes. Functionally, we have found that loss of CLP36 or disruption of the α-actinin-4-CLP36 complex significantly impairs RhoA signaling and traction force generation in podocytes (Figs. 6 and 7). Thus, CLP36 is important for maintenance of the integrity of the actin cytoskeleton and generation of traction force, which are likely critical for glomerular filtration barrier function (10, 44–46).
Genetic studies in human patients with FSGS have identified mutations in the actin-binding as well as non-actin-binding regions of α-actinin-4 (16, 18). Biochemical analyses have shown that the mutations in the actin-binding region (e.g. K255E) increase actin binding, which likely causes podocyte damage in patients bearing these mutations (16, 18–25). Mutations in non-actin binding regions such as R310Q or Q348R do not alter actin-binding activity (18). It was not clear how these mutations can contribute to defects in podocytes. The studies presented in this paper demonstrate that R310Q or Q348R mutation inhibits the interaction of α-actinin-4 with CLP36 (Fig. 5). This finding, together with our findings that (i) inhibition of the α-actinin-4-CLP36 interaction reduces the level of CLP36 and (ii) reduction of the level of CLP36 compromises RhoA signaling and generation of traction force in podocytes provide a plausible “loss-of-function” mechanism through which the α-actinin-4 mutations (or α-actinin-4 deficiency) could contribute to podocyte defects and glomerular failure in human patients.
Acknowledgments
We thank Dr. Wolfgang Siess (University of Munich, Germany) for anti-CLP36 Ab and Dr. Deshun Ma, Dr. Xiaohua Shi, and Torin Yeager for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants DK54639 (to C. W.) and DK079912 (to M. K.).
- FSGS
- focal segmental glomerulosclerosis
- Ab
- antibody.
REFERENCES
- 1. Möller C. C., Pollak M. R., Reiser J. (2006) Adv. Chronic Kidney Dis. 13, 166–173 [DOI] [PubMed] [Google Scholar]
- 2. Pollak M. R. (2008) Curr. Opin. Nephrol. Hypertens. 17, 138–142 [DOI] [PubMed] [Google Scholar]
- 3. Machuca E., Benoit G., Antignac C. (2009) Hum. Mol. Genet. 18, R185–194 [DOI] [PubMed] [Google Scholar]
- 4. Löwik M. M., Groenen P. J., Levtchenko E. N., Monnens L. A., van den Heuvel L. P. (2009) Eur. J. Pediatr. 168, 1291–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiang C. K., Inagi R. (2010) Nat. Rev. Nephrol. 6, 539–554 [DOI] [PubMed] [Google Scholar]
- 6. McCarthy H. J., Saleem M. A. (2011) Nephron Exp. Nephrol. 118, e1–8 [DOI] [PubMed] [Google Scholar]
- 7. Jalanko H., Patrakka J., Tryggvason K., Holmberg C. (2001) Ann. Med. 33, 526–533 [DOI] [PubMed] [Google Scholar]
- 8. Pavenstädt H., Kriz W., Kretzler M. (2003) Physiol. Rev. 83, 253–307 [DOI] [PubMed] [Google Scholar]
- 9. Padiyar A., Sedor J. R. (2005) Curr. Mol. Med. 5, 497–507 [DOI] [PubMed] [Google Scholar]
- 10. Faul C., Asanuma K., Yanagida-Asanuma E., Kim K., Mundel P. (2007) Trends Cell Biol. 17, 428–437 [DOI] [PubMed] [Google Scholar]
- 11. Michaud J. L., Kennedy C. R. (2007) Clin. Sci. 112, 325–335 [DOI] [PubMed] [Google Scholar]
- 12. Wiggins R. C. (2007) Kidney Int. 71, 1205–1214 [DOI] [PubMed] [Google Scholar]
- 13. Patrakka J., Tryggvason K. (2009) Nat. Rev. Nephrol. 5, 463–468 [DOI] [PubMed] [Google Scholar]
- 14. Cheng H., Harris R. C. (2010) Int. J. Biochem. Cell Biol. 42, 1380–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Honda K., Yamada T., Endo R., Ino Y., Gotoh M., Tsuda H., Yamada Y., Chiba H., Hirohashi S. (1998) J. Cell Biol. 140, 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaplan J. M., Kim S. H., North K. N., Rennke H., Correia L. A., Tong H. Q., Mathis B. J., Rodríguez-Pérez J. C., Allen P. G., Beggs A. H., Pollak M. R. (2000) Nat. Genet. 24, 251–256 [DOI] [PubMed] [Google Scholar]
- 17. Kos C. H., Le T. C., Sinha S., Henderson J. M., Kim S. H., Sugimoto H., Kalluri R., Gerszten R. E., Pollak M. R. (2003) J. Clin. Invest. 111, 1683–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weins A., Kenlan P., Herbert S., Le T. C., Villegas I., Kaplan B. S., Appel G. B., Pollak M. R. (2005) J. Am. Soc. Nephrol. 16, 3694–3701 [DOI] [PubMed] [Google Scholar]
- 19. Yao J., Le T. C., Kos C. H., Henderson J. M., Allen P. G., Denker B. M., Pollak M. R. (2004) PLoS Biol. 2, e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Michaud J. L., Lemieux L. I., Dubé M., Vanderhyden B. C., Robertson S. J., Kennedy C. R. J. (2003) J. Am. Soc. Nephrol. 14, 1200–1211 [DOI] [PubMed] [Google Scholar]
- 21. Michaud J. L., Chaisson K. M., Parks R. J., Kennedy C. R. (2006) Kidney Int. 70, 1054–1061 [DOI] [PubMed] [Google Scholar]
- 22. Weins A., Schlondorff J. S., Nakamura F., Denker B. M., Hartwig J. H., Stossel T. P., Pollak M. R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 16080–16085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cybulsky A. V., Takano T., Papillon J., Bijian K., Guillemette J., Kennedy C. R. J. (2009) Am. J. Physiol. Renal Physiol. 297, F987–995 [DOI] [PubMed] [Google Scholar]
- 24. Henderson J. M., Alexander M. P., Pollak M. R. (2009) J. Am. Soc. Nephrol. 20, 961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Michaud J. L., Hosseini-Abardeh M., Farah K., Kennedy C. R. (2009) Cell Motil. Cytoskeleton 66, 166–178 [DOI] [PubMed] [Google Scholar]
- 26. Dandapani S. V., Sugimoto H., Matthews B. D., Kolb R. J., Sinha S., Gerszten R. E., Zhou J., Ingber D. E., Kalluri R., Pollak M. R. (2007) J. Biol. Chem. 282, 467–477 [DOI] [PubMed] [Google Scholar]
- 27. Bauer K., Kratzer M., Otte M., de Quintana K. L., Hagmann J., Arnold G. J., Eckerskorn C., Lottspeich F., Siess W. (2000) Blood 96, 4236–4245 [PubMed] [Google Scholar]
- 28. Vallenius T., Luukko K., Mäkelä T. P. (2000) J. Biol. Chem. 275, 11100–11105 [DOI] [PubMed] [Google Scholar]
- 29. Tu Y., Wu S., Shi X., Chen K., Wu C. (2003) Cell 113, 37–47 [DOI] [PubMed] [Google Scholar]
- 30. Rastaldi M. P., Ferrario F., Yang L., Tunesi S., Indaco A., Zou H., D'Amico G. (1996) J. Am. Soc. Nephrol. 7, 2419–2427 [DOI] [PubMed] [Google Scholar]
- 31. Saleem M. A., O'Hare M. J., Reiser J., Coward R. J., Inward C. D., Farren T., Xing C. Y., Ni L., Mathieson P. W., Mundel P. (2002) J. Am. Soc. Nephrol. 13, 630–638 [DOI] [PubMed] [Google Scholar]
- 32. Tamura N., Ohno K., Katayama T., Kanayama N., Sato K. (2007) Biochem. Biophys. Res. Commun. 364, 589–594 [DOI] [PubMed] [Google Scholar]
- 33. Quick Q., Skalli O. (2010) Exp. Cell Res. 316, 1137–1147 [DOI] [PubMed] [Google Scholar]
- 34. Yang Y., Guo L., Blattner S. M., Mundel P., Kretzler M., Wu C. (2005) J. Am. Soc. Nephrol. 16, 1966–1976 [DOI] [PubMed] [Google Scholar]
- 35. Zhang Y., Tu Y., Gkretsi V., Wu C. (2006) J. Biol. Chem. 281, 12397–12407 [DOI] [PubMed] [Google Scholar]
- 36. Wang J. H., Li B. (2009) Methods Mol. Biol. 586, 301–313 [DOI] [PubMed] [Google Scholar]
- 37. Butler J. P., Toliã-Nŗrelykke I. M., Fabry B., Fredberg J. J. (2002) Am. J. Physiol. Cell Physiol. 282, C595–605 [DOI] [PubMed] [Google Scholar]
- 38. Zhang W., Gunst S. J. (2006) J. Physiol. 572, 659–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bendix P. M., Koenderink G. H., Cuvelier D., Dogic Z., Koeleman B. N., Brieher W. M., Field C. M., Mahadevan L., Weitz D. A. (2008) Biophys. J. 94, 3126–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shao H., Wang J. H., Pollak M. R., Wells A. (2010) PLoS One 5, e13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chrzanowska-Wodnicka M., Burridge K. (1996) J. Cell Biol. 133, 1403–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hall A., Nobes C. D. (2000) Philos. Trans. R Soc. Lond. B Biol. Sci. 355, 965–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shiu Y. T., Li S., Marganski W. A., Usami S., Schwartz M. A., Wang Y. L., Dembo M., Chien S. (2004) Biophys. J. 86, 2558–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Endlich N., Endlich K. (2006) Eur. J. Cell Biol. 85, 229–234 [DOI] [PubMed] [Google Scholar]
- 45. Saleem M. A., Zavadil J., Bailly M., McGee K., Witherden I. R., Pavenstadt H., Hsu H., Sanday J., Satchell S. C., Lennon R., Ni L., Bottinger E. P., Mundel P., Mathieson PW. (2008) Am. J. Physiol. Renal Physiol. 295, F959–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mathieson P. W. (2010) Nephrol. Dial. Transplant. 25, 1772–1773 [DOI] [PubMed] [Google Scholar]