

Abstract
Maintenance of cardiac structure and Z-disc signaling are key factors responsible for protecting the heart in a setting of stress, but how these processes are regulated is not well defined. We recently demonstrated that PI3K(p110α) protects the heart against myocardial infarction. The aim of this study was to determine whether PI3K(p110α) directly regulates components of the Z-disc and cardiac structure. To address this question, a unique three-dimensional virtual muscle model was applied to gene expression data from transgenic mice with increased or decreased PI3K(p110α) activity under basal conditions (sham) and in a setting of myocardial infarction to display the location of structural proteins. Key findings from this analysis were then validated experimentally. The three-dimensional virtual muscle model visually highlighted reciprocally regulated transcripts associated with PI3K activation that encoded key components of the Z-disc and costamere, including melusin. Studies were performed to assess whether PI3K and melusin interact in the heart. Here, we identify a novel melusin-PI3K interaction that generates lipid kinase activity. The direct impact of PI3K(p110α) on myocyte structure was assessed by treating neonatal rat ventricular myocytes with PI3K(p110α) inhibitors and examining the myofiber morphology of hearts from PI3K transgenic mice. Results demonstrate that PI3K is critical for myofiber maturation and Z-disc alignment. In summary, PI3K regulates the expression of genes essential for cardiac structure and Z-disc signaling, interacts with melusin, and is critical for Z-disc alignment.
Keywords: Cardiac Muscle, Heart Failure, Insulin-like Growth Factor (IGF), Mechanotransduction, Phosphatidylinositol 3-kinase, Z-disc, Costamere
Introduction
An important question in cardiac biology is what makes one heart stronger, or more capable of resisting stress, than another. Maintenance of cardiac structure and Z-disc signaling are considered key factors responsible for protecting the heart in a setting of stress. Mutations of genes encoding proteins of the costamere and Z-disc have been linked with cardiomyopathies in animals and humans (1, 2). Costameres are specialized membrane junctions that physically connect the Z-disc to the basal lamina outside of the cell. The Z-disc is well recognized for its role in maintaining structural integrity and, more recently, has emerged as a “hot spot” or nodal hub of cardiomyocyte signaling, converting mechanical signals into chemical signals, leading to a transcriptional response and induction of a cardiac phenotype such as hypertrophy (1). Despite progress in this area, the mechanisms responsible for regulating components of the costamere and Z-disc are not well defined (2).
The p110α isoform of phosphoinositide 3-kinase (PI3K(p110α)/PIK3CA) has cardioprotective properties (3). We recently demonstrated that cardiac expression of a constitutively active (ca)3 PI3K transgene (increased PI3K activity) provided protection in a mouse model of myocardial infarction (MI), whereas a reduction in cardiac PI3K activity (utilizing a dominant negative (dn) PI3K transgene) accelerated the progression of heart failure (4). This makes targeting the PI3K pathway an attractive approach for developing new treatment strategies for heart failure.
PI3K(p110α) is a key mediator of insulin-like growth factor 1 (IGF1)-induced and exercise-induced beneficial physiological cardiac hypertrophy (5). Unlike pathological hypertrophy/enlargement, physiological cardiac hypertrophy is not an independent predictor of cardiovascular mortality and has protective properties (3). IGF1 administered directly onto neonatal cardiomyocytes in vitro increased mRNA expression and translation of contractile proteins, implicating a possible link between activation of the IGF1-PI3K axis and regulation of the expression of cardiac muscle structural components (6).
The primary aim of this study was to determine whether PI3K(p110α) specifically regulates cardiac structural components. We hypothesized that regulation of cardiac structure by PI3K(p110α) may explain why caPI3K hearts are protected against a cardiac insult, whereas dnPI3K hearts are more susceptible to heart failure. To address this question, we subjected microarray data from PI3K transgenic mice under basal conditions (sham) and in a setting of MI (4) to a recently developed tool for visualizing changes in gene expression spatially across a three-dimensional virtual muscle model (VMus3D) (7). The VMus3D tool is a simple, fast approach that highlights differential expression of genes related to muscle structure, identifying regulatory/structural relationships otherwise hidden from more generalist mining approaches. Key findings from this analysis were then validated experimentally.
EXPERIMENTAL PROCEDURES
Experimental Design
The VMus3D model (described below) was applied to previously reported gene expression data (4) (GEO accession number GSE 7487) from cardiac-specific transgenic mouse models with increased or decreased PI3K activity under basal conditions (sham) and in response to MI. The experimental design encompassed a comparison of six models that lie on a spectrum ranging from a physiological “protective” model (i.e. caPI3K sham) to an accelerated heart failure phenotype (i.e. dnPI3K MI). Such a design optimizes the ability to accurately link molecular signatures with specific phenotypes.
The molecular phenotypes of the six groups are as follows: 1) Non-transgenic (Ntg/control, FVB/N background) sham: Normal cardiac function and gene signature. 2) dnPI3K sham: Normal cardiac function at baseline, gene signature reflecting a heart susceptible to stress. 3) caPI3K sham: Normal cardiac function at baseline, physiological gene signature reflecting a heart protected against stress. 4) Ntg MI: Poor cardiac function, heart failure/pathological gene signature. 5) dnPI3K MI: Very poor cardiac function, gene signature reflecting accelerated heart failure in comparison to Ntg MI. 6) caPI3K MI: Better cardiac function than Ntg MI, a gene signature reflecting a slowing of the heart failure process compared with Ntg MI.
VMus3D Modeling
Affymetrix probe set data generated were normalized using the robust multiarray average method (8, 9), then entered into VMus3D for visualizing gene expression changes in terms of location of their protein product in muscle structure (further details are presented in the supplemental material and Ref. 7). Visualization was limited to genes with p values ≤ 0.05 and colored using VMus3D according to their significance and the direction of fold change being positive or negative. A two-by-two contingency table was constructed to compare reciprocally expressed genes observed using VMus3D in sham and infarct settings. Significance was assessed using the Barnard test and performed in R (10, 11).4
Gene lists were selected on the basis of the patterns recognized using VMus3D, as described by criteria presented in the supplemental material. The resulting lists were subjected to gene set enrichment via DAVID (12).
Protein Analysis and Kinase Activity Assays
Heart lysates were prepared as described (4). Western blot analysis: For IGF1R, blots were probed with an anti-IGF1R β (Santa Cruz Biotechnology, Inc., sc-713, 1:1000) followed by an anti-GAPDH (Santa Cruz Biotechnology, Inc., sc-32233,1:5000). For Akt and ERK1/2, blots were probed with phospho-(Ser-473)Akt (#9271), Akt (#9272), phospho-ERK1/2 (#9101) and ERK1/2 (#9102) (all from Cell Signaling Technology, Inc.).
IRS1 Immunoprecipitation
Immunoprecipitation was performed as described previously (13) using 1.5 mg of heart lysate, protein A-Sepharose, and an anti-IRS1 antibody (Millipore, #06–248, 0.25 mg/ml). Blots were probed with the anti-IRS1 antibody (1:2000).
PI3K Activity/Lipid Kinase Activity
PI3K activity was assessed in mouse ventricular tissue (13, 14). Heart tissue lysate (1 mg) was immunoprecipitated with an anti-p85 antibody (0.5 μl, Millipore, #06–195) or an anti-melusin antibody (5 μg) (15) and subjected to an in vitro lipid kinase assay using phosphatidylinositol as a substrate. Part of the immunoprecipitated enzyme was subjected to Western blotting and probed with the anti-p85 antibody (1:5000) and anti-melusin antibody (1 μg/ml).
Interaction between the regulatory subunit of PI3K (p85) and melusin
Heart lysate (5 mg) from wild-type, melusin-null (16), and melusin-overexpressing mice (17) was immunoprecipitated with an anti-melusin antibody (15 μg) (15) followed by an immunoblot with an anti-p85 antibody (1:1000, Cell Signaling Technology, Inc., #4257) and anti-melusin antibody (0.1 μg/ml).
Cell Culture
Neonatal rat ventricular myocyte (NRVM) culture was performed as reported previously (18) and approved by an Institutional Animal Ethics Committee that conforms with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. NRVM were treated with two PI3K(p110α) inhibitors (PIK75 (19) 0–100 μm or A66 (20, 21) 0–50 μm, both from Symansis, Auckland, New Zealand) with and without IGF1 (10 nm, Novozymes Biopharma AU Limited, CM001) for 18 h. Following experimental treatments, cells were either collected for protein lysate isolation and immunoblotting (18) or plated on laminin-coated glass coverslips and fixed in 4% paraformaldehyde for subsequent analysis by immunofluorescence confocal microscopy (18).
Immunofluorescence Confocal Microscopy of NRVM and Heart Sections
NRVM and heart sections (8 μm) from PI3K transgenic mice were analyzed by immunofluorescence staining with antibodies to α-actinin (Sigma-Aldrich), phalloidin (FITC-conjugates, Molecular Probes), DAPI, and Cy2/Cy3-conjugated secondary antibodies (Chemicon) as described previously (18, 22). Prior to staining, hearts from caPI3K, dnPI3K, and Ntg mice were fixed in HistoChoice® tissue fixative (Amersco) for 4 h at 4 °C and then frozen in Tissue-Tek® O.C.Ttm compound (Sakura Finetechnical).
Statistical Analysis
Statistical significance for gene expression data and VMus3D are described under “VMus3D Modeling” and in the supplemental data. Data from protein analysis are presented as mean ± S.E. Significance was determined using one-way analysis of variance (p < 0.05) followed by the Fisher's protected least significance post hoc test (p < 0.05).
RESULTS
VMus3D Highlighted PI3K(p110α)-regulated Transcripts Encoding Key Components of the Costamere and Z-disc
We reported previously that dnPI3K mice (cardiac PI3K activity decreased by 77%) and caPI3K mice (cardiac PI3K activity increased 6.5-fold) displayed normal cardiac function under sham conditions but displayed distinct phenotypes in response to MI for 8 weeks (4). In a setting of MI, caPI3K mice were protected with better cardiac function than Ntg mice, whereas dnPI3K mice were more susceptible to heart failure progression with poorer cardiac function than Ntg mice (4). VMus3D was used to overlay gene expression data onto structural components of cardiac muscle using data mined from Ntg and PI3K transgenic mice under sham and MI settings (4) (Fig. 1). This highlighted a distinct reciprocal pattern of gene expression between the PI3K transgenic models under sham conditions (Fig. 1C, blue, up-regulation in caPI3K; red, down-regulation in dnPI3K; p < 0.05), suggesting that PI3K activity can regulate components of the costamere and Z-disc under basal conditions. This was consistent with the hypothesis that PI3K activity predisposes how the heart will respond to a subsequent cardiac insult such as MI and prompted us to perform a reciprocal expression analysis (dnPI3K versus caPI3K) across the full microarray dataset.
FIGURE 1.

VMus3D visualization of costamere and Z-disc gene expression. Schematic with labeled components of the Z-disc (A) and costamere (B). A small section of A (highlighted in gray) has been enlarged to more readily identify individual components. Gene expression in mice with increased PI3K (caPI3K) and decreased PI3K (dnPI3K) under sham (C) and MI conditions (D) compared with Ntg. The coloring indicates genes reciprocally expressed (p < 0.05) according to PI3K activity in the sham model. Blue, positive fold change; yellow, no change; red, negative fold change.
Genes encoding muscle structural/associated proteins that were differentially and reciprocally expressed in the PI3K models under sham conditions included dystroglycan (Dag1), filamin C (Flnc), ankyrin repeat domain 23 (Ankrd23/Darp), Rho-associated coiled-coil containing protein kinase 2 (Rock2), crystallin, α B (Cryab), Cd151, integrin β 1 binding protein 2 (Itgb1bp2/melusin), Lim domain binding 3 (Ldb3/cypher), and synaptopodin 2 (Synpo2/myopodin) (supplemental Equations 2 and 3 and Table I, p ≤ 0.05, Fig. 1). Each of these genes were up-regulated in caPI3K (highlighted in blue, Fig. 1) and down-regulated in dnPI3K (highlighted in red, Fig. 1) relative to Ntg.
Experimental Evidence for a Novel Interaction between Melusin and PI3K in the Heart
Melusin (Itgb1bp2) is known to interact with the cytoplasmic domain of β1-integrin, is enriched in costameres, and has been shown previously to play a critical role in sensing mechanical stress (16). The tail domain of melusin is responsible for the interaction with β1-integrin, whereas the amino-terminal portion of melusin has multiple proline-rich motifs and tyrosine phosphorylation sites that could bind to SH3- and SH2-containing proteins (23). Class IA PI3Ks (including the p110α catalytic subunit) interact with adaptor subunits containing SH2 domains, and the regulatory subunit (85-kDa adaptor) contains an SH3 domain (24), suggesting the possibility of an interaction between PI3K and melusin. To assess whether PI3K and melusin interact in the heart and the potential significance of such a complex, heart tissue lysate was immunoprecipitated with an anti-melusin antibody followed by an in vitro lipid kinase assay using PtdIns as a substrate. Lipid kinase activity was observed in the heart lysate immunoprecipitated with the anti-melusin or anti-p85 antibody (positive control) but not the negative control (Fig. 2A). To further validate an interaction between melusin and PI3K in the heart, experiments in hearts from wild-type, melusin-null, and melusin-overexpressing mice were performed. Melusin-overexpressing mice allow melusin interactors to be more readily identified, whereas the melusin-null mouse is an ideal control for specificity. In the current study, the p85 regulatory subunit of PI3K was clearly detected in a melusin immunoprecipitate obtained from melusin-overexpressing transgenic mouse heart extracts (TG), and a weaker signal for p85 was present in WT heart lysates. In contrast, no signal for p85 was detected in heart lysates from melusin-null mice (KO, Fig. 2B).
FIGURE 2.
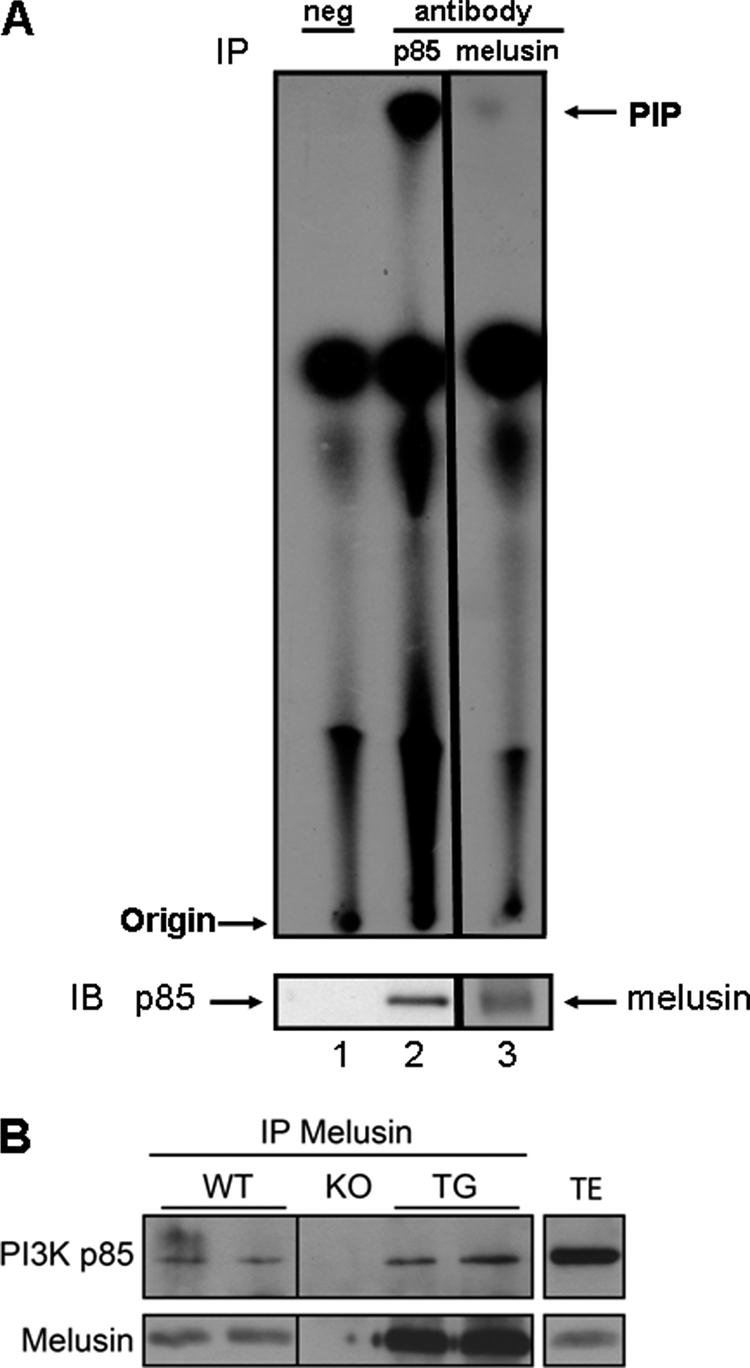
Interaction between PI3K and melusin in the heart. A, heart lysate was immunoprecipitated (IP) with anti-p85 antibody (lane 2, positive control), anti-melusin antibody (lane 3), or no antibody (lane 1, negative control) and subjected to a lipid kinase assay using phosphatidyl inositol as a substrate. PIP, PtdIns 3-phosphate represents PI3K activity. Part of the immunoprecipitate was subjected to Western blotting (IB) and probed with anti-p85 or anti-melusin. B, heart lysate from WT, melusin-null (KO), and melusin-overexpressing transgenic mice (TG) was immunoprecipitated with anti-melusin antibody followed by immunoblotting with anti-p85 and anti-melusin. Heart total extract (TE) was run as a reference for the molecular weight of p85. Results were confirmed in four independent experiments.
Enrichment of Genes Related to the IGF1 Pathway
To gain further insight as to how PI3K might sense biomechanical stress and regulate muscle structure and Z-disc signaling, we performed a gene set enrichment analysis to investigate the global effects of PI3K perturbation (supplemental Equation 3 and Table II). Analysis against all “pathways” listed in DAVID identified the following pathways (p < 0.05): Ribosome (KEGG 0310), IGF1R signaling (BIOCARTA), focal adhesion (KEGG 04510), insulin signaling (KEGG 04910), IGF1 signaling (BIOCARTA), IL-2 receptor β chain in T cell activation (BIOCARTA), and glycerophospholipid metabolism (KEGG 00564). Enrichment of the IGF1-related pathways was of particular interest, given that IGF1 plays an important role in mediating cardiac protection (5), and PI3K(p110α) is a critical downstream mediator of IGF1R-induced physiological heart growth (13).
By microarray, IGF1R and IRS1 mRNA expression were elevated in hearts of Ntg in a setting of MI compared with sham (Fig. 3A). IGF1R and IRS1 mRNA expression were lower in caPI3K sham hearts and higher in dnPI3K sham hearts (Fig. 3A). IGF1R mRNA expression did not significantly increase in caPI3K or dnPI3K mice in response to MI. To validate that changes at the level of gene expression reflected protein levels, we assessed IGF1R and IRS1 by Western blotting and immunoprecipitation, respectively. Consistent with our gene expression data, we observed parallel changes in protein expression of IGF1R and IRS1 in the sham and MI models (Fig. 3, B and C).
FIGURE 3.

IGF1R and IRS1 gene and protein expression. A, IGF1R and IRS1 gene expression (log2) obtained by microarray. *, p < 0.05 versus Ntg sham; #, p < 0.05 versus caPI3K sham. B, left panel, Western blot analysis of IGF1R and GAPDH in hearts from Ntg, caPI3K, and dnPI3K mice subjected to sham (Sh) or MI. Right panel, quantitative analysis of IGF1R relative to GAPDH. Mean values for Ntg Sh were normalized to 1. n = 3 for Sh groups, n = 4 for MI groups. *, p < 0.0001 versus Ntg Sh and caPI3K Sh; ^, p < 0.001 versus Ntg MI; §, p < 0.001 versus caPI3K MI; #, p < 0.01 versus Ntg MI and caPI3K MI. C, left panel, immunoprecipitated IRS1. Right panel, quantitative analysis of IRS1. Mean values for Ntg were normalized to 1. n = 3 for each group. *, p < 0.05 versus Ntg Sh; ^, p < 0.05 versus caPI3K Sh; #, p < 0.05 versus caPI3K MI.
Direct Evidence of an Impact of PI3K on Cardiac Myocyte Structure
The VMus3D model and subsequent gene set enrichment analysis suggested that IGF1-PI3K(p110α) signaling directly regulates cardiac myocyte structure. To test this hypothesis, we performed two studies. 1) We assessed the impact of two PI3K inhibitors on the myofiber structure in NRVM and 2) examined the myofiber morphology of hearts from Ntg, caPI3K, and dnPI3K under basal conditions.
1) Inhibition of PI3K(p110α) prevents normal formation of mature myofibers under basal conditions and in response to IGF1: NRVM were treated independently with two PI3K(p110α) inhibitors (PIK75 and A66) in the presence or absence of IGF1. PIK75 has been used previously to study the role of PI3K(p110α) but has some off-target activity (19, 25). A66 is considered a more highly specific and selective inhibitor for p110α (20, 21). The specificity of both inhibitors at different doses (0.1–100 μm) was tested in NRVM with or without IGF1. The ratio of pAkt/total Akt was reduced by more than 50% in IGF1 stimulated NRVM with 0.1 μm PIK75 or 0.5 μm A66 and was abolished completely in IGF1-stimulated NRVM treated with 1 μm PIK75 or 10 μm A66 (Fig. 4, A and B). The PI3K(p110α) inhibitors had no significant impact on pERK/total ERK at these concentrations (Fig. 4, A and B). At substantially higher concentrations of PIK75 (10 and 100 μm), an increase in ERK phosphorylation stimulated by IGF1 was not observed (Fig. 4A). A66 had no effect on pERK/total ERK, even at 50 μm (Fig. 4B).
FIGURE 4.

NRVM treated with PI3K(p110α) inhibitors (PIK75 or A66) in the absence and presence of IGF1. A and B, Western blot analyses of pAkt, total Akt, pERK, and ERK in cell lysates from NRVM treated with or without IGF1 in the presence of PIK75 (0–100 μm) or A66 (0–50 μm). C and D, immunofluorescence of control NRVM (dimethyl sulfoxide, DMSO) and NRVM treated with PIK75, A66, and/or IGF1 for 18 h. Anti-α actinin (a–f) stains sarcomeric bands and Z-lines, phalloidin-FITC (g–l) stains contractile thin filaments, and DAPI (m–r) stains nuclei. s–x, merged images. Scale bar = 20 μm. Experiments were repeated twice with identical findings. Representative images from a single experiment are shown.
Myofiber formation and sarcomere organization was examined by immunofluorescence. α-actinin was used to highlight the sarcomeric bands and Z-discs, and phalloidin was used to stain the contractile actin thin filaments. Sarcomeres in the control myocytes ran in multiple axes within the same cell and were generally punctate and non-striated (Fig. 4, C and D, dimethyl sulfoxide (DMSO)). Control cells also had an irregular rounded morphology. In contrast, myocytes treated with the PI3K(p110α) inhibitors alone had an elongated stellate morphology, and sarcomeres tended to run along a longitudinal axis rather than multiple axes (Fig. 4, C and D, b and c, h and i, and t and u). IGF1 stimulation resulted in an increase in NRVM size, and this was associated with the formation of mature myofibers (increase in the organization of actin myofibrils as assessed by phalloidin staining, with striated sarcomeres running in multiple axes; Fig. 4, C and D, d, j, and v). The PI3K(p110α) inhibitors prevented the formation of mature myofibers in response to IGF1 stimulation (Fig. 4, C and D, e, k, w, f, l, and x). Further, A66 at 10 μm was associated with a reduction in phalloidin staining under basal conditions and in the presence of IGF1 (Fig. 4D, i and l).
2) Reduced Z-disc alignment in dnPI3K hearts: Myofiber morphology was assessed in heart sections from adult Ntg, dnPI3K, and caPI3K by immunofluorescence. There were greater numbers of myofilaments and thicker myofibrils in caPI3K hearts compared with Ntg hearts (Fig. 5). In contrast, there were fewer myofilaments and thinner myofibrils in dnPI3K hearts compared with Ntg hearts (Fig. 5). Z-disc alignment (α-actinin stain) appeared reduced in dnPI3K hearts (Fig. 5). Phalloidin staining intensity of actin was markedly reduced in sections from dnPI3K heart (Fig. 5) and is consistent with a reduction in phalloidin staining observed in NRVM treated with A66 (Fig. 4D).
FIGURE 5.

Z-band alignment in myocytes from PI3K transgenic mice. Representative cross sections from ventricles of Ntg, caPI3K, and dnPI3K mice. Anti-αactinin (a–c) stains sarcomeric bands and Z-lines, phalloidin-FITC (d–f) stains contractile thin filaments, and DAPI (g–i) stains nuclei. j–l, merged images. Scale bar = 20 μm. n = 4 in each group.
DISCUSSION
The development of cardiomyopathy in humans and mouse models with mutations/defects in Z-disc proteins highlight the need to understand the molecular mechanisms that regulate Z-disc biology (2). A greater knowledge of the basic mechanisms responsible will be important for the development of improved therapeutic strategies for the heart. Z-disc proteins share “sticky” domains that are capable of mediating multiple protein-protein interactions and play an essential role in integrating structure and signaling in the complex three-dimensional network (2). Phosphatases and kinases, including calcineurin and protein kinase C, have been shown previously to reside at the Z-disc (2). However, whether PI3K(p110α) directly regulated cardiac structure and components of the Z-disc was unknown. The goal of this study was to examine whether PI3K(p110α) regulates structural components of cardiac muscle to protect the heart. We demonstrated previously that mice with increased PI3K activity (caPI3K) develop physiological cardiac hypertrophy, whereas mice with decreased PI3K activity have smaller hearts (14, 26). Cardiac function was normal in both models under basal/sham conditions, but under settings of stress (e.g. pressure overload, dilated cardiomyopathy, or MI), caPI3K mice were protected compared with Ntg, whereas dnPI3K mice developed heart failure more rapidly (4, 26–28). We hypothesized that applying the VMus3D model to a robust gene data set from well defined models (i.e. genetic models with reciprocal regulation of PI3K in control (sham) and stress settings (MI)) might enable us to uncover reciprocal expression profiles of key elements of the Z-disc that predispose how the heart responds to a subsequent cardiac insult such as MI.
Functional Consequences of Changes in PI3K-regulated Transcripts Encoding Costamere and Z-disc Proteins
Under basal/sham conditions, genes encoding muscle structural/associated proteins that were up-regulated in hearts of caPI3K mice and down-regulated in hearts of dnPI3K mice included dystroglycan (Dag1), filamin C (Flnc), ankyrin repeat domain 23 (Ankrd23/Darp), Rock2, crystallin α B, Cd151, melusin, cypher, and synaptopodin 2 (Synpo2, Myopodin) (Fig. 6). Thus, here we have observed a super-response of muscle structural/mechanosensors in mice with increased activity of PI3K(p110α) i.e. caPI3K. There is compelling evidence to demonstrate that altered expression of many of these genes would have a significant impact on functionality of the Z-disc and, subsequently, cardiac function. Dag1 links dystrophin to the extracellular matrix. Cardiomyocyte-specific deletion of Dag1 in mice led to dilated cardiomyopathy that was associated with depressed cardiac function (29). Flnc is a member of the filamin family, expressed in heart and skeletal muscle (30). Filamins stabilize actin filaments and link them to the cell membrane. Filamin mutations in humans have been associated with muscle dysfunction (30). Furthermore, Flnc was identified as a substrate of PI3K activated Akt in mouse heart (31). Ankrd23 belongs to the muscle ankyrin repeat protein (MARP) family and is a structural/regulatory signaling protein that interacts with titin (32). In skeletal muscle, the muscle ankyrin repeat protein family was shown to be protective against eccentric contraction-induced injury (33), suggesting that it plays an important role in maintaining normal structure and function. Finally, cypher is a cytoskeletal protein localized in the sarcomeric Z-disc that was shown to be essential for cardiac sarcomeric structure/stability. Mice with cardiac-specific deletion of cypher or global cypher knockout mice displayed severe cardiac defects and heart failure (34, 35).
FIGURE 6.

PI3K-regulated components of the costamere and Z-disc. Model highlighting components of the costamere and Z-disc that are regulated by PI3K. Expression of genes that were up-regulated in caPI3K hearts and down-regulated in dnPI3K hearts are highlighted in yellow. The model also highlights the interaction between PI3K and melusin in the heart.
Novel Interaction between PI3K and Melusin in the Adult Heart
The reciprocal regulation of melusin in the caPI3K and dnPI3K transgenic mice was of particular interest because melusin-null (16) and melusin-overexpressing transgenic mice (17) display similar phenotypes as dnPI3K and caPI3K transgenic mice, respectively. Both dnPI3K mice and melusin-null mice displayed normal cardiac structure and function under basal conditions but developed dilated cardiomyopathy in response to pressure overload (16, 26, 27). In contrast, melusin-overexpressing and caPI3K transgenic mice displayed a mild physiological hypertrophic phenotype associated with an increase in myocyte size, Akt phosphorylation, and normal cardiac function (14, 17, 27). Further, both models (melusin-overexpressing and caPI3K transgenics) were protected in a setting of pressure overload (17, 27). Collectively, these similarities prompted us to assess whether PI3K and melusin form a complex together in the heart. To our knowledge, we have shown for the first time that the regulatory subunit of PI3K, i.e. p85, directly interacts with melusin in the adult heart and generates lipid kinase activity.
The IGF1-PI3K(p110α) Pathway Is an Essential Regulator of Cardiac Structure
Previous studies have demonstrated that G-protein-coupled receptors, including the angiotensin II type 1 receptor, can sense biomechanical stress and regulate components of the Z-disc (2). It has also been shown that cyclic stretch of NRVM stimulated secretion of IGF1 (36) and that treatment of NRVM with IGF1 increased mRNA expression and translation of contractile proteins (6). It was previously unclear whether the IGF1-PI3K(p110α) axis played a critical role in regulating components of the cardiac Z-disc. In this study, a gene enrichment analysis highlighted overrepresentation of components of the IGF1 pathway. Gene and protein expression levels of IGF1R and IRS1 were increased in hearts of Ntg in a setting of MI compared with sham. Given that the IGF1R pathway plays a critical role in mediating cardiac protection (5), we speculate that IGF1R and IRS1 increase in response to stress (e.g. MI) as a mechanism for protecting the heart. IGF1R and IRS1 expression levels were lower in caPI3K sham hearts and higher in dnPI3K sham hearts. Further, any increases in expression of IGF1R/IRS1 in a setting of MI were smaller or absent in the PI3K transgenic mice. We hypothesize that in a heart susceptible to stress (i.e. dnPI3K sham), the heart attempts to compensate by increasing IGF1R signaling, but because PI3K is a critical effector of the IGF1R pathway, the dnPI3K transgene prevents an increase in structural components and Z-disc signaling that would otherwise provide protection. By contrast, it is not necessary for IGF1R signaling to be elevated in the caPI3K heart because structural components are enhanced already. Our VMus3D model highlighted distinct reciprocal regulation of structural components of the costamere and Z-disc that support this hypothesis, i.e. up-regulation in caPI3K and down-regulation in dnPI3K.
To ascertain whether PI3K(p110α) had a direct impact on cardiac structure, we used two independent approaches. First, we examined the impact of two PI3K(p110α) inhibitors on the structure of NRVM under basal conditions and in the presence of IGF1. Second, we examined the structure of cardiac sections from caPI3K and dnPI3K mice in comparison to Ntg. Both studies demonstrated that PI3K(p110α) has an impact on myofiber morphology and is critical for Z-disc alignment. PI3K(p110α) inhibitor-treated NRVM (without stimulation) displayed sarcomeres that typically ran in a longitudinal axis rather than multiple axes. IGF1 treatment was associated with the formation of mature myofibers, which was prevented in the presence of the PI3K(p110α) inhibitors. caPI3K hearts contained greater numbers of myofilaments and thicker myofibrils than Ntg, whereas dnPI3K hearts contained fewer myofilaments and thinner myofibrils. These findings are consistent with the development of physiological hypertrophy in caPI3K transgenic mice and the small heart phenotype of dnPI3K transgenic mice (14). Z-disc alignment was reduced in dnPI3K hearts, and phalloidin staining intensity of actin was markedly reduced in sections from dnPI3K hearts. This finding is consistent with reduced phalloidin staining of actin in NRVM pretreated with A66. Decreased phalloidin staining in dnPI3K mice is considered reflective of dysregulated sarcomere morphology, as opposed to these mice lacking actin filaments in their hearts, because there was no evidence of differentially expressed genes representative of the actin family in dnPI3K hearts. Collectively, the above findings suggest a defect in myofibril formation and Z-disc alignment in myocytes with reduced PI3K activity.
Targeting the PI3K Pathway with Caution: Heart Failure versus Cancer
Manipulating components of the PI3K pathway has been recognized as the most active drug development in the cancer field (37). PI3K is commonly constitutively activated because of a mutation in many cancers (38). Our previous work demonstrated that dnPI3K mice were more susceptible to the development of atrial fibrillation and/or heart failure than control mice when exposed to pathological insults, including pressure overload (26, 27), dilated cardiomyopathy (28), and MI (4). These findings raise potential concerns for the effects of PI3K inhibitors on the hearts of cancer patients. The very distinct effects of PI3K activation on components of the costamere and Z-disc under basal conditions (sham) in this study provide important new insights as to why inhibition of PI3K makes the heart susceptible to a cardiac insult and highlights that caution must be taken when considering the administration of PI3K inhibitors to patients.
In summary, understanding the very complex process of mechanotransduction in cardiomyocytes is of utmost importance to our knowledge of how the heart responds in a setting of stress and will be critical for the development of more effective treatment strategies for heart failure. Here we have used a novel approach to visualize genes expressing proteins that form part of the cardiac contractile apparatus that are regulated by PI3K. To our knowledge, we have shown for the first time that PI3K can directly affect components of the costamere and Z-disc, PI3K complexes with melusin in the heart, and that PI3K has a direct impact on cardiac myofiber maturation and Z-disc alignment. Collectively, our data suggest that activation of PI3K is a key mechanism that enables structural remodeling of the costamere and Z-disc to protect the heart in a setting of stress.
Supplementary Material
This work was supported by postgraduate scholarships from Griffith University and the Commonwealth Scientific and Industrial Research Organisation Food Futures National Research Flagship (to A. J. W.) and by National Health and Medical Research Council (NHMRC) Project Grant 367600 (to J. R. M.). This work was supported in part by the Victorian Government Operational Infrastructure Support Program. This work was also supported by Australian Research Council Future Fellowship FT0001657 (to J. R. M.), Honorary NHMRC Senior Research Fellowship 586604 (to J. R. M.), NHMRC Peter Doherty Fellowship 351012 (to R. C. Y. L.), University of New South Wales Vice Chancellor Research Fellowship (to R. C. Y. L.), and by an University of Melbourne Roper fellowship (to D. C. H. N.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables I–III and additional methods, equations, and references.
T. Galili, personal communication.
- ca
- constitutionally active
- MI
- myocardial infarction
- dn
- dominant negative
- IGF1
- insulin-like growth factor 1
- VMus3D
- three-dimensional virtual muscle model
- Ntg
- non-transgenic
- NRVM
- neonatal rat ventricular myocyte(s).
REFERENCES
- 1. Samarel A. M. (2005) Am. J. Physiol. Heart Circ. Physiol. 289, H2291–2301 [DOI] [PubMed] [Google Scholar]
- 2. Frank D., Frey N. (2011) J. Biol. Chem. 286, 9897–9904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bernardo B. C., Weeks K. L., Pretorius L., McMullen J. R. (2010) Pharmacol. Ther. 128, 191–227 [DOI] [PubMed] [Google Scholar]
- 4. Lin R. C., Weeks K. L., Gao X. M., Williams R. B., Bernardo B. C., Kiriazis H., Matthews V. B., Woodcock E. A., Bouwman R. D., Mollica J. P., Speirs H. J., Dawes I. W., Daly R. J., Shioi T., Izumo S., Febbraio M. A., Du X. J., McMullen J. R. (2010) Arterioscler. Thromb. Vasc. Biol. 30, 724–732 [DOI] [PubMed] [Google Scholar]
- 5. McMullen J. R. (2008) Clin. Exp. Pharmacol. Physiol. 35, 349–35418290876 [Google Scholar]
- 6. Ito H., Hiroe M., Hirata Y., Tsujino M., Adachi S., Shichiri M., Koike A., Nogami A., Marumo F. (1993) Circulation 87, 1715–1721 [DOI] [PubMed] [Google Scholar]
- 7. Waardenberg A. J., Reverter A., Wells C. A., Dalrymple B. P. (2008) BMC Syst. Biol. 2, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li C., Wong W. H. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 31–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nishiyama A., Kambe F., Kamiya K., Seo H., Toyama J. (1998) Cardiovasc. Res. 40, 343–351 [DOI] [PubMed] [Google Scholar]
- 10. Barnard G. A. (1947) Biometrika 34, 123–138 [DOI] [PubMed] [Google Scholar]
- 11. Deleted in proof.
- 12. Dennis G., Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A. (2003) Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 13. McMullen J. R., Shioi T., Huang W. Y., Zhang L., Tarnavski O., Bisping E., Schinke M., Kong S., Sherwood M. C., Brown J., Riggi L., Kang P. M., Izumo S. (2004) J. Biol. Chem. 279, 4782–4793 [DOI] [PubMed] [Google Scholar]
- 14. Shioi T., Kang P. M., Douglas P. S., Hampe J., Yballe C. M., Lawitts J., Cantley L. C., Izumo S. (2000) EMBO J. 19, 2537–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sbroggiò M., Ferretti R., Percivalle E., Gutkowska M., Zylicz A., Michowski W., Kuznicki J., Accornero F., Pacchioni B., Lanfranchi G., Hamm J., Turco E., Silengo L., Tarone G., Brancaccio M. (2008) FEBS Lett. 582, 1788–1794 [DOI] [PubMed] [Google Scholar]
- 16. Brancaccio M., Fratta L., Notte A., Hirsch E., Poulet R., Guazzone S., De Acetis M., Vecchione C., Marino G., Altruda F., Silengo L., Tarone G., Lembo G. (2003) Nat. Med. 9, 68–75 [DOI] [PubMed] [Google Scholar]
- 17. De Acetis M., Notte A., Accornero F., Selvetella G., Brancaccio M., Vecchione C., Sbroggiò M., Collino F., Pacchioni B., Lanfranchi G., Aretini A., Ferretti R., Maffei A., Altruda F., Silengo L., Tarone G., Lembo G. (2005) Circ. Res. 96, 1087–1094 [DOI] [PubMed] [Google Scholar]
- 18. Ng D. C., Ng I. H., Yeap Y. Y., Badrian B., Tsoutsman T., McMullen J. R., Semsarian C., Bogoyevitch M. A. (2011) J. Biol. Chem. 286, 1576–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knight Z. A., Gonzalez B., Feldman M. E., Zunder E. R., Goldenberg D. D., Williams O., Loewith R., Stokoe D., Balla A., Toth B., Balla T., Weiss W. A., Williams R. L., Shokat K. M. (2006) Cell 125, 733–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun M., Hillmann P., Hofmann B. T., Hart J. R., Vogt P. K. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 15547–15552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jamieson S. M., Flanagan J. U., Kolekar S., Buchanan C., Kendall J. A., Lee W. J., Rewcastle G. W., Denny W. A., Singh R., Dickson J., Baguley B., Shepherd P. R. (2011) Biochem. J. 438, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ng D. C., Gebski B. L., Grounds M. D., Bogoyevitch M. A. (2008) Cell Motil. Cytoskeleton 65, 40–58 [DOI] [PubMed] [Google Scholar]
- 23. Brancaccio M., Guazzone S., Menini N., Sibona E., Hirsch E., De Andrea M., Rocchi M., Altruda F., Tarone G., Silengo L. (1999) J. Biol. Chem. 274, 29282–29288 [DOI] [PubMed] [Google Scholar]
- 24. Vanhaesebroeck B., Leevers S. J., Panayotou G., Waterfield M. D. (1997) Trends Biochem. Sci. 22, 267–272 [DOI] [PubMed] [Google Scholar]
- 25. Chaussade C., Rewcastle G. W., Kendall J. D., Denny W. A., Cho K., Grønning L. M., Chong M. L., Anagnostou S. H., Jackson S. P., Daniele N., Shepherd P. R. (2007) Biochem. J. 404, 449–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McMullen J. R., Shioi T., Zhang L., Tarnavski O., Sherwood M. C., Kang P. M., Izumo S. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 12355–12360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McMullen J. R., Amirahmadi F., Woodcock E. A., Schinke-Braun M., Bouwman R. D., Hewitt K. A., Mollica J. P., Zhang L., Zhang Y., Shioi T., Buerger A., Izumo S., Jay P. Y., Jennings G. L. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 612–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pretorius L., Du X. J., Woodcock E. A., Kiriazis H., Lin R. C., Marasco S., Medcalf R. L., Ming Z., Head G. A., Tan J. W., Cemerlang N., Sadoshima J., Shioi T., Izumo S., Lukoshkova E. V., Dart A. M., Jennings G. L., McMullen J. R. (2009) Am. J. Pathol. 175, 998–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Michele D. E., Kabaeva Z., Davis S. L., Weiss R. M., Campbell K. P. (2009) Circ. Res. 105, 984–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou A. X., Hartwig J. H., Akyürek L. M. (2010) Trends Cell Biol. 20, 113–123 [DOI] [PubMed] [Google Scholar]
- 31. Murray J. T., Campbell D. G., Peggie M., Mora A., Alfonso M., Cohen P. (2004) Biochem. J. 384, 489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller M. K., Bang M. L., Witt C. C., Labeit D., Trombitas C., Watanabe K., Granzier H., McElhinny A. S., Gregorio C. C., Labeit S. (2003) J. Mol. Biol. 333, 951–964 [DOI] [PubMed] [Google Scholar]
- 33. Barash I. A., Bang M. L., Mathew L., Greaser M. L., Chen J., Lieber R. L. (2007) Am. J. Physiol. Cell Physiol. 293, C218–227 [DOI] [PubMed] [Google Scholar]
- 34. Zheng M., Cheng H., Li X., Zhang J., Cui L., Ouyang K., Han L., Zhao T., Gu Y., Dalton N. D., Bang M. L., Peterson K. L., Chen J. (2009) Hum. Mol. Genet. 18, 701–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou Q., Chu P. H., Huang C., Cheng C. F., Martone M. E., Knoll G., Shelton G. D., Evans S., Chen J. (2001) J. Cell Biol. 155, 605–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shyu K. G., Ko W. H., Yang W. S., Wang B. W., Kuan P. (2005) Cardiovasc. Res. 68, 405–414 [DOI] [PubMed] [Google Scholar]
- 37. Cheng H., Force T. (2010) Circ. Res. 106, 21–34 [DOI] [PubMed] [Google Scholar]
- 38. McMullen J. R., Jay P. Y. (2007) Cell Cycle 6, 910–913 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.