

Abstract
Pendrin and prestin both belong to a distinct anion transporter family called solute carrier protein 26A, or SLC26A. Pendrin (SLC26A4) is a chloride-iodide transporter that is found at the luminal membrane of follicular cells in the thyroid gland as well as in the endolymphatic duct and sac of the inner ear, whereas prestin (SLC26A5) is expressed in the plasma membrane of cochlear outer hair cells and functions as a unique voltage-dependent motor. We recently identified a motif that is critical for the motor function of prestin. We questioned whether it was possible to create a chimeric pendrin protein with motor capability by integrating this motility motif from prestin. The chimeric pendrin was constructed by substituting residues 160–179 in human pendrin with residues 156–169 from gerbil prestin. Non-linear capacitance and somatic motility, two hallmarks representing prestin function, were measured from chimeric pendrin-transfected human embryonic kidney 293 cells using the voltage clamp technique and photodiode-based displacement measurement system. We showed that this 14-amino acid substitution from prestin was able to confer pendrin with voltage-dependent motor capability despite the amino acid sequence disparity between pendrin and prestin. The molecular mechanism that facilitates motor function appeared to be the same as prestin because the motor activity depended on the concentration of intracellular chloride and was blocked by salicylate treatment. Radioisotope-labeled formate uptake measurements showed that the chimeric pendrin protein retained the capability to transport formate, suggesting that the gain of motor function was not at the expense of its inherent transport capability. Thus, the engineered pendrin was capable of both transporting anions and generating force.
Keywords: Membrane Proteins, Molecular Motors, Neuroscience, Protein Motifs, Protein Structure, Hair Cells, Molecular Motor, Pendrin, Prestin, Transporter
Introduction
Pendrin, also known as sodium-independent chloride-iodide transporter, is found at the luminal membrane of follicular cells in the thyroid gland. It transports iodine from the cytoplasm to the follicle lumen, an activity that is necessary for production of thyroid hormone. Pendrin is also expressed in the endolymphatic duct and sac, in the utricle and saccule, and in the external sulcus of the inner ear (1). Impaired pendrin function promotes a progressive increase in endolymph volume followed by an enlargement of the membranous labyrinth and surrounding osseous structures, which leads to degeneration of inner ear sensory cells (2). Mutations in this gene (3–5) are associated with Pendred syndrome (6), which is an autosomal recessive disorder characterized by severe to profound bilateral sensorineural hearing impairment, vestibular dysfunction, temporal bone abnormalities, development of euthyroid goiter, and impaired iodide organification.
Pendrin belongs to a distinct anion transporter family called solute carrier protein 26A (SLC26A).2 11 members of this family have been identified so far (7). Individual members of this family serve two fundamentally distinct functions. Most members, including pendrin (SLC26A4), transport monovalent and divalent anions across a variety of epithelia. Prestin (SLC26A5), found in the plasma membrane of cochlear outer hair cells (8, 9), is the only member in this family that functions as a voltage-dependent motor with the capability to perform rapid electromechanical and mechanoelectrical conversion on a microsecond time scale (10, 11). Prestin-based somatic motility of cochlear outer hair cells is responsible for cochlear amplification in mammals (12–15).
Using a comparative and evolutionary approach (16) and consensus amino acid sequence analyses, we recently identified an amino acid motif within the sulfate transporter domain of prestin. This motif appears to constitute the critical structural/functional basis for the voltage-dependent motor capability of prestin. Amino acid substitution using this motif from gerbil prestin was able to convert zebrafish and chicken orthologs into voltage-dependent motors.3 Because pendrin is one of the paralogs of prestin and shares 40% amino acid homology with prestin, we questioned whether it was possible to create a modified pendrin protein that gains voltage-dependent motor function by adopting this motif from prestin. Because pendrin is an anion transporter, we also examined whether the gain of function was at the expense of its transport function. The goal was to determine whether the region being substituted in pendrin was critical to its transporter function. We show that substitution of 14 amino acids is able to confer prestin-like motor capability to pendrin. The gain of motor function is not at the expense of its inherent transport capability. Thus, this is the first engineered protein that can function as an anion transporter and a molecular motor.
EXPERIMENTAL PROCEDURES
Substitution of Amino Acid Residues of Pendrin with Amino Acid Residues from Prestin
Gerbil (Meriones unguiculatus) prestin (gPres) and human (Homo sapiens) pendrin (A4) were used for substitution of amino acids. The equivalent regions in gPres and A4 sequences were identified by amino acid sequence alignment using BLAST. To construct chimeric pendrin, A4(g14), residues 160–179 (HFLVSSSNGTVLNTTMIDTA) of A4 were replaced by residues 156–169 (IVIPGGVNATNGTE) from gPres (Fig. 1A). To do this, cDNAs of gPres and A4 were cloned into the pEGFP-N1 vector to generate the C-terminal EGFP-tagged fusion proteins. Unique restriction enzyme recognition sites, ScaI and HindIII, were identified in the native pendrin protein coding sequence. DNA synthesis (GenScript USA, Inc., Piscataway, NJ) was used to create the chimeric pendrin fragment and substituted into the human pendrin nucleotide sequence using the mentioned restriction sites. The correct orientation, reading frame, and amino acid composition of all the constructs were confirmed by sequence analyses. The unmodified A4 has 780 amino acids. After domain substitution with 14 amino acid residues from gPres, the chimeric pendrin, designated as A4(g14), has 774 amino acids (Fig. 1B).
FIGURE 1.

Consensus amino acid sequences of gPres and A4 and diagram of the construction of the chimeric pendrin containing the 14-amino acid motif from gPres. A, 14 amino acids from gPres were used to replace 20 amino acids in A4 to generate A4(g14). The amino acid residues being swapped between gPres and A4 are highlighted by the red box. Identical residues in the consensus sequences are shown in red. B, schematic drawings of the constructs of gPres, A4, and A4(g14). After substitution, A4(g14) has 774 amino acids (a.a.).
Site-directed mutagenesis was performed to mutate three amino acid residues (P159V, G160A, and G161A) in the motility motif of both gPres and A4(g14), using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). These two oligonucleotide sets were as follows: 1) gPres(m), forward (5′-TCGTCATCGTGGCAGGAGTGAACGCAACC-3′) and reverse (5′-GGTTGCGTTCACTCCTGCCACGATGACGATGTCATCG-3′); 2) A4(g14m), forward (5′-CGACGAAATCGTCATCGTGGCAGCAGTGAACGCAACCAACG-3′) and reverse (5′-CGTTGGTTGCGTTCACTGCTGCCACGATGACGATTTCGTCG-3′). The correct orientation, reading frame, and amino acid composition of all the constructs were confirmed by sequence analyses.
Cell Culture and Transient Transfection
Human embryonic kidney (HEK) 293 cells were cultured in DMEM solution (Invitrogen), supplemented with 10% fetal bovine serum. For electrophysiological studies, the cells were passaged into 35-mm dishes 24 h before transfection. A4 or its mutants (4 μg) were introduced into the dishes using 10 μl of Lipofectamine (Invitrogen) following the protocol from the manufacturer.
Nonlinear Capacitance (NLC) Measurements
NLC was measured from the HEK cells with clear membrane-associated EGFP expression 24–48 h after transfection. During whole-cell patch clamp recordings, cells were bathed in the extracellular solution containing (in mm): 120 NaCl, 2 MgCl2, 2 CoCl2, 20 triethanolamine, 10 HEPES, 10 4-aminopyridine. Recording pipettes were pulled from borosilicate glass with resistances between 2.5 and 5.0 MΩ and filled with a solution containing (in mm): 140 CsCl, 2 MgCl2, 10 EGTA, 10 HEPES. When intracellular Cl− needed to be replaced, the following intracellular medium was used for the solution in the patch electrode (in mm): 150 sodium pentane sulfonate, 10 HEPES, 1 EGTA. When 100 mm salicylate was applied to the bath solution through a perfusion pipette, the solution contained (in mm): 20 NaCl, 100 sodium salicylate, 2 MgCl2, 2 CoCl2, 20 triethanolamine, 10 HEPES, 10 4-AP. All solutions used in this study were adjusted to pH 7.35, 300–310 mosm. Data included in this study were from the cells whose membrane resistance, Rm, was greater than 300 MΩ after whole-cell configuration was established. Series resistance, ranging from 10 to 17 MΩ, was compensated off-line. The capacitive currents were sampled at 100 kHz and low-pass filtered at 5 kHz. The capacitive currents were amplified by Axopatch 200B (Molecular Devices). Membrane capacitance was measured using a two-sine-wave voltage stimulus protocol (10-mV peak at both 390.6 Hz and 781.2 Hz) with subsequent fast Fourier transform-based admittance analysis (18) from a holding potential of 0 mV. Data were acquired using jClamp (Scisoft, New Haven, CT) and analyzed with Igor (WaveMetrics, Portland, OR).
NLC can be described as the first derivative of a two-state Boltzmann function relating non-linear charge movement to voltage (19, 20). The capacitance function is described as
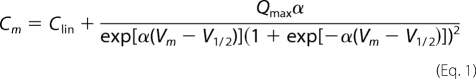 |
Four parameters (Qmax, V½, Clin, and z) were obtained from the equation, where Qmax is maximum the charge transfer, V½ is the voltage at which the maximum charge is equally distributed across the membrane, or equivalently, the peak of the voltage-dependent capacitance, Clin is linear capacitance, and α = ze/kT is the slope of the voltage dependence of charge transfer where k is the Boltzmann's constant, T is absolute temperature, z is the valence of the charge movement, and e is the electron charge. The linear capacitance, Clin, is proportional to the surface area of the membrane (the size of the cell). To compare the magnitude of NLC obtained from different cells with different sizes, we normalized NLC by Clin of the cells. For the same reason, Qmax was also normalized by Clin.
Somatic Motility Measurements
Voltage-dependent somatic motility of the transfected HEK 293 cells was measured and calibrated by a photodiode-based system (21, 22). A suction pipette or microchamber was used to mechanically hold the cell and to deliver voltage commands. Microchambers were fabricated from 1.5-mm thin-wall glass tubes (World Precision Instruments, Inc., Sarasota, FL). The microchamber had a series resistance of ∼0.3–0.4 MΩ. When the cell was 50% inserted into the pipette, the input resistance was 3–4 MΩ. The electrical stimulus was a 100-Hz sinusoidal voltage burst with 100 ms in duration. Voltage commands of 400 mV (peak-to-peak) were used. Because the cells were ∼50% inserted into the microchamber, the voltage drops on the extruded segment were estimated to be 50% of the voltage applied, or 200 mV (21). The photodiode system had a cutoff (3 db) frequency of 1100 Hz. The sampling frequency was 5 kHz. With an averaging of 200 trials and low-pass filtering set at 200 Hz, cellular motion as low as 5 nm could be detected. All the electrophysiological experiments were performed at the room temperature (20 ± 2 °C).
Transporter Function Assessment
To measure the transporter function of A4(g14), conventional radioisotope technique was used. 14C-Labeled formate (Moravek Biochemicals, Inc., Brea, CA) was used as the substrate as described in a previous study (23, 24). HEK cells transfected with A4, gPres, and A4(g14) were tested. Untransfected and EGFP-transfected HEK cells were used as negative controls. Fluorescence-activated flow cytometry was used to isolate EGFP-positive cells before transport capability was examined. For cell sorting, HEK cells were detached and separated with trypsin (Invitrogen) treatment 24 h after transfection. The cells were then centrifuged and resuspended in the DMEM solution with 10% FBS. Cytometry analysis and sorting were performed using a FACStar Plus (BD Biosciences) with a water-cooled argon laser tuned to 488 nm. Green fluorescence was detected with a 530/30-nm band-pass filter. Cell sorting was based on the fluorescence intensity of the transfected cells after calibration using normal untreated HEK cells. Cells with fluorescence intensity 4-fold greater than the non-fluorescent cells were collected. Data acquisition and analysis were performed with the CellQuest software. 600,000 events (cells) were collected for each sample. The collected cells were resuspended in culture medium and allowed to attach to culture dishes for 6 h. The floating dead cells were removed by changing the medium, and the remaining (attached) cells were detached and replated into a 24-well plate after cell counting with a hemacytometer. The cells were cultured for another 42 h to ensure firm attachment before the formate uptake was measured.
To measure [14C]formate uptake, cells in the 24-well culture cluster were first incubated for 30 min in the solution containing (in mm): 130 NaCl, 20 HEPES, 5 KCl, 5 glucose, 2 CaCl2, and 1 MgCl2 (pH 7.3 and 305 osm/liter). Cells were then incubated in room temperature for 12 min in the solution containing (in mm): 140 potassium gluconate, 20 HEPES, and 5 glucose (pH 7.3 and 305 osm/liter). [14C]Formate was added in this solution with a concentration of 20 μm. Cells were then washed three times with cold potassium gluconate solution without [14C]formate. Cells were lysed with 200 μl of 0.5 m NaOH and neutralized with 0.5 m HCl. The lysate was used for the liquid scintillation counting to determine the [14C]formate uptake. The concentration (in pmol) of the 14C-labeled formate was determined from each well estimated to contain ∼200,000 cells. In each run, three wells were used and assayed for each plasmid. The experiments were repeated in three separate runs. Therefore, the data in each group were collected from a sample size (n) of 3 × 3 = 9 trials for each plasmid and control.
RESULTS
Gain of NLC in the Chimeric Pendrin
Membrane targeting is essential for the expression of function of a membrane-bound protein. It has been demonstrated that gPres and A4 show robust membrane-targeted expression when expressed in the HEK 293 cells.3 To demonstrate that A4(g14) was expressed in the membrane, we examined the expression of EGFP-tagged A4(g14) and a membrane-based dye, di-8-amino-naphthyl-ethenyl-pyridinium (di-8-ANEPPS), using confocal microscopy (Zeiss LSM 510, Thornwood, NY). Di-8-ANEPPS is a member of the ANEP (aminoaphthyl ethenyl pyridinium) class of membrane potential dyes. Confocal images taken from transfected HEK cells substantiated that the EGFP-tagged gPres, A4 and A4(g14), and the di-8-ANEPPS were co-localized in the membrane (Fig. 2A). Only the cells with strong cell surface EGFP expression were selected for the experiments. To minimize variation of expression levels among cells transfected with different constructs, we selected cells with similar EGFP intensities in the membrane for the electrophysiological experiments described below.
FIGURE 2.

Heterogenic expression and NLC measured from gPres, A4, and A4(g14). A, representative confocal images of attached and detected HEK cells transfected by A4 and A4(g14) with EGFP tag (in green). The membrane of transfected cells was labeled by di-8-ANEPPS (di-8, in red), a membrane-bound dye. Co-localization of fluorescent signals indicates the membrane-associated expression of the proteins (merge, orange). Bar: 10 μm. B, examples (color-coded thin lines) of NLC obtained from HEK cells transfected with gPres, A4, and A4(g14), respectively. The capacitance-voltage responses were fitted with the Boltzmann function (color-coded heavy lines). Clin was subtracted. pF, picofarads. C, four parameters derived from curve fittings with Boltzmann's function. Data are expressed as mean ± S.D. n = 18, 13, and 14 for gPres, A4, and A4(g14), respectively. *, p < 0.05, **, p < 0.01 (Student's t test).
The voltage-sensing and motor functions of mammalian prestin are manifested by two hallmarks: NLC and electromotility. NLC and electromotility are fully coupled (19, 20, 25, 26) and can be characterized by a simple two-state Boltzmann function with indistinguishable fitted parameters between the two (25). Because NLC can be easily and accurately measured experimentally, it is often used to characterize prestin function (10). NLC is characterized by a bell-shaped dependence on membrane potential with a peak between −80 and −20 mV in outer hair cells (20) and in prestin-transfected cell lines (8, 27). We measured NLC from gPres-, A4-, and A4(g14)-transfected cells. NLC was also measured from EGFP-expressing cells, transfected with the EGFP plasmid, as a negative control. 24 cells from EGFP-expressing cells were measured, and an example of the lack of NLC (a flat response) is depicted in Fig. 2B (black line). In contrast, gPres-expressing cells showed a robust bell-shaped capacitance-voltage relation with a peak capacitance of about 1.2 picofarads near −67 mV (Fig. 2B, thin red line). Four parameters (Qmax, Clin, V½, and z) were obtained from curve-fitting of NLC response using the first derivative of the Boltzmann function (heavy red line). Qmax is the maximum charge transfer, Clin is the linear capacitance, V½ is the voltage at which the maximum charge is equally distributed across the membrane, or equivalently, the peak of the voltage-dependent capacitance, and z is the valence of the charge movement. Because HEK cells varied in sizes that were reflected by the Clin value, we normalized NLC and Qmax obtained from each cell by its Clin. The normalized mean values and standard deviations of the four parameters from the 18 gPres-expressing cells are plotted in Fig. 2C. Using the identical voltage protocol, we measured NLC from 13 A4-transfected cells. A small NLC response with the peak near −46 mV was observed (Fig. 2B, blue lines). Parameters obtained from curve-fitting showed that the Qmax/Clin, NLC/Clin, and z value were all significantly less than those of gPres (Fig. 2C). In contrast, when NLC from A4(g14)-expressing cells was measured, a significant gain of magnitude of NLC was observed (Fig. 2B, green lines). Accompanying the ∼300% gain in magnitude was the shift of the voltage-dependent curve toward the positive potential with reference to the curve of A4. 14 A4(g14)-expressing cells were measured, and Student's t test showed that the Qmax/Clin, NLC/Clin, and z values were all significantly greater (p < 0.01) than those of A4 (Fig. 2C).
Intracellular anions, such as Cl−, play a critical role in voltage-dependent motor activity of prestin (27). Removal of intracellular Cl− eliminates or reduces NLC, motility, and voltage-dependent stiffness of outer hair cells (27–31). We examined whether NLC of A4(g14) was also affected by the removal/reduction of intracellular Cl−, as observed with prestin. A tracking procedure was used to continuously monitor the peak capacitance (Cm) after obtaining whole-cell configuration (32). With this method, the reduction in NLC and the time course of the change were monitored. Fig. 3A shows an example from such recording. As shown in the inset, a substantial reduction of Cm was observed within the first 10 s after the membrane was ruptured and the intracellular Cl− began to be replaced by pentane sulfonate in the patch electrode. The Cm value stabilized near 1 min. We measured NLC from six A4(g14)-expressing and nine gPres-expressing cells at 2 min after membrane rupture. As Fig. 3A shows, the magnitude of NLC of either gPres-expressing or A4(g14)-expressing cells was significantly reduced when the intracellular Cl− concentration was reduced/replaced. The reduction of NLC was accompanied by a shift of voltage dependence toward more negative potentials. Although the residual NLC response seen in A4(g14) and gPres might be due to incomplete wash of intracellular Cl− or from transport activity, the drastic reduction in magnitude suggests that NLC seen in A4(g14) was sensitive to intracellular Cl− concentration.
FIGURE 3.

Changes of NLC after removal of intracellular Cl−, application of salicylate, and mutations. A, examples of NLC obtained from gPres- and A4(g14)-expressing cells before and after intracellular Cl− was replaced by pentane sulfonate. The inset shows the time course of the change of peak membrane capacitance (Cm) of an A4(g14)-expressing cell after the membrane was ruptured. B, examples of NLC recorded before, during, and after (washout) 100 mm salicylate perfusion. pF, picofarads. C, effects of P-V/G-A mutations on NLC of gPres and A4(g14). In A–C, heavy lines represent curve fitting using the Boltzmann function. D, NLC parameters of gPres (n = 18), gPres(m) (n = 7), A4(g14) (n = 14), and A4(g14m) (n = 8) derived from curve fittings. Data are expressed as mean ± S.D.; **, p < 0.01 (Student's t test).
Salicylate, one of the most widely used drugs, is known to induce reversible reduction of NLC and motility in outer hair cells (32–34). Salicylate is thought to directly interact with the voltage sensor/motor (27, 32). We examined NLC change of A4(g14) in response to salicylate treatment. 100 mm salicylate was perfused to the cell through a pipette (with a tip diameter of 5–10 μm) positioned 20 μm away from the cell. As in the example shown in Fig. 3B, salicylate perfusion induced significant reduction of NLC. The reduction in NLC was reversible after salicylate was washed out in the bath.
Comparative and evolutionary-based analyses of the consensus sequence alignments of prestin and its orthologs show that non-mammalian orthologs lack a prolyl residue and have only one glycyl residue in the equivalent region (16). A4 also lacks a prolyl residue and two glycyl residues in the equivalent region (Fig. 1A). A prolyl residue often acts as a structural disruptor in the middle of regular secondary structure elements such as α-helices and β-sheets, whereas a glycyl residue is more flexible than other amino acids. However, the combined Pro-Gly residues might also contribute to an α-helical structure (35). We questioned whether mutation of prolyl and glycyl resides in the motif would affect the magnitude and voltage dependence of NLC in A4(g14) and gPres. Substitutions were made to replace Pro with Val and the adjacent two Gly residues with Ala. The prolyl and glycyl residues and their substitutions (Val and Ala) all have non-polar and electrically neutral side chains. The mutant proteins, designated as gPres(m) and A4(g14m), exhibited robust membrane targeting 24 h after transfection. We measured NLC from seven gPres(m) cells and eight A4(g14m) cells. Examples of such recordings are presented in Fig. 3C. Four parameters related with NLC are presented in Fig. 3D. For either mutated protein, the magnitude of NLC was not significantly different from the unmodified counterpart (p > 0.05 for either case). However, there was an ∼20-mV shift of V½ toward the negative voltage for both mutated proteins. The shift was statistically significant (p < 0.01 for both gPres(m) and A4(g14m) proteins).
Gain of Motor Function in the Modified Pendrin
Substitution of the motif from gPres conferred A4(g14) with significantly augmented NLC. We questioned whether the gain of NLC was accompanied by a concurrent gain of motor capability. We measured electromotility of A4(g14)-transfected HEK cells using the microchamber technique (8, 21, 36). Because a spherical cell cannot effectively change its surface area, we produced a dumbbell-shaped cell by drawing the cell into the suction pipette or the microchamber (Fig. 4A). An estimated voltage of 200 mV (peak-to-peak) was applied to the extruded membrane segment. This voltage magnitude was large enough to generate a saturated response because the resting membrane potential of the A4(g14)-transfected HEK cells was between −10 and 0 mV, and the V½ was between −70 and −20 mV. When the voltage stimulation was applied to the extruded segment of the gPres-transfected HEK cells, a large cycle-by-cycle response following the voltage stimulation was seen (Fig. 4B). We measured motility from eight gPres-transfected cells. The response magnitude was 85 ± 27 nm. In contrast, none of the 12 A4-transfected cells measured exhibited any time-registered responses with the system resolution of 5 nm. Interestingly, 11 A4(g14)-transfected HEK cells displayed unambiguous motility (supplemental Video 1) when the cells were stimulated with the same voltage protocol. The motility magnitude was 51 ± 19 nm, significantly less (p < 0.01) than the magnitude of 85 ± 27 nm (n = 8) seen in the gPres-transfected cells. However, this was consistent with the observation that the magnitude of NLC of A4(g14)-expressing cells was significantly less than the cells with gPres expression. Nevertheless, substitution of 14 amino acids from gPres clearly rendered A4 with motor capability.
FIGURE 4.

Electromotility measurements of gPres, A4, and A4(g14), respectively. A, measurement of somatic motility of the HEK cell using the microchamber technique. The cell was about 50% inserted into the microchamber, and the length change of the extruded segment was measured in response to the voltage waveform shown in the bottom panel of B. B, examples of motility responses measured from gPres-, A4-, and A4(g14)-transfected HEK cells. EGFP-transfected cells were used as the negative control.
Transport Capability of the Modified Pendrin Was Retained
Pendrin is a transporter that mediates the exchange of anions (Cl−, HCO3−, OH−, I−, or formate) across the plasma membrane (7, 37), whereas mammalian prestin is a molecular motor with diminished transport capability (24, 27). We questioned whether the region being replaced in A4 was structurally and functionally important to its transport function. In other words, was the gain of motor function of A4(g14) at the expense of transport function? We measured anion transport activity of A4 and A4(g14) by using radioactively labeled formate uptake assays (23, 24) after EGFP-positive cells were sorted out by fluorescence-activated flow cytometry. A4-transfected cells were used as a positive control, whereas cells transfected with the pEGFP plasmid only were used as a negative control. Formate uptake by untreated (non-transfected) cells was also measured to rule out a possible membrane permeability change after Lipofectamine treatment. EGFP-transfected cells had a formate uptake of about 81 ± 12 pmol/∼200,000 cells (Fig. 5). A similar number (∼200,000) of untreated cells showed similar uptake levels.
FIGURE 5.

Transport activity of gPres, A4, and A4(g14), respectively. 14C-Labeled format uptake was measured after cell sorting using flow cytometry. Untransfected and EGFP vector-transfected cells were used as controls. Data are expressed as mean ± S.D.
Cells transfected with A4 showed a formate uptake of 239 ± 8 pmol/∼200,000 cells. This was a 295% increase over the background level (Fig. 5). The formate uptake of A4(g14)-transfected cells was 234 ± 21 pmol, an increase of ∼289% from the background. No statistical significance (p > 0.05) was found between them. To verify that the formate uptake observed in A4 and A4(g14) was not an artifact due to a nonspecific change in membrane permeability, an anion transporter blocker, DIDS, was added to the solution (23). As shown, formate uptake was significantly reduced across all the groups. Comparison between A4- and A4(g14)-transfected groups revealed no statistical difference (p > 0.05), suggesting that DIDS inhibition was equally effective to A4 and A4(g14).
DISCUSSION
We showed that substitution of a span of 14 amino acids from gerbil prestin was able to confer pendrin, a chloride-iodide transporter, with motor function. This is the first time, to our knowledge, that it was demonstrated that a minor modification in amino acids can render a transport protein with motor capability. This is astonishing because of the significant disparity in the amino acid context within which it was placed. If prestin comprises two conceptually essential functional domains, the voltage sensor and the actuator, concurrent acquisition of NLC and motility indicates that the substituted sequence contains the same structural determinant for the these two functional domains. We noted that the magnitude of NLC and motility of A4(g14) was still significantly less than that of gPres. This is not surprising because the primary sequences of pendrin and prestin are sufficiently different. It is conceivable that the full expression of motor function would require additional regions and/or structures in the molecule to work together.
The motor activity of the modified pendrin appears to be based on the same mechanism(s) as that of prestin because NLC of A4(g14) was sensitive to removal of intracellular Cl− and could be blocked by salicylate, which may compete for the Cl− binding site in the molecule (27). Presently, there is no crystallographic structure information available for any member of the SLC26A proteins. However, based on the 12- or 10-transmembrane loop model (38, 39), this motif is located in the second external loop or in the intracellular loop. It would be quite unusual for the motif to be located outside of the transmembrane domain. It is well recognized that the charged residue(s) serving as voltage sensor for voltage-dependent channels are usually located within the transmembrane domain. If this domain is present in the extracellular or intracellular loop, this unusual location could be consistent with the notion that prestin does not have an intrinsic voltage sensor (27). The membrane topology of pendrin is quite controversial (37). In the 12-loop model where N and C termini are both located in the intracellular side, the substituted segment would be in the second extracellular loop. Regardless of what the topology of pendrin is and where the substituted segment is located, one thing that is quite certain is that substitution and reduction of the total amino acids from 780 to 774 did not affect the structural integrity and inherent transport function of pendrin. The fact that the gain of motor function is not accompanied by a loss of transport function suggests that the amino acids and the region being replaced in pendrin are not critical for its transport function. The implication is that the transport and motor activities are independent, and each has its own structural bases in the chimeric pendrin. This also supports the notion that the motor and transporter mechanisms may not have to be mechanistically related, although prestin and pendrin evolved from a common ancestral gene (16).
Some recent evidence suggests that prestin can transport anions (23) and/or is an electrogenic transporter of bicarbonate and chloride ions (40). Consensus sequence analysis shows that prestin and pendrin have the highest homology in the sulfate transporter region. The two proteins differ most in the regions at the N and C termini, especially in the two charge clusters in the C terminus (8). In the substituted region, pendrin and prestin differ significantly in the composition and number of amino acids (Fig. 1). Pendrin lacks one prolyl and two glycyl residues and contains a span of 17 amino acids for the equivalent 11-amino acid (IPGGVNATNGT) motif in prestin (Fig. 1A). The lack of motor capability in pendrin and prestin orthologs suggests that the composition and number of amino acid residues in this region are important for motor function. It appears that prolyl and glycyl residues impart some alterations in NLC properties and thus likely have a minor impact on the structure and/or function of prestin and A4(g14). This was demonstrated by the substitution of prolyl and glycyl residues with valyl and alanyl residues, which did not affect the membrane expression or the magnitude of NLC, although a shift of voltage dependence occurred for both mutant proteins (Fig. 3C). We should point out that prolyl, glycyl, valyl, and alanyl residues all have non-polar and neutral side chains, whereas the size of their side chains differs. Thus, it is not entirely surprising that mutations of prolyl and glycyl residues did not reduce or abolish NLC.
In all previous studies, loss or reduction of NLC after amino acid mutations was used as a criterion to search for amino acids that are critical for the voltage sensor and for the roles of C and N termini (27, 31, 39, 41, 42). However, loss of function after mutation may not be an unequivocal demonstration of the role of the amino acid(s) for a specific function. For example, mutation in amino acid residue Cys-499 resulted in a significant reduction of NLC and somatic motility (15). It is unclear, however, why mutation of this single amino acid can cause reduction of NLC and motility. On the contrary, we measured gain of function, which can occur only if that segment is responsible for the function. Questions may be raised about whether the gain of function was due to improved protein expression in the membrane after modification. We, in fact, noticed that unmodified A4 appeared to have more expression in the membrane than A4(g14) based on the level of EGFP intensity in the membrane. More importantly, the gain seen in A4(g14) was accompanied by a significant shift in voltage dependence (Fig. 2). An increase in the membrane expression level alone would not cause the shift of voltage dependence (43). Furthermore, the gain of NLC was accompanied by concurrent gain of somatic motility. Collectively, all these data suggest that the gain of function does not originate simply from an increase in the amount of protein expression in the membrane and that fundamental changes must have occurred in the modified molecule that confer it with new capability. We should point out that currently, there is no technically feasible and reliable way to quantify the membrane expression at the individual cell level.
A recent study by Schaechinger et al. (44) showed that a substitution of a fragment of 54 amino acids from rat prestin can render zebrafish prestin with motor capability. This fragment is located between amino acid residues 86 and 140, a region that encompasses the sulfate transport motif (8). The amino acid composition in this fragment is highly similar between rat prestin and zebrafish prestin, with a difference of only seven amino acid residues that are scattered along the length of this peptide fragment. It is difficult to explain why this difference could turn zebrafish prestin into a mammalian prestin-like motor. The motif we identified is not located in the same region as that reported by Schaechinger et al. (44). Although it is difficult to reconcile the two findings, it is possible that there may be several areas in the prestin molecule that are important for electromechanical conversion.
Prestin is the only protein identified that can perform rapid electromechanical and mechanoelectrical conversion on a microsecond time scale. The force generated by one prestin molecule is on the same order of magnitude as a myosin motor (8). Its piezoelectricity is 4 orders of magnitude greater than the best man-made material (17, 29). We showed that a minor change in amino acid sequence allowed pendrin to gain NLC and motility, the two hallmarks for the prestin motor. Although it represents the first of several required steps toward mechanistic understanding of how prestin works, it is also the first engineered pendrin that can function as an anion transporter and a voltage-dependent motor (force generator). This may also represent an important first step for modification and eventual synthesis of prestin and pendrin for a variety of potential applications in protein chemistry, nanotechnology, and medicine in the future.
Supplementary Material
Acknowledgments
We thank Dr. Garrett Soukup and Dr. Greg Perry for assistance in radioisotope experiments and flow cytometry and Qian Zhang and Shuping Jia for some technical assistance. We thank Dr. Frank Dowd for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health NIH Grants R01 DC008649 (to K. B.) and R01 DC004696 (to D. H.) from the NIDCD. The Integrated Biological Imaging Facility, supported by the Creighton University Medical School, was constructed with support from C06 Grant RR17417-01 from the NCRR, National Institutes of Health.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Video 1.
X. Tan, J. L. Pecka, J. Tang, O. E. Okoruwa, S. Lovas, D. Z. He, and K. W. Beisel, manuscript in preparation.
- SLC26A
- solute carrier protein 26A
- gPres
- gerbil prestin
- A4
- human pendrin
- NLC
- nonlinear capacitance
- MΩ
- megaohms
- di-8-ANEPPS
- di-8-amino-naphthyl-ethenyl-pyridinium
- DIDS
- 4,4-diisothiocyanatostilbene-2,2′-disulfonic acid
- EGFP
- enhanced green fluorescent protein.
REFERENCES
- 1. Everett L. A., Morsli H., Wu D. K., Green E. D. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 9727–9732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Choi B. Y., Madeo A. C., King K. A., Zalewski C. K., Pryor S. P., Muskett J. A., Nance W. E., Butman J. A., Brewer C. C., Griffith A. J. (2009) J. Med. Genet. 46, 856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sheffield V. C., Kraiem Z., Beck J. C., Nishimura D., Stone E. M., Salameh M., Sadeh O., Glaser B. (1996) Nat. Genet. 12, 424–426 [DOI] [PubMed] [Google Scholar]
- 4. Coyle B., Coffey R., Armour J. A., Gausden E., Hochberg Z., Grossman A., Britton K., Pembrey M., Reardon W., Trembath R. (1996) Nat. Genet. 12, 421–423 [DOI] [PubMed] [Google Scholar]
- 5. Everett L. A., Glaser B., Beck J. C., Idol J. R., Buchs A., Heyman M., Adawi F., Hazani E., Nassir E., Baxevanis A. D., Sheffield V. C., Green E. D. (1997) Nat. Genet. 17, 411–422 [DOI] [PubMed] [Google Scholar]
- 6. Pendrin V. (1896) Lancet 148, 532 [Google Scholar]
- 7. Mount D. B., Romero M. F. (2004) Pflugers Arch. 447, 710–721 [DOI] [PubMed] [Google Scholar]
- 8. Zheng J., Shen W., He D. Z., Long K. B., Madison L. D., Dallos P. (2000) Nature 405, 149–155 [DOI] [PubMed] [Google Scholar]
- 9. Belyantseva I. A., Adler H. J., Curi R., Frolenkov G. I., Kachar B. (2000) J. Neurosci. 20, RC116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dallos P., Fakler B. (2002) Nat. Rev. Mol. Cell Biol. 3, 104–111 [DOI] [PubMed] [Google Scholar]
- 11. He D. Z., Zheng J., Kalinec F., Kakehata S., Santos-Sacchi J. (2006) J. Membr. Biol. 209, 119–134 [DOI] [PubMed] [Google Scholar]
- 12. Brownell W. E., Bader C. R., Bertrand D., de Ribaupierre Y. (1985) Science 227, 194–196 [DOI] [PubMed] [Google Scholar]
- 13. Ashmore J. F. (1987) J. Physiol. 388, 323–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liberman M. C., Gao J., He D. Z., Wu X., Jia S., Zuo J. (2002) Nature 419, 300–304 [DOI] [PubMed] [Google Scholar]
- 15. Dallos P., Wu X., Cheatham M. A., Gao J., Zheng J., Anderson C. T., Jia S., Wang X., Cheng W. H., Sengupta S., He D. Z., Zuo J. (2008) Neuron 58, 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Okoruwa O. E., Weston M. D., Sanjeevi D. C., Millemon A. R., Fritzsch B., Hallworth R., Beisel K. W. (2008) Evol. Dev. 10, 300–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dong X. X., Ospeck M., Iwasa K. H. (2002) Biophys. J. 82, 1254–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Santos-Sacchi J., Wu M., Kakehata S. (2001) Hear. Res. 159, 69–73 [DOI] [PubMed] [Google Scholar]
- 19. Ashmore J. F. (1989) in Cochlear Mechanisms Structure, Function, and Models (Kemp D., Wilson J. P. eds) pp. 107–113, Plenum Press, New York [Google Scholar]
- 20. Santos-Sacchi J. (1991) J. Neurosci. 11, 3096–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He D. Z. (1997) J. Neurosci. 17, 3634–3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jia S., He D. Z. (2005) Nature Neurosci. 8, 1028–1034 [DOI] [PubMed] [Google Scholar]
- 23. Bai J. P., Surguchev A., Montoya S., Aronson P. S., Santos-Sacchi J., Navaratnam D. (2009) Biophys. J. 96, 3179–3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tan X., Pecka J. L., Tang J., Okoruwa O. E., Zhang Q., Beisel K. W., He D. Z. (2011) J. Neurophysiol. 105, 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang X., Yang S., Jia S., He D. Z. (2010) Brain Res. 1333, 28–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Homma K., Dallos P. (2011) J. Biol. Chem. 286, 2297–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oliver D., He D. Z., Klöcker N., Ludwig J., Schulte U., Waldegger S., Ruppersberg J. P., Dallos P., Fakler B. (2001) Science 292, 2340–2343 [DOI] [PubMed] [Google Scholar]
- 28. He D. Z., Jia S., Dallos P. (2003) J. Neurosci. 23, 9089–9096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He D. Z., Jia S., Zuo J., Latham S., Riordan G. P., Kachar B. (2010) Cytoskeleton 67, 43–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rybalchenko V., Santos-Sacchi J. (2003) J. Physiol. 547, 873–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rybalchenko V., Santos-Sacchi J. (2008) Biophys. J. 95, 4439–4447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kakehata S., Santos-Sacchi J. (1996) J. Neurosci. 16, 4881–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shehata W. E., Brownell W. E., Dieler R. (1991) Acta Otolaryngol. 111, 707–718 [DOI] [PubMed] [Google Scholar]
- 34. Tunstall M. J., Gale J. E., Ashmore J. F. (1995) J. Physiol. 485, 739–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sarojini V., Balaji Rao R., Ragothama S., Balaram P. (2010) J. Pept. Sci. 16, 430–436 [DOI] [PubMed] [Google Scholar]
- 36. Albert J. T., Winter H., Schaechinger T. J., Weber T., Wang X., He D. Z., Hendrich O., Geisler H. S., Zimmermann U., Oelmann K., Knipper M., Göpfert M. C., Oliver D. (2007) J. Physiol. 580, 451–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dossena S., Rodighiero S., Vezzoli V., Nofziger C., Salvioni E., Boccazzi M., Grabmayer E., Bottà G., Meyer G., Fugazzola L., Beck-Peccoz P., Paulmichl M. (2009) J. Mol. Endocrinol. 43, 93–103 [DOI] [PubMed] [Google Scholar]
- 38. Zheng J., Long K. B., Shen W., Madison L. D., Dallos P. (2001) Neuroreport 12, 1929–1935 [DOI] [PubMed] [Google Scholar]
- 39. Navaratnam D., Bai J. P., Samaranayake H., Santos-Sacchi J. (2005) Biophys. J. 89, 3345–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muallem D., Ashmore J. (2006) Biophys. J. 90, 4035–4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zheng J., Du G. G., Matsuda K., Orem A., Aguiñaga S., Deák L., Navarrete E., Madison L. D., Dallos P. (2005) J. Cell Sci. 118, 2987–2996 [DOI] [PubMed] [Google Scholar]
- 42. Rajagopalan L., Patel N., Madabushi S., Goddard J. A., Anjan V., Lin F., Shope C., Farrell B., Lichtarge O., Davidson A. L., Brownell W. E., Pereira F. A. (2006) J. Neurosci. 26, 12727–12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seymour M. L., Rajagopalan L., Volk M. J., Liu H., Brownell W. E., Pereira F. A. (2011) 34th Meeting of the Association for Research in Otolaryngology, Baltimore, MD, February 19–23, 2011, Abstract 70, The Association for Research in Otolaryngology, Mount Royal, NJ [Google Scholar]
- 44. Schaechinger T. J., Gorbunov D., Halaszovich C. R., Moser T., Kügler S., Fakler B., Oliver D. (2011) EMBO J. 30, 2793–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.