

Abstract
Nicotinic acetylcholine receptor (nAChR) α4 and β2 subunits assemble in two alternate stoichiometries to produce (α4β2)2α4 and (α4β2)2β2, which display different agonist sensitivities. Functionally relevant agonist binding sites are thought to be located at α4(+)/β2(−) subunit interfaces, but because these interfaces are present in both receptor isoforms, it is unlikely that they account for differences in agonist sensitivities. In contrast, incorporation of either α4 or β2 as auxiliary subunits produces isoform-specific α4(+)/α4(−) or β2(+)/β2(−) interfaces. Using fully concatenated (α4β2)2α4 nAChRs in conjunction with structural modeling, chimeric receptors, and functional mutagenesis, we have identified an additional site at the α4(+)/α4(−) interface that accounts for isoform-specific agonist sensitivity of the (α4β2)2α4 nAChR. The additional site resides in a region that also contains a potentiating Zn2+ site but is engaged by agonists to contribute to receptor activation. By engineering α4 subunits to provide a free cysteine in loop C at the α4(+)α4(−) interface, we demonstrated that the acetylcholine responses of the mutated receptors are attenuated or enhanced, respectively, following treatment with the sulfhydryl reagent [2-(trimethylammonium)ethyl]methanethiosulfonate or aminoethyl methanethiosulfonate. The findings suggest that agonist occupation of the site at the α4(+)/(α4(−) interface leads to channel gating through a coupling mechanism involving loop C. Overall, we propose that the additional agonist site at the α4(+)/α4(−) interface, when occupied by agonist, contributes to receptor activation and that this additional contribution underlies the agonist sensitivity signature of (α4β2)2α4 nAChRs.
Keywords: Cysteine-mediated Cross-linking, Ion Channels, Neuroscience, Nicotinic Acetylcholine Receptors, Receptor Structure-Function, MTS Reagents, Concatenated Receptors, Loop C
Introduction
The α4β2 nAChR2 is the predominant nAChR subtype in the brain where it constitutes one of the most important modulatory receptor systems influencing activities such as cognition, mood, consciousness, and nociception (1). The α4β2 nAChR is essential for nicotine self-administration (2) and has been implicated in autosomal dominant nocturnal frontal lobe epilepsy, depression, and age-related neurodegenerative diseases (1).
nAChR α4 and β2 subunits assemble in two alternate stoichiometries, (α4β2)2α4 and (α4β2)2β2 (Fig. 1A), whose relative ratio can be altered by chronic exposure to nicotine or autosomal dominant nocturnal frontal lobe epilepsy mutations (3). The (α4β2)2α4 and (α4β2)2β2 nAChRs display different desensitization kinetics (4), unitary conductance (4), calcium permeability (5), and sensitivity to chronic exposure to nicotine (4, 6, 7) and to modulation by Zn2+ (8). The (α4β2)2β2 nAChR has high sensitivity to ACh relative to the (α4β2)2α4 nAChR (4, 7, 9). They also differ in sensitivity to other agonists, including cytisine and sazetidine-A (9, 10). These agonists activate currents at (α4β2)2α4 and (α4β2)2β2 nAChRs with different potencies but also with strikingly different efficacies.
FIGURE 1.

ACh binding sites at (α4β2)2α4 nAChR. A, schematic representations showing the subunit arrangement and interfaces on the (α4β2)2α4 (left panel) and (α4β2)2β2 (right panel) nAChRs. In both receptors, the α4(+)/β2(−) interfaces contain agonist sites. B, alignment of conserved aromatic residues contributing to agonist binding in human (h) α4β2 nAChRs and Torpedo (tc) α1 and δ nAChR subunits. C, the left-hand panel is a side-on view of the α4(+)/α4(−) interface showing the location of conserved agonist-binding aromatic residues α4Tyr-126, α4Trp-182, α4Tyr-223, and α4Tyr-230 on the (+) face of the binding site and α4Trp-88 (homologous to β2Trp-82) on the (−) face of the site. The conserved residues form an aromatic box that is homologous to the agonist site located at the α4(+)/β2(−) interface and are part of loops A, B, C, and D that are considered structural signatures of the agonist binding site of Cys loop receptors. Note that loop C, which is thought to be a dynamic region that undergoes an inward capping motion upon agonist binding, is present in the primary face of the putative agonist site. The right-hand panel is a side-on view of α4(+)/β2(−) showing the α/β agonist site with the signature A, B, C, and D loops as well as the conserved aromatic residues. D, schematic representation of the linear sequence of the concatenated (α4β2)2α4 nAChR showing the orientation of the (+) and (−) faces of the subunits.
The structural determinants that bestow isoform-specific agonist sensitivity to the alternate α4β2 nAChRs have not been fully elucidated. The α4β2 nAChRs are pentameric structures that belong to the Cys loop family of ligand-gated ion channels that includes the muscle nAChR and the GABAA receptor. By analogy to the muscle α1γα1δβ1 nAChR (11), it is presumed that the α4(+)/β2(−) subunit interfaces harbor the agonist site (Fig. 1A). As such interfaces are present in both isoforms, it is unlikely that they confer signature agonist sensitivities to the alternate α4β2 nAChRs. In contrast, the auxiliary (fifth) subunit can be either α4 or β2, thus leading to a different composition of non-agonist binding interfaces (see Fig. 1A). In the (α4β2)2β2 receptor, there are two β2(+)/α4(−) interfaces and a β2(+)/β2(−) interface, whereas in the (α4β2)2α4 receptor, there are two β2(+)/α4(−) interfaces and an α4(+)/α4(−) interface (see Fig. 1A). As β2(+)/β2(−) and α4(+)/α4(−) interfaces are isoform-specific, they are likely candidates for conferring distinctive agonist sensitivity signatures to the (α4β2)2β2 and (α4β2)2α4 nAChRs. Our recent work showed that the α4(−)/α4(+) interface contains a Zn2+ potentiating site within a region that is homologous to a Zn2+ inhibiting site located on β2(+)/α4(−) interfaces in both receptor forms (8). Furthermore, crystal structures of acetylcholine-binding protein (AChBP) lacking vicinal cysteines in loop C and bound to allosteric ligands have suggested that modulators may bind sites at non-α interfaces in heteromeric nAChRs through interactions with conserved aromatic residues (12). Therefore, because of the conservation of agonist-binding aromatic residues in α4 and β2 nAChR subunits, it is plausible to speculate that additional agonist binding sites may be present at the β2(+)/β2(−) and α4(+)/α4(−) interfaces that are responsible for the isoform-specific agonist sensitivity of the alternate forms of the α4β2 nAChR.
In this study, we focused our attention on the α4(+)/α4(−) interface of the (α4β2)2α4 nAChR and its possible role in the agonist sensitivity signature of this α4β2 nAChR isoform. Using homology modeling, we first identified a putative agonist binding site at the α4(+)/α4(−) interface that is contributed by conserved agonist-binding aromatic residues. Obliteration of the site by exchanging the auxiliary α4 subunit for a chimeric subunit consisting of the N-terminal domain of the β2 subunit and the remaining part of the α4 subunit produced receptors with an agonist sensitivity comparable with that of the (α4β2)2β2 isoform. The putative site is capable of binding agonist as suggested by functional assays of mutant receptors engineered by alanine substitution of conserved aromatic residues contributing to the consensus agonist sites located at α4(+)/β2(−) subunit interfaces. Subsequently, using mutated receptors with a free cysteine residue in loop C together with covalent modifications with methanethiosulfonate (MTS) reagents, we demonstrated that the additional site contributes directly to channel gating. We propose that the additional site at the α4(+)/α4(−) defines the agonist sensitivity of the (α4β2)2α4 nAChR isoform.
EXPERIMENTAL PROCEDURES
Mutagenesis and Expression in Oocytes
Mutant α4 subunits were created as described previously (7, 8). The fully concatenated form of the (α4β2)2α4 isoform, construct β2_α4_β2_α4_α4, was engineered as described previously (9). Briefly, the signal peptide and start codon were removed from all the subunits but the first (a β2 subunit), and the subunits were bridged by Ala-Gly-Ser linkers. Only the last subunit in the construct (an α4 subunit) contained a stop codon. The subunits were subcloned into a modified pCI plasmid vector (Promega) using unique restriction enzyme sites flanking the N and C termini of each subunit. Constructs were assayed for integrity by determining the ACh sensitivity of constructs co-expressed with an excess of β2 or α4 monomers carrying the LT reporter mutation (L9′T in the second transmembrane domain). No changes were observed in comparison with constructs expressed alone. This indicates that the constructs did not degrade into lower order concatamers or monomers as such degradation products would incorporate the β2LT or α4LT monomers into receptors of higher sensitivity to ACh than the intact constructs (13). Henceforth, concatenated receptors will be referred to as β2_α4_β2_α4_α4, whereas (α4β2)2α4 or (α4β2)2β2 will be used to denote receptors assembled from loose α4 and β2 subunits when α4 subunits are in excess or shortage, respectively, over β2 subunits. For clarity, mutations in the concatenated receptors are shown as superscript positioned in the (+) or (−) face of the mutated subunit (e.g. in β2_W182Aα4_β2_α4_α4 the mutation W182A is located in the (+) face of the α4 subunit occupying the second position of the linear sequence of the concatamer; see supplemental Fig. 1 for experimental evidence showing the spatial orientation of the subunits in the concatamer). A subunit made of the N terminus of the β2 subunit and the remaining part of the α4 subunit was ligated to β2_α4_β2_α4 to construct the chimera β2_α4_β2_α4_β2/α4. Chimeric β2/α4 subunit was synthesized by GeneArt (Regensburg, Germany) and comprised the N terminus of β2 (1 to Arg-231) and the remaining part of the α4 subunit (Arg-241 to Ile-628). The residue numbering we use below includes the signal peptide sequence. To obtain the position in the mature form, subtract 28 for α4 and 26 for β2. Non-linked or concatenated α4β2 nAChRs were expressed in Xenopus oocytes as described previously (8, 9).
Oocyte Electrophysiology
Oocyte isolation and two-electrode voltage clamp recordings on oocytes were carried out as described previously (7–9). Concentration-response curves (CRCs) for agonists were obtained by normalizing agonist-induced responses to the control responses induced by a near-maximum effective agonist concentration as described previously (7–10). A minimum interval of 5 min was allowed between agonist applications to ensure reproducible recordings. The agonist CRC data were first fitted to the one-component Hill equation,
where EC50 represents the concentration of agonist inducing 50% of the maximal response (Imax), x is the agonist concentration, and nH is the Hill coefficient. When agonists induced biphasic receptor activation, the CRC data were fitted with a two-component Hill equation,
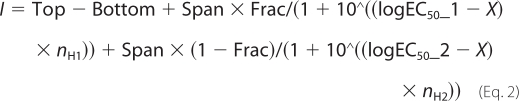 |
where Top and Bottom are the plateaus at the right and left ends of the curve in the same units as I; logEC50_1 and logEC50_2 are the concentrations that give half-maximal high sensitivity or low sensitivity stimulatory effects, respectively; nH1 and nH2 are the Hill coefficients; Frac is the proportion of maximal response due to the higher sensitivity component; and Span is a fitted coefficient between 0 and 1 that gives the weight of the first component. To determine the effects of the α4β2-selective competitive antagonist dihydro-β-erythroidine (DhβE) on the ACh responses of wild type (WT) or mutant receptors, the antagonist was included in the ACh and perfusing (Ringer's) solution. The responses to ACh obtained in the presence of the antagonists were normalized to control ACh responses (responses to ACh evoked in the absence of the antagonist).
Modification of Substituted Cysteines by MTS Reagents
MTS reagents aminoethyl methanethiosulfonate (MTSEA) and [2-(trimethylammonium)ethyl]methanethiosulfonate (MTSET) (Toronto Research Chemicals) were used to modify covalently a free Cys residue in loop C. The free Cys residue was created by dismantling the disulfide bond between vicinal Cys-225 and Cys-226 at the tip of loop C through serine substitution of Cys-226. The effect of the MTS reagents was assessed as follows. Oocytes expressing receptors with a free cysteine in loop C or WT receptors were first challenged with a control ACh concentration every 5 min until a stable response was obtained. Oocytes were then perfused with Ringer's solution containing MTSEA (2.5 mm) or MTSET (1 mm) for 180 s after which time the impaled cells were washed with Ringer's solution for 60 s. After washing, ACh was applied every 5 min until the amplitude of the responses was constant. The average of the current amplitudes prior to application of MTS reagents was the control response current (Iinitial), and the average of current amplitudes after rinsing was the average response after MTS application (Iafter MTS). The effect of the MTS reagents was estimated using the following equation: Percent change = ((Iafter MTS/Iinitial) − 1) × 100. The covalent effects of MTS were confirmed by exposing the oocytes to the reducing reagent dithioerythritol (DTT) at 5 mm for 60 s (MTSET-treated oocytes) or at 1 mm for 180 s (MTSEA-treated oocytes). In all cells tested, the response after DTT treatment was comparable with the one prior to the MTSET or MTSEA application, confirming that the functional changes observed were due to the covalent modification of Cys-225 by the MTS reagents. MTS reagents had no functional effects on WT β2_α4_β2_α4_α4 (see Fig. 6E).
FIGURE 6.

Effects of MTS reagents on mutant β2_α4_β2_α4_α4 nAChRs. A, structure of MTS reagents used to covalently modify free Cys-225 in loop C. B, β2_α4_β2_α4_C226Sα4 receptors displayed biphasic ACh responses. For comparison, we show the ACh CRC for WT. Data were analyzed by nonlinear regression as described under “Experimental Procedures.” Each point represents the mean (± S.E.) of at least four oocytes. C, representative current traces of the responses to ACh (800 or 3 μm) from two different oocytes expressing β2_α4_β2_α4_C226Sα4 receptors before and after modification by MTSET. DTT was applied at the end of the experiments to confirm that the effects observed were due to the covalent modification of Cys-225. D, representative current traces of the responses to ACh (800 or 3 μm) from two different oocytes expressing β2_α4_β2_α4_C226Sα4 receptors before and after modification by MTSEA. DTT reversed the effects of MTSEA. E, bar graph summary of the percentage of change (mean ± S.E.) in the amplitude of the ACh currents after MTS treatment on WT and mutant receptors. *, values are significantly different from amplitudes of the ACh responses prior to MTS treatment (p < 0.001 (one-way analysis of variance)).
Structure Homology Modeling
Sequences of the human α4 and β2 nAChR subunits were obtained from the ExPASy proteomics server with accession numbers P43681 (α4) and P17787 (β2). The homopentameric Lymnaea stagnalis AChBP structure (Protein Data Bank code 1UW6) was used to generate models of the extracellular domain of the (α4β2)2α4 receptor as described previously (8).
Statistical Analysis
Data analyses were performed using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA). Data were pooled from at least three different batches of oocytes. An F-test determined whether the one-site or biphasic model best fit the data; the simpler one-component model was preferred unless the extra sum-of-squares F-test had a value of p less than 0.05. log EC50 values for ACh or ACh plus DhβE and changes in current-response amplitudes in response to mutations or MTS application were analyzed using one-way analysis of variance with a Dunnett or Bonferroni post hoc correction for the comparison of all mutated receptors to determine significance between WT and mutant receptors. Significance levels between mutant receptors were determined using unpaired t tests. Data are plotted as mean ± S.E. Fit parameter values are the best fitting values with the S.E. values estimated from the fit.
RESULTS
The conservation of aromatic residues contributing to the agonist binding site in nAChR subunits together with homology modeling of the extracellular domain of the (α4β2)2α4 nAChR suggested an additional agonist site at the α4(+)/α4(−) interface (Fig. 1, B and C). The primary (+) face of this putative agonist site would be contributed by the auxiliary α4 subunit, whereas the complementary (−) face would be contributed by an α4 subunit that simultaneously provides the primary face of the consensus agonist site at an α4(+)/β2(−) interface. The putative site is homologous to the agonist site at the α4(+)/β2(−) interfaces, having the key conserved agonist-binding residues α4Tyr-126 (loop A), α4Trp-182 (loop B), on the primary face of the binding site. Importantly, the (+) site also contains a segment of amino acids containing a pair of vicinal cysteines (α4Cys-225 and α4Cys-226) flanked by the conserved aromatic residues α4Tyr-223 and α4Tyr-230. This region is homologous to loop C in the (+) face of the agonist site at α4(+)/β2(−) interfaces. This segment is not present in the β2(+)/α4(−) interface as neither the vicinal cysteines nor the residue equivalent to α4Tyr-223 are conserved in β2 (Fig. 1B) (11). Presently, it is believed that agonist binding triggers a capping motion of loop C that traps bound agonist in the binding site, which then leads to channel gating via molecular interactions in the coupling interface (14–16). On its complementary side, α4Trp-88 (loop D) is in a position homologous to β2Trp-82 in the agonist sites at α4(+)/β2(−) interfaces (Fig. 1C). The homologous residues in Torpedo and muscle nAChR are α1Tyr-93, α1Trp-149, α1Tyr-190, α1Tyr-198, and residues Trp-55 and Trp-57 located in the complementary ϵ/γ or δ subunits of the binding site (11).
To determine whether the α4(+)/α4(−) interface influences the agonist sensitivity of (α4β2)α4 nAChR, we first exchanged the N-terminal region of the β2 subunit for the N-terminal domain of the α4 subunit at the auxiliary subunit position (i.e. changed from an extracellular α4(+)/α4(−) interface to an extracellular β2(+)/α4(−) interface; Fig. 2A) and then tested the sensitivity of the chimeric receptor to ACh. To obviate uncertainties about subunit assembly, stoichiometry, and positions especially concerning placement of mutant or chimeric subunits in the additional as opposed to the consensus agonist binding positions when engaging heterologous expression from loose subunits, we conducted our studies using as a template the β2_α4_β2_α4_α4 nAChR construct, a fully concatenated form of the (α4β2)2α4 nAChR that replicates the functional properties of (α4β2)2α4 nAChRs (9). Importantly, β2_α4_β2_α4_α4 nAChRs are stable and produce homogeneous channels when expressed in Xenopus oocytes (9). We have shown previously that the α4 subunit is involved either in the inhibitory effects of Zn2+ (due to ion binding at β2(+)/α4(−) interfaces) or in the potentiating effects of Zn2+ (due to ion binding at the α4(+)/α4(−) interfaces) (8). Using a mutation that impairs sensitivity of (α4β2)2α4 receptors to inhibition or potentiation by Zn2+ (α4H195A) (8), we determined that the α4 subunits located in the fourth and fifth positions of the linear sequence of the concatenated receptor contribute to the Zn2+ potentiating site (supplemental Fig. 1). The (+) face of the site is contributed by the α4 subunit in the fifth position, which is therefore the auxiliary subunit in the concatamer β2_α4_β2_α4_α4 receptor. The inhibitory Zn2+ sites are located at the interface between the second (an α4 subunit) and third (a β2 subunit) positions and between the fifth (the auxiliary α4 subunit) and first (a β2 subunit) subunits of the concatamer receptor (supplemental Fig. 1). From these findings, we inferred that the consensus agonist sites at α/β interfaces are located at the interface between the first β2 and second α4 subunits and between the third β2 and fourth α4 subunits of the concatamer receptor.
FIGURE 2.

Agonist sensitivity of chimeric β2_α4_β2_α4_β2/α4 nAChR. A, schematic of the chimeric β2_α4_β2_α4_β2/α4 nAChR. B, ACh CRC at β2_α4_β2_α4_β2/α4 nAChRs. The ACh CRCs for WT nAChRs are shown for comparison. Data were fit by nonlinear regression analysis as described under “Experimental Procedures.” C, representative traces of the responses of chimeric β2_α4_β2_α4_β2/α4 nAChRs to EC100 of ACh (1 mm), cytisine (Cyt) (100 μm), and sazetidine-A (Saz) (1 μm). D, the CRC for sazetidine-A. The data were analyzed as described under “Experimental Procedures.” Each point in B and D represents the mean (± S.E.) of at least three oocytes.
Oocytes expressing chimeric β2_α4_β2_α4_β2/α4 or WT β2_α4_β2_α4_α4 concatamer receptors were functionally characterized using two-electrode voltage clamp. In oocytes expressing chimeric β2_α4_β2_α4_β2/α4 nAChRs (Fig. 2A), ACh activated current responses in a concentration-dependent manner but with a potency that was about 10-folder greater than for ACh action at WT β2_α4_β2_α4_α4 nAChRs (chimeric receptor ACh EC50 = 8.2 ± 0.5 μm (n = 4); WT ACh EC50 = 78 ± 8 μm (n = 6; p < 0.001) (Fig. 2B and Table 1). Interestingly, the ACh sensitivity of the chimeric receptor was not statistically different from that for the (α4β2)2β2 isoform (3.4 ± 1 μm; Table 2) (7, 9) or for its concatenated version β2_α4_β2_α4_β2 (EC50 = 3.2 ± 1 μm (n = 4); Fig. 2B and Table 1) (9). This finding suggested that ablation of the putative agonist site at the α4(+)/α4(−) interface by substitution with an extracellular β2(+)/α4(−) interface produces an assembly with ACh sensitivity approaching that of (α4β2)2β2 nAChRs. We examined this possibility by testing the functional effects of cytisine and sazetidine-A on β2_α4_β2_α4_β2/α4 nAChRs. Cytisine is a partial agonist at (α4β2)2α4 nAChRs or its concatenated template, but it has negligible agonist efficacy (less than 0.06%) at non-linked or concatenated (α4β2)2β2 nAChRs (7, 9). In contrast, sazetidine-A displays poor efficacy at loose or concatenated (α4β2)2α4 receptors (less than 0.05%) (9, 10) but is a potent, full agonist at non-linked or concatenated (α4β2)2β2 nAChRs (9, 10). Cytisine failed to elicit currents at β2_α4_β2_α4_β2/α4 nAChRs even at 100 μm, a concentration producing maximal effects at β2_α4_β2_α4_α4 nAChRs (Fig. 2C) (9). Sazetidine-A behaved as a full agonist at β2_α4_β2_α4_β2/α4 nAChRs and displayed a potency (EC50 = 16 ± 9 nm (n = 3)) that was comparable with that at loose or concatenated (α4β2)2β2 nAChR (EC50 = 7 ± 1 nm; Ref. 9) (Fig. 2, C and D). These findings show that removal of the α4(+)/α4(−) interface produces an (α4β2)2β2 nAChR-like agonist sensitivity and are thus consistent with the suggestion that the α4(+)/α4(−) interface in (α4β2)2α4 nAChR may underlie the agonist sensitivity signature of the isoform (α4β2)2α4 receptor. In light of this finding, henceforth, we will refer to the auxiliary α4 subunit as the fifth subunit.
TABLE 1.
Summary of ACh effects on WT and mutant β2_α4_β2_α4_α4 nAChRs
Data are the mean ± S.E. for 4–10 experiments. ACh EC50 values, Hill coefficients, and high sensitivity fractions (HF) are indicated.
| Receptor type | ACh EC50 | nH | ACh EC50_1 | nH1 | ACh EC50_2 | nH2 | HF |
|---|---|---|---|---|---|---|---|
| μm | μm | μm | |||||
| β2α4β2α4β2 | 3.2 ± 1 | 1.2 ± 0.5 | |||||
| β2α4β2α4α4 | 78 ± 8 | 0.72 ± 0.02 | |||||
| β2α4β2α4β2/α4 | 8.2 ± 0.5 | 1.1 ± 0.1 | |||||
| β2W182Aα4β2W182Aα4W182Aα4 | 684 ± 63a | 1.37 ± 0.1 | |||||
| β2W182Aα4β2α4α4 | 1.51 ± 0.02a | 0.55 ± 0.1 | 566 ± 62a | 2.5 ± 0.7 | 0.63 ± 0.1 | ||
| β2α4β2W182Aα4α4 | 1.23 ± 0.03a | 0.70 ± 0.2 | 668 ± 63a | 1.98 ± 0.7 | 0.64 ± 0.07 | ||
| β2α4β2α4W182Aα4 | 28 ± 2a | 1.4 ± 0.2 | 3483 ± 25a | 3.8 ± 1.2 | 0.66 ± 0.06 | ||
| β2W182Aα4β2W182Aα4α4 | 5.2 ± 0.9a | 0.73 ± 0.1 | 591 ± 65a | 1.1 ± 0.2 | 0.13 ± 0.08 | ||
| β2W182Aα4β2α4W182Aα4 | 7.8 ± 4a | 1.5 ± 0.9 | 599 ± 51a | 3.8 ± 0.8 | 0.28 ± 0.09 | ||
| β2α4β2W182Aα4W182Aα4 | 3.6 ± 1a | 0.8 ± 0.2 | 502 ± 68a | 1.3 ± 0.1 | 0.14 ± 0.07 | ||
| β2Y230Aα4β2α4α4 | 3.10 ± 0.6a | 0.76 ± 0.2 | 540 ± 43a | 3.9 ± 2 | 0.35 ± 0.07 | ||
| β2α4β2Y230Aα4α4 | 3.3 ± 0.3a | 0.63 ± 0.05 | 538 ± 51a | 3.7 ± 0.8 | 0.37 ± 0.1 | ||
| β2α4β2α4Y230Aα4 | 1.64 ± 0.07a | 1.1 ± 0.6 | 251 ± 61a | 2.0 ± 0.8 | 0.15 ± 0.06 | ||
| β2C226Sα4β2α4α4 | 2.19 ± 0.5a | 1.6 ± 0.3 | 352 ± 43a | 1.3 ± 0.2 | 0.17 ± 0.06 | ||
| β2α4β2C226Sα4α4 | 1.61 ± 0.1a | 1.4 ± 0.2 | 527 ± 65a | 1.1 ± 0.3 | 0.16 ± 0.06 | ||
| β2α4β2α4C226Sα4 | 1.12 ± 0.5a | 1.1 ± 0.4 | 385 ± 79a | 1.2 ± 0.6 | 0.13 ± 0.09 |
a Values are significantly different from WT; p < 0.05 (one-way analysis of variance).
TABLE 2.
Concentration-response data for ACh activation of WT and mutant α4β2 nAChR isoforms
The maximal current of the mutant receptors was normalized to that of WT receptors to estimate the relative maximal ACh responses of mutant receptors. Data represent the mean ± S.E. for seven to nine experiments. ND, not determined; NR, no functional responses elicited by ACh.
| (α4β2)2α4 |
(α4β2)2β2 |
|||||
|---|---|---|---|---|---|---|
| Relative maximal current | EC50 | nH | Relative maximal current | EC50 | nH | |
| μA | μm | μA | μm | |||
| WT | 1 | 81 ± 14 | 0.72 ± 0.07 | 1 | 3.4 ± 1 | 0.68 ± 0.07 |
| α4Y126A | 0.056 ± 0.004 | ND | ND | 0.063 ± 0.008 | ND | ND |
| α4W182A | 0.87 ± 0.08 | 634 ± 78 | 1.3 ± 0.5 | 0.76 ± 0.05 | 500 ± 78 | 1.6 ± 0.4 |
| α4Y223A | NR | ND | ND | NR | ND | ND |
| α4Y230A | 0.23 ± 0.04 | 478 ± 99 | 1.3 ± 0.2 | 0.15 ± 0.07 | 504 ± 112 | 1.5 ± 0.5 |
| β2W82A | 0.46 ± 0.12 | 256 ± 67 | 1 ± 0.4 | NR | ND | ND |
Mutant Agonist Sites Produce Biphasic ACh Effects
Given that the putative site at the α4(+)/α4(−) interface is homologous to the agonist binding sites at the α4(+)/β2(−) interfaces, we postulated that the site at the α4(+)/α4(−) interface influences receptor function possibly by binding agonist. We targeted the conserved aromatic residue α4Trp-182 in the α4(+)/α4(−) interface for substitution with alanine, aiming to demonstrate that ACh binds this site. α4Trp-182 is equivalent to muscle α1Trp-149 and establishes cation-π interactions with the quaternary ammonium group of ACh (17) and affects binding affinity as well as gating (18, 19). As shown in Table 2, alanine substitution of α4Trp-182 on loose subunit (α4β2)2α4 nAChRs reduced sensitivity to activation by ACh without impacting functional expression.
Alanine substitution of Trp-182 in the fifth subunit in β2_α4_β2_α4_α4 receptors should decrease the sensitivity of the receptor to ACh. Based on the finding that obliteration of the additional site in chimeric β2_α4_β2_α4_β2/α4 nAChRs increased ACh sensitivity, we anticipated that impairment of the site at the α4(+)/α4(−) by alanine substitution of α4Trp-182 would produce sufficient separation between the ACh EC50 values of the intact and impaired binding sites to generate a biphasic ACh CRC comprising a high sensitivity and a low sensitivity component. The high sensitivity component would be contributed by the unaltered agonist sites at the α4(+)/β2(−) interfaces, and as suggested by the ACh sensitivity of the chimeric β2_α4_β2_α4_β2/α4 receptor, the ACh EC50 should approximate the ACh EC50 for high sensitivity α4β2 receptors. The mutated site would contribute to the low sensitivity component, and the EC50 value should be much greater than that for WT. Introducing α4W182A into the (+) face of the putative agonist site at the α4(+)/α4(−) interface produced mutant β2_α4_β2_α4_W182Aα4 nAChRs. The CRC for ACh at mutant β2_α4_β2_α4_W182Aα4 receptors was biphasic (p < 0.003; n = 10) with a component that comprised about 65% of the curve and had an ACh EC50 value that was about 3-fold lower than that for the WT β2_α4_β2_α4_α4 receptor (Fig. 3A and Table 1). The second CRC component became evident at ACh concentrations higher than 500 μm and displayed an ACh sensitivity that was 45-fold lower than that for WT β2_α4_β2_α4_α4 receptors (p < 0.0001; n = 10) (Table 1). We next explored whether impairment of the binding sites at the α4(+)/β2(−) interfaces produced similar effects. Fig. 3, B and C, show that the effects of ACh on β2_W182Aα4_β2_α4_α4 or β2_α4_β2_W182Aα4_α4 receptors were biphasic (p < 0.001; n = 6). The relative abundance of the CRC component with the highest sensitivity for ACh was similar for all three mutant receptors. However, the ACh sensitivity of the high sensitivity component in the CRC for β2_W182Aα4_β2_α4_α4 or β2_α4_β2_W182Aα4_α4 receptors was ∼78-fold greater than that for WT, making it comparable with the ACh sensitivity of the isoform (α4β2)2β2 or chimeric β2_α4_β2_α4_β2/α4 receptors, both of which lack the α4(+)/α4(−) interface (Table 1). In contrast, when the mutation was in the fifth subunit (i.e. β2_α4_β2_α4_W182Aα4), the ACh sensitivity for the equivalent CRC component was increased by only 3-fold relative to WT (Table 1). The ACh CRC component with the lowest ACh sensitivity also differed in both types of mutant receptors. For receptors with mutant α4(+)/β2(−) interfaces, the ACh sensitivity of this component was 8-fold lower than WT (Table 1), whereas for β2_α4_β2_α4_W182Aα4 nAChRs it was 45-fold lower than that for WT (Table 1). For all three mutant receptors, the Hill coefficient of the component with lowest ACh sensitivity increased to 2–3 relative to WT, whereas changes in the Hill coefficient of the CRC component with the highest ACh sensitivity were not significant in comparison with WT. To test whether the biphasic ACh CRC reflected the ACh sensitivity of a homogeneous population of receptors with two intact agonist sites and one impaired agonist site, we introduced the mutation simultaneously into all three α4 subunits of the concatamer to produce triple mutant β2_W182Aα4_β2_W182Aα4_W182Aα4 receptor. We expected that the ACh CRC for this receptor would be monophasic and that the estimated ACh EC50 would be similar to that for non-linked (α4W182Aβ2)2α4W182A nAChRs. Fig. 3D shows that when the W182A was simultaneously introduced into the α4(+)/α4(−) and the two α4(+)/β2(−) interfaces (triple mutant receptor) in the β2_α4_β2_α4_α4 receptor the ACh CRC was monophasic (p < 0.001; n = 10) and had an ACh EC50 value that was no different from that obtained for non-linked (α4W182Aβ2)2α4W182A nAChRs (Tables 1 and 2).
FIGURE 3.

Effect of α4W182A subunit on ACh sensitivity of β2_α4_β2_α4_α4 nAChR. When the W182A mutation was introduced into the fifth (A), second (B), or fourth (C) positions of the concatenated receptor, biphasic ACh CRCs were obtained. When the mutation was present in all α4 subunits of the β2_α4_β2_α4_α4 nAChR, the ACh CRC was monophasic (D). Data were fit by nonlinear regression analysis as described under “Experimental Procedures.” Each point represents the mean (± S.E.) of at least four oocytes.
Next, we introduced α4W182A simultaneously into the (+) face of two subunit interfaces to create mutant β2_α4_β2_W182Aα4_W182Aα4, β2_W182Aα4_β2_α4_W182Aα4, or β2_W182Aα4_β2_W182Aα4_α4 receptors. As shown in Fig. 4, introducing W182A simultaneously into two α4 subunits of β2_α4_β2_α4_α4 receptors still produced biphasic ACh CRC (p < 0.007; n = 6–8) (Fig. 4) regardless of the combination of binding sites mutated. In comparison with single mutant receptors, the relative abundance of the CRC component with the highest sensitivity to ACh was reduced in all three mutants (p < 0.001; n = 6–12) (Figs. 3 and 4 and Table 1). For all three double mutant receptors, the ACh sensitivity of the high sensitivity component was enhanced as compared with WT but decreased as compared with the equivalent ACh EC50 for single mutant receptors (p < 0.001; n = 6–8) (Table 1). The relative abundance of the CRC fraction with the highest ACh sensitivity was higher when the mutation was incorporated into the second and fifth subunits, but this difference was not statistically significant (Fig. 4 and Table 1). The ACh EC50 estimated for the component with the lowest ACh sensitivity was similar for all three double mutant receptors and was not different from the ACh EC50 for (α4W182Aβ2)2β2 receptors (EC50 = 500 ± 78 μm; Table 2). Overall, therefore, the findings with the single and double W182A mutant β2_α4_β2_α4_α4 receptors together with the effects of α4W182A on non-linked α4β2 nAChR function (Table 2) indicate that the ACh CRC component with the highest sensitivity for ACh is contributed predominantly by unaltered binding sites, whereas the mutated sites contribute predominantly to the CRC component with the lowest sensitivity for ACh.
FIGURE 4.

Activation of β2_α4_β2_α4_α4 receptors containing two α4W182A mutant subunits. Simultaneous introduction of α4Trp-182 into two sites to produce β2_W182Aα4_β2_W182A α4_α4 (A), β2_W182Aα4_β2_α4_W182Aα4 (B), or β2_α4_β2_W182Aα4_W182Aα4 (C) led to biphasic ACh CRCs. Data were fit by nonlinear regression analysis as described under “Experimental Procedures.” Each point represents the mean (± S.E.) of at least four oocytes.
DhβE Inhibits α4(+)/α4(−) Site
The finding that introducing W182A into the fifth subunit produces an ACh CRC fraction with diminished sensitivity for ACh suggests that the α4(+)/α4(−) interface is capable of binding agonist. DhβE displays high affinity (low nm Ki values) competitive antagonism for α4β2 nAChRs (7, 9). Demonstrating that the site at the α4(+)/α4(−) interface is inhibited by DhβE would further support the suggestion that the site is capable of binding agonist. We assessed this by determining the ACh sensitivity of mutant β2_α4_β2_α4_W182Aα4 in the presence and absence of 1 μm DhβE. Because α4Trp-182 contributes to inhibition of α4β2 nAChR by DhβE (20) and as suggested by the present studies binds agonist in the putative site at the α4(+)/α4(−) interface, we anticipated DhβE to decrease the sensitivity of both mutant α4(+)/α4(−) and unaltered α4(+)/β2(−) sites albeit with different potency. As shown in Fig. 5, A and B, inhibition of the ACh responses of β2_α4_β2_α4_W182Aα4 nAChRs by DhβE reached a plateau at ACh concentrations higher than 800 μm, producing a monophasic ACh CRC lacking the component with the lowest ACh sensitivity detected in the absence of the antagonist. As shown in Fig. 5, C–E, the ACh CRC for receptors with mutant α4(+)/β2(−) binding sites (i.e. β2_W182Aα4_β2_α4_α4 or β2_α4_β2_W182Aα4_α4 nAChRs) also became monophasic in the presence of DhβE. However, by comparison with β2_α4_β2_α4_W182Aα4 receptors, DhβE inhibited more potently the responses elicited by concentrations of ACh lower than 10 μm, a concentration range activating non-mutated agonist sites.
FIGURE 5.

Effects of antagonist DhβE on mutant β2_α4_β2_α4_α4 nAChRs. A, typical traces showing the effects of ACh on β2_α4_β2_α4_W182Aα4 nAChRs in the absence (black) and presence of 1 μm DhβE (red). B, CRC for the effects of DhβE on the ACh responses of β2_α4_β2_α4_W182Aα4 nAChRs. Data were fit by nonlinear regression analysis as described under “Experimental Procedures.” C, representative traces showing the effects of 1 μm DhβE (in red) on the ACh responses (in black) of β2_α4_β2_W182Aα4_α4 nAChRs. D and E, ACh CRC in the absence or presence of 1 μm DhβE obtained for β2_α4_β2_α4_W182Aα4 or β2_W182Aα4_β2_α4_α4 nAChRs, respectively. Data were fit by nonlinear regression analysis as described under “Experimental Procedures.” Each point in B, D, and E represents the mean (± S.E.) of at least four oocytes.
Loop C in Fifth Subunit Affects Receptor Function
Loop C in the agonist site of Cys loop ligand-gated ion channels does not bind agonists directly, but agonist binding elicits loop C closure, and this is considered to lead to channel gating via molecular interactions in the coupling interface (14, 21–23). To assess the significance of loop C on the function of the putative site at the α4(+)/α4(−) interface, we introduced α4Tyr-230 into the (+) face of the site and then assayed its effects on the function of β2_α4_β2_α4_α4 receptors. α4Tyr-230 is equivalent to muscle α1Tyr-198 in the C terminus of loop C. It has been shown to affect gating but not agonist binding affinity in muscle nAChR (19), and in the (α4β2)2α4 receptors, it decreases the sensitivity to ACh and functional expression (Table 2). We hypothesized that if the site at the α4(+)/α4(−) interface had the capability of contributing to receptor activation, alanine substitutions of Tyr-230 should impair channel gating, leading to a biphasic ACh CRC comprising a fraction contributed by agonist sites with unaltered loop C and a component contributed by mutant loop C. As shown in Table 1 (see also supplemental Fig. 2), β2_α4_β2_α4_Y230Aα4 nAChRs produced a biphasic ACh CRC, comprising a component with an ACh EC50 comparable with the ACh EC50 of the (α4β2)2β2 nAChRs and a component with reduced ACh sensitivity as compared with the high sensitivity fraction. The high sensitivity component represented ∼15% of the CRC (Table 1). Incorporating α4Y230A into either of the α4(+)/β2(−) ACh binding sites produced comparatively similar effects except that the high sensitivity fraction was 2 times greater (p < 0.001; n = 10) (Table 1 and supplemental Fig. 2, A–C).
Modification of Loop C with MTS Reagents
Demonstrating that conformational changes in loop C in the fifth subunit alters the ACh responses of the β2_α4_β2_α4_α4 nAChR would strengthen the conclusion that the putative agonist binding site in the α4(+)/α4(−) interface contributes directly to channel gating. Loop C conformational transitions can be inferred from changes in the agonist responses of receptors brought about by covalent modification of substituted (22) or free (24) Cys residues in loop C by MTS reagents. Accordingly, we substituted serine for Cys-226 at the tip of loop C to dismantle the disulfide bond between Cys-225 and Cys-226 to make Cys-225 accessible to MTS reagents (MTSET or MTSEA; Fig. 6A). Substitution of serine for Cys-226 in any of the three α4 subunits of the β2_α4_β2_α4_α4 nAChR yielded biphasic ACh CRCs that were comparable with those produced by the α4Y230A mutation (Fig. 6B, Table 1, and supplemental Fig. 2, D–F). These findings are in accord with recent studies of muscle nAChR showing that removal of the Cys bridge in loop C decreases agonist sensitivity without abolishing loop C function (24). To assess the effects of MTS reagents on the function of intact and mutated sites, we performed the experiments using 800 or 3 μm ACh. 800 μm ACh produced near maximal activation of the ACh CRC fraction with the lowest ACh sensitivity, whereas 3 μm ACh produced almost maximal activation of the ACh CRC fraction with the highest ACh sensitivity (Table 1 and supplemental Fig. 2, D–F). We have shown above that ACh produces a biphasic CRC at β2_α4_β2_α4_α4 receptors with one mutant agonist site and that the CRC component with the lowest ACh sensitivity is contributed predominantly by the mutated site, whereas the component with the highest sensitivity component reflects predominantly the activation of intact binding sites. As shown in Fig. 6C, when the current responses of β2_α4_β2_α4_C226Sα4 nAChRs were activated by 800 μm ACh, MTSET decreased the amplitude of the ACh responses by ∼30%. In accord with a covalent interaction between α4Cys-225 and MTSET, exposure to DTT reversed the effects of MTSET (Fig. 6C). By comparison, MTSET had no effects on the amplitude of the responses evoked by 3 μm ACh (Fig. 6C). These results were expected because 3 μm ACh activated predominantly intact agonist sites, and MTSET produced similar effects on the ACh of β2_C226Sα4_β2_α4_α4 or β2_α4_β2_C226Sα4_α4 mutant nAChRs (Fig. 6E). Exposure to MTSEA did not have any apparent effect on the current responses elicited by 800 μm ACh in β2_α4_β2_α4_C226Sα4 nAChRs (Fig. 6D). However, when the responses were elicited by lower concentrations of ACh (3 μm) after MTSEA treatment, the amplitude of the ACh responses was enhanced markedly, and this effect was reversed by DTT (Fig. 6D). This result demonstrated that MTSEA enhanced the ACh responses of the mutant receptor by covalently modifying Cys-225. Comparable effects were observed when β2_C226Sα4_β2_α4_α4 or β2_α4_β2_C226Sα4_α4 were exposed to MTSEA (Fig. 6E). Although we observed differences in the effects of MTS at each agonist site, the differences were not significant.
DISCUSSION
The α4 and β2 subunits of the nAChR assemble into alternate forms that differ markedly in sensitivity to activation by agonists. Because α4β2 nAChRs receptors contain two identical agonist binding sites at the α(+)/β(−) interfaces, a long standing question has been what structural features confer agonist sensitivity to the alternate forms of the α4β2 nAChR. Here, we provide evidence that an additional site at the α4(+)/α4(−) interface in the (α4β2)2α4 nAChR underlies the agonist sensitivity signature of the isoform (α4β2)2α4 nAChR. The additional site binds ACh and when engaged by ACh contributes to channel gating through a coupling pathway that includes loop C. The findings have important structural and functional implications about the role of subunit composition in the structural-functional properties of the α4β2 nAChR and the manner by which agonists activate heteromeric Cys loop ligand-gated ion channels.
The agonist site at the α4(+)/α4(−) is a major determinant of the agonist sensitivity of the (α4β2)2α4 nAChR. Its removal increases ACh sensitivity and sazetidine-A efficacy and obliterates cytisine efficacy, all of which are functional signatures of the isoform (α4β2)2β2 nAChR. Moreover, when agonist binding by the α4(+)/α4(−) site is weakened by α4W182A, ACh produces biphasic agonist responses in accord with the functional behavior of β2_α4_β2_α4_α4 receptors containing unaltered and mutated agonist sites. The high sensitivity component of the curve represents the contribution of unaltered agonist sites at the α4(+)/β2(−) interfaces, whereas the mutated site contributes to the low sensitivity component as demonstrated by the differential sensitivity of these components to ACh or the antagonist DhβE. To our knowledge, this is the first time that biphasic agonist CRCs have been obtained from receptors with fixed stoichiometry. This is possible because in concatenated pentamers the agonist sites can be selectively impaired or ablated. A similar phenomenon occurs when the ACh binding sites of muscle nAChR are impaired individually. In muscle nAChR, the two agonist sites are located at the α/γ and α/δ interfaces and are thus structurally different (11). Selective mutagenesis of the sites is therefore possible in muscle nAChR assembled from non-linked subunits, and when the mutant receptors are expressed heterologously, exposure to the nicotinic competitive antagonist tubocurarine produces a biphasic 125I-bungarotoxin CRC (25).
The additional agonist site clearly contributes to the activation of the receptor by agonist in a manner comparable with that of the consensus agonist site located at each of the α4(+)/β2(−) interfaces. When loop C in the primary face of the α4(+)/α4(−) was impaired through alanine substitution of Tyr-230 or serine substitution of Cys-226 or by covalent modification of a free Cys-225 by MTS reagents, the amplitude of the ACh responses was reduced (alanine substitution and MTSET) or enhanced (MTSEA) Although this site resides in the same interface where the signature potentiating Zn2+ site of the (α4β2)2α4 isoform is located (8), analogous to that of the benzodiazepine site in GABAA receptors (26), it is not a classic allosteric site. Neither Zn2+ nor benzodiazepines are capable of channel gating but appear to enhance receptor responses to agonist by increasing channel opening frequency (benzodiazepines; Ref. 27) or by increasing burst duration (Zn2+; Ref. 28).
MTS modification of a free cysteine engineered at the tip of loop C in the fifth subunit provided strong support for the idea that the putative site at the α4(+)/α4(−) interface contributes to channel gating. Interestingly, MTSET and MTSEA affected loop C function differentially. It is possible that MTS reagents lock loop C into different conformations depending on how the Cys-MTS moiety is oriented within the binding pocket. MTSET could attenuate ACh responses by stabilizing the mutated loop C into an extended, antagonist-bound-like conformation, thus reducing ACh efficacy. By comparison, MTSEA could enhance ACh responses by locking the mutated loop C into a capped, agonist-bound-like conformation, which would increase agonist efficacy, leading to the activation of the mutated sites at submaximal ACh concentrations. High resolution x-ray crystal structures of an MTS-Y53C mutant of the AChBP support this view (29). Loop C in MTSET-Y53C AChBP is in an extended, antagonist-bound conformation, which is consistent with the finding that α7W55C-MTSET nAChRs are unresponsive to ACh (29). Another reagent, methyl methanethiosulfonate, enhances the ACh responses of α7Y53C nAChR and locks loop C in methyl methanethiosulfonate-Y53C AChBP into a closed, agonist-bound-like conformation (29). Although MTSET and MTSEA may affect the contribution of loop C to receptor activation differentially, it is clear from their effects on the ACh responses of the mutant agonist sites that all three sites contribute to activation of (α4β2)2α4 nAChRs.
The sites at the α4(+)/β2(−) and α4(+)/α4(−) interfaces respond differentially to alanine substitutions in respect to sensitivity to ACh or DhβE. The sites also appeared to interact differently with the agonists cytisine and sazetidine as suggested by the remarkable changes in the efficacy of these ligands brought about by ablation of the α4(+)/α4(−) interface. This could arise from differences in the overall architecture of the agonist sites and the coupling regions. Although the primary face in the agonist site at α4(+)/α4(−) and α4(+)/β2(−) interfaces is contributed by identical amino acids, the complementary face is not. In the case of the agonist site at the α4(+)/α4(−), the complementary face is contributed by the (−) face of the adjacent α4 subunit, providing a likely structural determinant for differential agonist binding site interactions. Moreover, the coupling pathway leading to gating may also differ in both types of agonist sites, and this could affect the interfacial interactions implicated in binding and gating. Loops Cys, β1β2, β8β9, the end of β10, pre-M1 region, M2-M3 linker, and post-M4 have been shown to contribute to gating in the Cys loop receptor family (30, 31). Although these regions are conserved in the fifth subunit, the interactions of these regions with the complementary face of the binding sites are likely to vary, thus altering or introducing coupling steps that may lead to profoundly different effects on agonist-induced gating. In support of this possibility, it has been shown previously that single point mutations in the coupling regions of Cys loop receptors alter the effects of ligands on receptor function (for example, see Refs. 32–35).
How does the additional agonist site impact (α4β2)2α4 receptor function? The additional site appears to increase the efficacy of ACh. The biphasic nature of the ACh CRC of β2_α4_β2_α4_W182Aα4 suggests that the three binding sites must be fully engaged by ACh to attain maximal receptor activation, although occupancy of two agonist sites allows receptor activation. This possibility is consistent with our previous findings that the maximal ACh current responses of (α4β2)2β2, which has only two agonist sites, are smaller than those of the (α4β2)2α4 receptors (7, 9, 10). Interestingly, the W182A mutation produced similar effects when incorporated in any of the three agonist sites of the β2_α4_β2_α4_α4 receptor, suggesting that ACh efficacy in (α4β2)2α4 receptors is determined by the number of agonist sites engaged by ACh. This is comparable with the activation of homomeric α7/5HT3 chimeric receptors that requires occupancy of three binding sites to maximally stabilize the open channel as long as one site is at a subunit separated from the other two (36). In the case of the β2_α4_β2_α4_α4 receptor, the agonist sites are arranged non-consecutively as well (Fig. 1A and supplemental Fig. 1).
Sensitivity to activation by ACh was also affected by the number of agonist sites contributing to β2_α4_β2_α4_4 activation. Sensitivity to ACh increased through either ablation of the α4(+)/α4(−) interface or through alanine substitution in the α4(+)/α4(−) or either of the α4(+)/β2(−) sites. In contrast, the ACh sensitivity of homomeric α7/5HT3 chimeric receptors appears to increase by occupancy of three non-consecutive agonist sites (36). It may be that the agonist site at the α4(+)/α4(−) interface has less affinity for ACh than the sites at the α4(+)/β2(−) interfaces, which could decrease the overall ACh sensitivity of the receptor. Unlike the agonist sites in homomeric receptors, the ACh sites at (α4β2)2α4 receptors are not structurally identical. This could lead to each type of site having different gating effects, leading to overall changes in agonist sensitivity, in comparison with α4β2 nAChR activated by two identical agonist sites. In accord with this possibility, we found that the Trp-182 mutation affected the site at the α4(+)/α4(−) and α4(+)/β2(−) interfaces differentially. Furthermore, the pattern of changes in receptor function of the single or double agonist binding mutants brought about by alanine substitution of agonist site residues as observed in terms of relative abundance of the components of the ACh CRC, sensitivity to ACh, and changes in the nH coefficient was influenced by the subunit interface location of the mutated agonist sites. This suggests complex interactions between the agonist sites that may also impact the overall ACh sensitivity of the (α4β2)2α4 receptor. Studies of the microscopic currents of the (α4β2)2α4 nAChR should aid the understanding of how the additional agonist site impacts the activation of this (α4β2) nAChR isoform in comparison with (α4β2)2β2 as well as homomeric Cys loop receptors.
Supplementary Material
Acknowledgments
We thank R. J. Lukas and P. Whiteaker (Barrow Institute for Neurology, Phoenix, AZ) for useful comments on the work and manuscript.
This work was supported by grants from the Royal Society and Brookes University.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
- nAChR
- nicotinic acetylcholine receptor
- ACh
- acetylcholine
- AChBP
- acetylcholine-binding protein
- MTS
- methanethiosulfonate
- MTSET
- [2-(trimethylammonium)ethyl]methanethiosulfonate
- MTSEA
- aminoethyl methanethiosulfonate
- CRC
- concentration-response curve
- DhβE
- dihydro-β-erythroidine.
REFERENCES
- 1. Gotti C., Clementi F. (2004) Prog. Neurobiol. 74, 363–396 [DOI] [PubMed] [Google Scholar]
- 2. Maskos U., Molles B. E., Pons S., Besson M., Guiard B. P., Guilloux J. P., Evrard A., Cazala P., Cormier A., Mameli-Engvall M., Dufour N., Cloëz-Tayarani I., Bemelmans A. P., Mallet J., Gardier A. M., David V., Faure P., Granon S., Changeux J. P. (2005) Nature 436, 103–107 [DOI] [PubMed] [Google Scholar]
- 3. Son C. D., Moss F. J., Cohen B. N., Lester H. A. (2009) Mol. Pharmacol. 75, 1137–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nelson M. E., Kuryatov A., Choi C. H., Zhou Y., Lindstrom J. (2003) Mol. Pharmacol. 63, 332–341 [DOI] [PubMed] [Google Scholar]
- 5. Tapia L., Kuryatov A., Lindstrom J. (2007) Mol. Pharmacol. 71, 769–776 [DOI] [PubMed] [Google Scholar]
- 6. Kuryatov A., Luo J., Cooper J., Lindstrom J. (2005) Mol. Pharmacol. 68, 1839–1851 [DOI] [PubMed] [Google Scholar]
- 7. Moroni M., Zwart R., Sher E., Cassels B. K., Bermudez I. (2006) Mol. Pharmacol. 70, 755–768 [DOI] [PubMed] [Google Scholar]
- 8. Moroni M., Vijayan R., Carbone A., Zwart R., Biggin P. C., Bermudez I. (2008) J. Neurosci. 28, 6884–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carbone A. L., Moroni M., Groot-Kormelink P. J., Bermudez I. (2009) Br. J. Pharmacol. 156, 970–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zwart R., Carbone A. L., Moroni M., Bermudez I., Mogg A. J., Folly E. A., Broad L. M., Williams A. C., Zhang D., Ding C., Heinz B. A., Sher E. (2008) Mol. Pharmacol. 73, 1838–1843 [DOI] [PubMed] [Google Scholar]
- 11. Unwin N. (2005) J. Mol. Biol. 346, 967–989 [DOI] [PubMed] [Google Scholar]
- 12. Hansen S. B., Taylor P. (2007) J. Mol. Biol. 369, 895–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Groot-Kormelink P. J., Broadbent S. D., Boorman J. P., Sivilotti L. G. (2004) J. Gen. Physiol. 123, 697–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hansen S. B., Sulzenbacher G., Huxford T., Marchot P., Taylor P., Bourne Y. (2005) EMBO J. 24, 3635–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee W. Y., Sine S. M. (2005) Nature 438, 243–247 [DOI] [PubMed] [Google Scholar]
- 16. Purohit P., Auerbach A. (2007) J. Gen. Physiol. 130, 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiu X., Puskar N. L., Shanata J. A., Lester H. A., Dougherty D. A. (2009) Nature 458, 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Akk G. (2001) J. Physiol. 535, 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Akk G., Auerbach A. (1999) Br. J. Pharmacol. 128, 1467–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iturriaga-Vásquez P., Carbone A., García-Beltrán O., Livingstone P. D., Biggin P. C., Cassels B. K., Wonnacott S., Zapata-Torres G., Bermudez I. (2010) Mol. Pharmacol. 78, 366–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mukhtasimova N., Free C., Sine S. M. (2005) J. Gen. Physiol. 126, 23–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Venkatachalan S. P., Czajkowski C. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 13604–13609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang H. L., Toghraee R., Papke D., Cheng X. L., McCammon J. A., Ravaioli U., Sine S. M. (2009) Biophys. J. 96, 3582–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mukhtasimova N., Lee W. Y., Wang H. L., Sine S. M. (2009) Nature 459, 451–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bren N., Sine S. M. (1997) J. Biol. Chem. 272, 30793–30798 [DOI] [PubMed] [Google Scholar]
- 26. Galzi J. L., Changeux J. P. (1994) Curr. Opin. Struct. Biol. 4, 554–565 [Google Scholar]
- 27. Rogers C. J., Twyman R. E., Macdonald R. L. (1994) J. Physiol. 475, 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsiao B., Mihalak K. B., Magleby K. L., Luetje C. W. (2008) J. Neurophysiol. 99, 999–1007 [DOI] [PubMed] [Google Scholar]
- 29. Brams M., Gay E. A., Sáez J. C., Guskov A., van Elk R., van der Schors R. C., Peigneur S., Tytgat J., Strelkov S. V., Smit A. B., Yakel J. L., Ulens C. (2011) J. Biol. Chem. 286, 4420–4428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiu X., Hanek A. P., Wang J., Lester H. A., Dougherty D. A. (2005) J. Biol. Chem. 280, 41655–41666 [DOI] [PubMed] [Google Scholar]
- 31. Gay E. A., Yakel J. L. (2007) J. Physiol. 584, 727–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Campos-Caro A., Sala S., Ballesta J. J., Vicente-Agulló F., Criado M., Sala F. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 6118–6123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Castaldo P., Stefanoni P., Miceli F., Coppola G., Del Giudice E. M., Bellini G., Pascotto A., Trudell J. R., Harrison N. L., Annunziato L., Taglialatela M. (2004) J. Biol. Chem. 279, 25598–25604 [DOI] [PubMed] [Google Scholar]
- 34. Perkins D. I., Trudell J. R., Crawford D. K., Asatryan L., Alkana R. L., Davies D. L. (2009) J. Biol. Chem. 284, 27304–27314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu X. Q., Zhang L., Stewart R. R., Weight F. F. (2003) J. Biol. Chem. 278, 46583–46589 [DOI] [PubMed] [Google Scholar]
- 36. Rayes D., De Rosa M. J., Sine S. M., Bouzat C. (2009) J. Neurosci. 29, 6022–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.