

Abstract
AIMS
Tolterodine and 5-hydroxymethyl tolterodine (5-HMT) are equipotent active moieties of tolterodine; 5-HMT is the singular active moiety of fesoterodine. Formation of 5-HMT from fesoterodine and tolterodine occurs via esterases and CYP2D6 respectively. This randomized, crossover, open-label, multiple-dose study in CYP2D6 extensive metabolizers (EMs) and poor metabolizers (PMs) compared the pharmacokinetics of fesoterodine vs. tolterodine extended release (ER).
METHODS
Subjects received fesoterodine and tolterodine ER with a ≥3-day washout period. Treatment comprised 4-mg once daily doses for 5 days escalated to 8-mg once daily for 5 days. Pharmacokinetics of active moieties were compared by drug, dose and genotype.
RESULTS
Active moiety exposures following fesoterodine and tolterodine ER increased proportional to dose in EMs and PMs. In EMs only, coefficients of variation for AUC and Cmax following fesoterodine (up to 46% and 48% respectively) were lower than those following tolterodine ER (up to 87% and 87% respectively). Following fesoterodine and tolterodine ER administration, active moiety exposures ranged up to sevenfold and 40-fold respectively. Mean urinary excretion of 5-HMT following fesoterodine 4 and 8 mg, respectively, was 0.44 and 0.89 mg in EMs and 0.60 and 1.32 mg in PMs. Following tolterodine ER 4 and 8 mg, it was 0.38 and 0.71 mg respectively (EMs only). Renal clearance was similar regardless of administered drug, dose or genotype.
CONCLUSIONS
Tolterodine, not 5-HMT, was the principal source of variability after tolterodine ER administration. Fesoterodine delivers 5-HMT with less variability than tolterodine, regardless of CYP2D6 status, with up to 40% higher bioavailability. The pharmacokinetics of fesoterodine were considerably less variable than TER.
Keywords: fesoterodine, overactive bladder, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Tolterodine and 5-hydroxymethyl tolterodine (5-HMT) are equipotent active moieties of tolterodine; 5-HMT is the singular active moiety of fesoterodine. The formation of 5-HMT from tolterodine occurs via CYP2D6, and some subjects are poor metabolizers CYP2D6. On the other hand, the formation of 5-HMT from fesoterodine occurs via ubiquitous esterases. Cross-study comparisons of data from phase 1 studies suggest that active moiety exposures are considerably more variable following tolterodine extended release vs. fesoterodine.
WHAT THIS STUDY ADDS
This head-to-head study confirmed the findings of reduced pharmacokinetic variability of fesoterodine and further delineates that tolterodine, and not 5-HMT, was the principal source of variability after administration of tolterodine extended release. The data suggest that fesoterodine delivers 5-HMT consistently, regardless of CYP2D6 status, with up to 40% higher bioavailability compared with tolterodine.
Introduction
Antimuscarinic agents are the mainstay of pharmacologic therapy for the treatment of overactive bladder (OAB) [1]. Tolterodine and fesoterodine are effective and well-tolerated antimuscarinics for the treatment of OAB symptoms [2]. Tolterodine is available as an immediate-release tablet for administration twice daily and as an extended-release (ER) capsule for once daily administration. Fesoterodine is available as an ER tablet for once daily use. The standard recommended daily dose of tolterodine is 4 mg daily, whereas fesoterodine is approved for initiation at 4 mg once daily and can be titrated to 8 mg once daily based on individual patient response.
Following oral administration, tolterodine is converted in the liver by the cytochrome P450 (CYP) 2D6 pathway to an active metabolite, 5-hydroxymethyl tolterodine (5-HMT). Both tolterodine and 5-HMT exert antimuscarinic activity in vitro[3]. Fesoterodine is a prodrug that is also converted to 5-HMT, and the antimuscarinic activity of fesoterodine is entirely attributable to 5-HMT [4]. Conversion of fesoterodine occurs rapidly via esterases and not the CYP2D6 pathway [5]. The elimination of the active metabolite, 5-HMT, is mediated by multiple pathways [3, 6], including two equally prominent hepatic metabolism pathways involving CYP2D6 and CYP3A4 enzymes. In addition, 5-HMT is excreted unchanged in the urine, accounting for about 16% of the fesoterodine dose [7].
The CYP2D6 enzyme shows a very high degree of interindividual variability that is primarily because of extensive genetic polymorphism [6] that results in poor metabolizers (PMs), intermediate metabolizers (IMs), extensive metabolizers (EMs) and ultrarapid metabolizers (UMs) of its substrates. Up to 10% of Caucasians and less than 1% of Asians are PMs, devoid of CYP2D6 activity, and thereby cannot utilize CYP2D6-dependent metabolic pathways for drug elimination. It is estimated that about 10% to 15% of the European population are CYP2D6 IMs [8]. The proportion of UMs in the Western European population is estimated to be about 5.5% [9].
Following tolterodine administration, patients can be exposed to two equipotent active moieties, tolterodine and 5-HMT, in widely varying ratios depending on CYP2D6 genetic polymorphism [10, 11] and/or variability arising from drug interactions with CYP2D6 [12]. On the contrary, patients receiving fesoterodine are exposed to 5-HMT as a singular active moiety, regardless of CYP2D6 genotype. This contrast was also observed in two separate phase 1 studies comparing tolterodine ER and fesoterodine in EMs and PMs [11, 13]. The Cmax of tolterodine varied between 1 and 60 nm whereas the Cmax of 5-HMT varied only between 3 and 13 nm. As expected CYP2D6 PMs had the highest tolterodine concentrations [14]. Although these comparisons are of sufficient scientific interest and value, they are limited by the cross-study comparison and dissimilar study designs. The present study was designed to provide a within-study comparison of the pharmacokinetic variability in EMs and PMs following administration of fesoterodine or tolterodine ER at 4 and 8 mg once daily. A secondary objective of the study was to estimate the urinary excretion of 5-HMT following administration of fesoterodine and tolterodine ER.
Methods
Study design
This was a randomized, open-label, two-way crossover, multiple-dose, within-subject dose escalation study in 30 healthy subjects identified as CYP2D6 EMs (n = 20) and PMs (n = 10) by genotyping. This study was conducted in compliance with the ethical principles of the Declaration of Helsinki and all International Conference Harmonization Good Clinical Practice guidelines. The protocol was approved by the institutional review board at the investigational centre participating in the study, and all subjects provided written informed consent before entering the study.
Subjects
Subjects were included if they were healthy adults between the ages of 18 and 55 years, inclusive, with a body mass index (BMI) of 18 to 30 kg m−2 and a total bodyweight >50 kg. Key exclusion criteria were evidence or history of clinically significant diseases or clinical findings at screening, any condition possibly affecting drug absorption or 12-lead ECG demonstrating QTc >450 ms at screening.
Subjects were genotyped for CYP2D6 status before enrolment. The alleles genotyped were *3, *4, *5, *6, *7, *8, *10, *14, *18, *21, *36, *41 and duplication. Subjects who were homozygous or with a combination of *3, *4, *5, *6, *7, *8, *14, *18 and *21 were classified as PMs. Other genotypes were classified as EMs.
Study procedures
Screening evaluation occurred within 28 days before the first dose of period 1. Day 0 was defined as the day before the first day of dosing (day 1) in each period. Subjects were admitted to the Clinical Research Unit (CRU) on day 0 (in the morning) and days 4 and 9 (in the evening) and remained there until after the ECG measurement at 4 h post dose on day 1 and day 6 or the last scheduled assessment on day 11 respectively. Subjects were required to return to the CRU on the mornings of days 2, 3, 4, 7, 8 and 9 for dose administration, after which they were able to leave the CRU.
Subjects received tolterodine ER 4 mg daily for 5 days followed by tolterodine ER 8 mg daily for 5 days (treatment A) and fesoterodine 4 mg daily for 5 days followed by fesoterodine 8 mg daily for 5 days (treatment B). Treatment order was randomized and there was at least a 3-day washout period between treatments. Subjects received study medication once daily in the morning on days 1 to 5 (4-mg dose) and on days 6 to 10 (8-mg dose) of each study period. Doses were administered after breakfast on days 1 to 4 and days 6 to 9, or after at least a 10-h overnight fast on days 5 and 10.
Subjects were required to fast (4-h period) before all clinical laboratory measurements. Subjects also fasted overnight on two occasions in each period for a minimum of 10 h from the evening before until the time of dosing on days 5 and 10. On each study day, lunch and dinner were served at approximately 4 and 10 h, respectively, after breakfast. The same meal schedule and menu was followed between the two treatment periods.
Blood and urine sample collection and analysis
In each period, pharmacokinetic (PK) blood samples (6 ml) were collected pre-dose on days 1, 4, 5, 6, 9 and 10, on day 5 at 1, 2, 3, 4, 5, 6, 8, 10, 12 and 15 h post dose and on day 10 at 1, 2, 3, 4, 5, 6, 8, 10, 12, 15, 24, 30 and 36 h post dose. Following tolterodine ER, blood samples were collected in tubes that contained no serum separator or other additives and were kept at room temperature until clotted. Following fesoterodine, blood samples were collected in tubes containing sodium heparin. Samples were centrifuged at approximately 1700 g for 10 min at 4°C. Samples were stored in screw-capped polypropylene tubes at approximately −20°C within 1 h of collection. Serum samples were analysed for tolterodine and 5-HMT at Eurofins AvTech Laboratories, Inc. (Portage, MI, USA) and plasma samples were analysed for 5-HMT at Advion BioSciences, Inc. (Ithaca, NY, USA).
Urine was collected over 24 h post dose on days 5 and 10. Voided volume was collected in 1-l polyethylene containers and stored at approximately 4°C. At the end of the collection period, the urine output from each subject was pooled, the total volume was recorded and a 10-ml sample was stored in polypropylene tubes at −70°C. Urine samples were analysed for 5-HMT at Advion BioSciences, Inc. (Ithaca, NY, USA). All plasma, serum and urine samples were analysed using validated analytical methods described previously [11, 13] and conducted in compliance with Pfizer standard operating procedures. All concentration calculations were based on the peak area ratios of tolterodine or 5-HMT to the internal standard. The calibration curves (0.10–60 ng ml−1, 0.10–10 ng ml−1, 0.02–20 ng ml−1 and 1.00–1000 ng ml−1 for tolterodine in serum, 5-HMT in serum, 5-HMT in plasma and 5-HMT in urine respectively), were characterized by regression coefficient, slope and intercept using a 1/x2-weighted (except for 5-HMT in plasma, 1/x-weighted was used) linear regression. Concentrations of tolterodine or 5-HMT in the quality control and study samples were determined by inverse prediction from the calibration curve. The mean precision estimates (expressed as the coefficient of variation) of the back-calculated calibration curve concentrations were ≤10.6% (serum tolterodine), ≤7.1% (serum 5-HMT), ≤5.6% (plasma 5-HMT) and ≤3.3% (urine 5-HMT). The corresponding accuracy (expressed as the % difference from the theoretical concentration) ranged from −1.3 to 2.0% (serum tolterodine), −0.8 to 1.0% (serum 5-HMT), −0.5 to 0.5% (plasma 5-HMT) and −4.3 to 5.0% (urine 5-HMT). The precision estimates of the quality control samples were ≤8.1% (serum tolterodine), ≤7.2% (serum 5-HMT), ≤3.3% (plasma 5-HMT) and ≤6.3% (urine 5-HMT). The corresponding accuracy ranged from 0.3 to 4.0% (serum tolterodine), −3.0 to 0.8% (serum 5-HMT), −7.1 to −4.5% (plasma 5-HMT) and −1.5 to 0.4% (urine 5-HMT).
Pharmacokinetic analysis
The pharmacokinetic parameters calculated for the comparison of active moieties of fesoterodine and tolterodine ER were area under the plasma concentration vs. time curve from pre-dose to 24 h post dose [AUC(0,24 h)], maximum observed plasma concentration (Cmax), time of maximum plasma concentration (tmax) and terminal phase half-life (t1/2). Additionally, for 5-HMT, the amount excreted in urine (Ae) and renal clearance (CLr) were determined after tolterodine ER and fesoterodine administration. Concentrations were converted to nm units (using molecular weights of 325.5 and 341.5, for tolterodine and 5-HMT respectively) for PK calculations and comparisons of the active moieties of the two treatments, 5-HMT for fesoterodine doses and 5-HMT plus tolterodine for tolterodine doses.
Statistical analysis
Assuming a dropout rate of approximately 20%, 30 subjects were enrolled into the study to ensure that 24 subjects completed the study. The sample size of 24 completed subjects was empirically selected to provide a sufficient number of subjects to enable meaningful pharmacokinetic variability characterization for the two treatments. Based on previous between-study comparisons, active moiety variability was associated specifically because of high tolterodine exposures in CYP2D6 PMs. Therefore, for a precise characterization of the range of active moiety exposures, PM and EM subjects were enrolled in a 1:2 ratio.
The PK parameters were summarized descriptively by moiety, treatment, dose and genotype (EMs and PMs). Plots of median predose concentrations for each treatment and dose level were visually examined to assess the attainment of steady state.
Clinical and laboratory safety assessment
Safety evaluations included clinical monitoring, subject-reported AEs including serious AEs, vital signs (heart rate and blood pressure), 12-lead ECGs and clinical laboratory tests.
Results
Study population
Thirty subjects were enrolled in this study: 20 EMs and 10 PMs of CYP2D6. The age range was 19 to 53 years and the mean age was 31.9 years. Of the subjects enrolled, the majority were men (n = 19/30) and White (n = 27/30). The mean weight was 77.4 kg; the mean height was 173.8 cm and the mean BMI was 25.5 kg m−2.
Active moiety pharmacokinetics
The median concentration vs. time profiles of tolterodine and 5-HMT following repeated doses of tolterodine ER and of 5-HMT following repeated doses of fesoterodine are shown in Figure 1A–C and the pharmacokinetic values are summarized in Table 1 by EMs and PMs. Following tolterodine ER administration, 5-HMT is not formed in PMs of CYP2D6, with the exception of quantifiable but very low (<0.5 ng ml−1) concentrations in some PMs at the 8-mg dose. Furthermore, there was a marked effect of the CYP2D6 genotype on tolterodine exposures (approximately 10-fold higher AUC and sixfold higher Cmax in PMs). In contrast, 5-HMT was formed in both EMs and PMs when fesoterodine was administered, and the exposure was affected only to a modest extent (1.5- to twofold higher Cmax and AUC in PMs). The plasma concentration vs. time data over 24 h post dose on day 5 (4-mg dose) and 36 h post dose on day 10 (8-mg dose) were not adequate for accurate characterization of the half-life of tolterodine, particularly in PMs. The apparent terminal half-life of 5-HMT following 4-mg and 8-mg doses of fesoterodine was well characterized, and the mean ± SD values were 10.4 ± 4.3 and 7.6 ± 2.4 h, respectively, in EMs and 10.4 ± 1.9 and 9.6 ± 2.0 h, respectively, in PMs. There was no apparent difference in 5-HMT half-life between doses or between the EMs and PMs.
Figure 1.
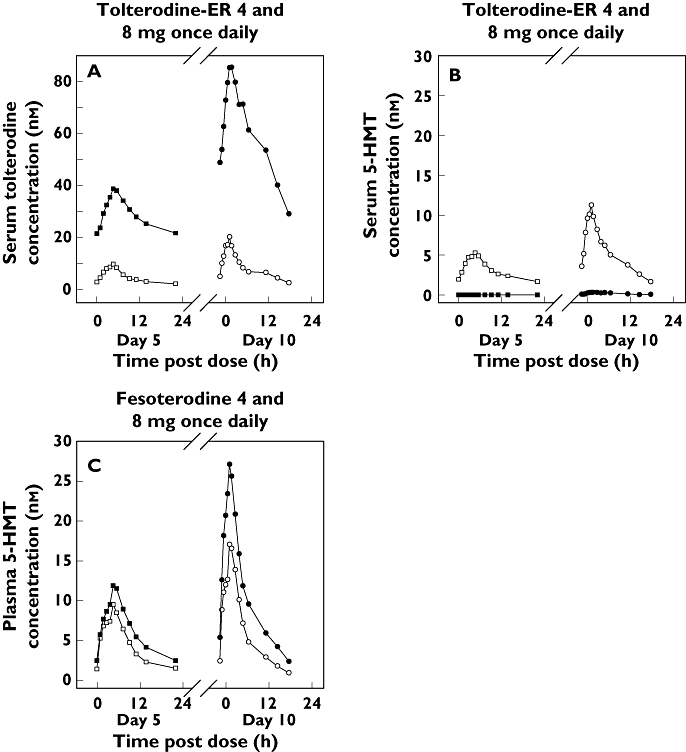
Median steady-state serum concentrations of tolterodine (A) and 5-HMT (B) following administration of tolterodine extended release (ER) and plasma concentrations of 5-HMT (C) following administration of fesoterodine. 4 mg (EM) ( ); 4 mg (PM) (
); 4 mg (PM) ( ); 8 mg (EM) (
); 8 mg (EM) ( ); 8 mg (PM) (
); 8 mg (PM) ( )
)
Table 1.
Active moiety pharmacokinetics at steady state following 4- and 8-mg once daily doses of fesoterodine vs. tolterodine extended release (ER)
| Tolterodine ER 4 mg | Tolterodine ER 8 mg | Fesoterodine 4 mg | Fesoterodine 8 mg | |||||
|---|---|---|---|---|---|---|---|---|
| Tolterodine | 5-HMT | Tolterodine + 5-HMT | Tolterodine | 5-HMT | Tolterodine + 5-HMT | 5-HMT | 5-HMT | |
| Parameter (units)* | EMs (n = 18) | EMs (n = 18) | EMs (n = 18) | EMs (n = 18) | ||||
| AUC(0,24 h) (nm h) (%CV) | 61.1 (116) | 66.1 (42) | 138.1 (83) | 139.1 (120) | 141.2 (42) | 299.8 (87) | 91.7 (46) | 179.6 (42) |
| Cmax (nm) (%CV) | 5.6 (120) | 5.2 (41) | 11.6 (87) | 13.5 (106) | 10.7 (43) | 25.8 (81) | 8.8 (48) | 16.4 (40) |
| tmax (h) (range) | 5 (2, 12) | 5 (2, 6) | 5 (2, 6) | 5 (1, 12) | 4.5 (3, 12) | 4 (1, 6) | 5 (2, 6) | 5 (4, 6) |
| Ae (mg) (%CV) | NA† | 0.38 (39) | NA† | NA† | 0.71 (30) | NA† | 0.44 (40) | 0.89 (36) |
| CLr (ml min−1) (%CV) | NA† | 259 (25) | NA† | NA† | 235 (29) | NA† | 214 (27) | 225 (24) |
| PMs (n = 10) | PMs (n = 10) | PMs (n = 9) | PMs (n = 8) | |||||
|---|---|---|---|---|---|---|---|---|
| AUC(0,24 h) (nm h) (%CV) | 601.7 (47) | NA‡ | 601.7 (47) | 1417.9 (44) | NA‡ | 1421.8 (44) | 136.0 (32) | 314.3 (29) |
| Cmax (nm) (%CV) | 35.7 (46) | NA‡ | 35.7 (46) | 80.9 (40) | NA‡ | 81.2 (40) | 11.9 (33) | 26.5 (27) |
| tmax (h) (range) | 5.5 (5, 8) | NA‡ | 5.5 (5, 8) | 6 (4, 12) | NA‡ | 6 (4, 12) | 5 (4, 6) | 5 (4, 6) |
| Ae (mg) (%CV) | NA† | NA‡ | NA† | NA† | NA‡ | NA† | 0.60 (30) | 1.32 (25) |
| CLr (ml min−1) (%CV) | NA† | NA‡ | NA† | NA† | NA‡ | NA† | 207 (38) | 199 (24) |
Geometric mean (%CV) for AUC(0,24 h), Cmax, and CLr; arithmetic mean (%CV) for Ae, and median (range) for tmax. Tolterodine and 5-HMT concentrations were expressed in nm units, using their molecular weights (325.5 and 341.5 respectively).
Urine samples were not assayed for tolterodine.
5-HMT not formed in PMs following tolterodine ER administration; quantifiable but very low (<0.5 ng ml−1) concentrations in some PMs at the 8-mg dose. EM, extensive metabolizer; 5-HMT, 5-hydroxymethyl tolterodine; PM, poor metabolizer; NA, Not applicable.
Based on comparison of mean AUC values in CYP2D6 EMs who form 5-HMT after administration of both drugs, the bioavailability of 5-HMT was about 39% and 27% higher from fesoterodine compared with tolterodine ER at the 4- and 8-mg doses respectively. The pharmacokinetics of both 5-HMT and tolterodine appear to be dose-proportional, with the following exception of apparent lack of proportionality. Because of the zero and near-zero exposures of 5-HMT in PMs given tolterodine ER, the geometric mean Cmax and AUC values did not appear to be dose-proportional when the data were pooled across genotypes, but were dose-proportional in EMs.
In order to highlight the intersubject variability, individual-subject active moiety concentrations, stratified by genotype and dose level following administration of tolterodine ER and fesoterodine, are shown in Figures 2 and 3. The systemic variability of 5-HMT was well controlled after administration of fesoterodine as well as tolterodine ER (metabolized to 5-HMT in EMs only). Figure 2A (panel A) shows that across the 4-mg and 8-mg tolterodine ER doses given to this group of EM and PM subjects, the active moiety concentrations varied over a >40-fold range. When the individual contributions from tolterodine (Figure 2B) and 5-HMT (Figure 2C) to the total active moiety concentrations following tolterodine ER doses were analysed separately, it was apparent that the concentrations varied much less for 5-HMT (<10-fold) than they did for tolterodine (>100-fold). This indicated that the principal source of variability in active moiety concentrations after tolterodine ER administration was from tolterodine and not 5-HMT. The lower variability of 5-HMT concentrations (ranging over <10-fold) was also maintained when it was formed via esterases and delivered by administration of fesoterodine 4- and 8-mg doses (Figure 3). Consistent with the mean data in Table 1, the individual profiles in Figure 3 demonstrate that fesoterodine administration resulted in bioavailability of 5-HMT in EMs and PMs at levels that were generally higher compared with those seen in EMs only following the same doses of tolterodine ER (Figure 2C).
Figure 2.
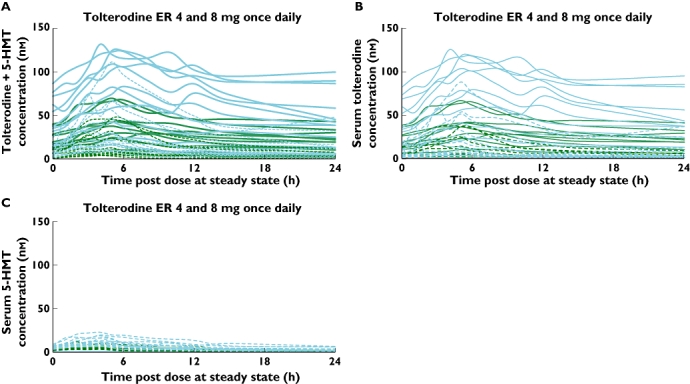
Individual subject steady-state serum concentrations of (A) tolterodine+5-HMT (B) tolterodine and (C) 5-HMT following administration of tolterodine ER (solid and dashed lines for CYP2D6 poor metabolizers and extensive metabolizers respectively; green and blue lines for 4- and 8-mg doses respectively). 5-HMT, 5-hydroxymethyl tolterodine; ER, extended release
Figure 3.
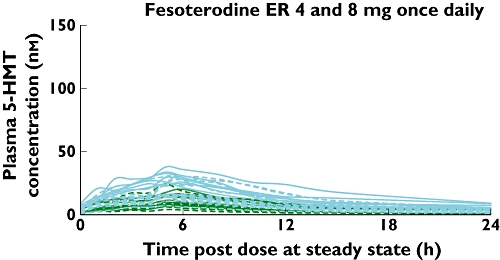
Individual subject steady-state plasma concentrations of 5-hydroxymethyl tolterodine (5-HMT) following administration of fesoterodine (solid and dashed lines for CYP2D6 poor metabolizers and extensive metabolizers respectively; green and blue lines for 4- and 8-mg doses respectively). ER, extended release
As shown in Table 2, the coefficients of variation for AUC and Cmax of 5-HMT following administration of fesoterodine 4 and 8 mg (up to 46% and 48% respectively) were all lower than those of tolterodine + 5-HMT following administration of tolterodine ER 4 and 8 mg (up to 87% and 87% respectively). At the same dose, mean exposure to active moieties (tolterodine and 5-HMT) after tolterodine ER was somewhat higher than the mean exposure to active moiety (5-HMT) after fesoterodine, whereas the exposure ranges were much narrower for fesoterodine. This is largely because of the contribution from excessively high tolterodine exposures in PMs (Figure 4, Table 2).
Table 2.
Span of active moiety exposures, stratified by CYP2D6 genotype at steady state following 4- and 8-mg once daily doses of fesoterodine vs. tolterodine extended release (ER)
| Active moiety AUC(0,24 h) (nm h)* | Active moiety Cmax (nm)* | |||||||
|---|---|---|---|---|---|---|---|---|
| Fesoterodine† | Tolterodine ER‡ | Fesoterodine† | Tolterodine ER‡ | |||||
| 4 mg | 8 mg | 4 mg | 8 mg | 4 mg | 8 mg | 4 mg | 8 mg | |
| EMs | ||||||||
| Geometric mean | 91.7 | 179.7 | 138.1 | 299.8 | 8.8 | 16.4 | 11.6 | 25.8 |
| Minimum | 34.8 | 70.3 | 30.4 | 80.6 | 3.7 | 6.7 | 3.7 | 8.2 |
| Maximum | 220.8 | 357.2 | 586.0 | 1290.0 | 23.9 | 29.9 | 46.7 | 111.0 |
| Maximum : minimum ratio§ | 6.3 | 5.1 | 19.3 | 16.0 | 6.5 | 4.4 | 12.6 | 13.5 |
| PMs | ||||||||
| Geometric mean | 136.0 | 314.3 | 601.7 | 1421.8 | 11.9 | 26.5 | 35.7 | 81.2 |
| Minimum | 77.3 | 159.9 | 186.0 | 432.0 | 6.4 | 12.6 | 10.5 | 24.2 |
| Maximum | 216.7 | 489.0 | 1210.0 | 2490.0 | 19.7 | 37.5 | 69.7 | 132.0 |
| Maximum : minimum ratio§ | 2.8 | 3.1 | 6.5 | 5.8 | 3.1 | 3.0 | 6.6 | 5.5 |
Tolterodine and 5-hydroxymethyl tolterodine (5-HMT) concentrations were expressed in nm units, using their molecular weights (325.5 and 341.5 respectively).
Active moiety: 5-HMT.
Active moiety: Tolterodine+5-HMT.
Index of the span of exposures. ER, extended release; EM, extensive metabolizer; PM, poor metabolizer.
Figure 4.
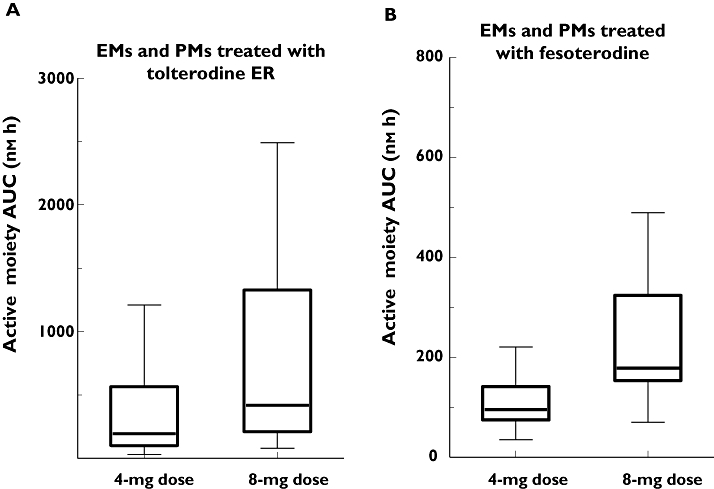
Box–Whisker plots of active moiety exposures, by dose and genotype, following administration of tolterodine extended release (ER) (A) and fesoterodine (B). EM, extensive metabolizer; PM, poor metabolizer
Following fesoterodine 4-mg and 8-mg doses, the mean urinary excretion of 5-HMT was about 0.44 and 0.89 mg, respectively, in EMs and 0.60 and 1.32 mg, respectively, in PMs. The urinary excretion of 5-HMT following fesoterodine was about 1.5-fold higher in PMs, similar to the higher systemic exposures (Table 1). When subjects were analysed separately by CYP2D6 genotype, the mean renal clearance of 5-HMT following 4- and 8-mg fesoterodine doses was 214 and 225 ml min−1 in EMs and 207 and 199 ml min−1 in PMs. The urinary excretion of 5-HMT (in PMs only) was about 0.38 and 0.71 mg and the renal clearance was 259 and 235 ml min−1 following 4- and 8-mg doses of tolterodine ER respectively.
Safety
There were no unusual safety signals observed during the study. In this study in healthy volunteers, both fesoterodine and tolterodine ER were well tolerated. The tolerability of fesoterodine 4 and 8 mg and tolterodine ER 4 and 8 mg was typical of that seen with antimuscarinic agents [2]. Dry mouth was reported in 7% and 14% of the tolterodine ER 4- and 8-mg groups, respectively, and 11% and 26% of the fesoterodine 4- and 8-mg groups. Constipation was reported in 3% and 4% of the tolterodine ER 4- and 8-mg groups and 4% and 4% of the fesoterodine 4- and 8-mg groups respectively. Headache was reported in 14% and 25% in the tolterodine ER 4- and 8-mg groups and 7% and 0% of the fesoterodine 4- and 8-mg groups respectively. All cases of dry mouth, constipation and headache were mild or moderate. Three serious AEs were reported, each leading to discontinuation from the study: one case of abdominal pain related to appendicitis (subject was EM, and the event occurred about 2 days after the last dose of tolterodine ER 8 mg in period 1), one case of myocardial infarction (subject was PM, and the event occurred about 7 days after the last dose of tolterodine ER 8 mg in period 1) and one case of chest pain (subject was EM, and the event occurred about 2 h after the third dose of tolterodine ER 4 mg in period 2). Only the SAE of chest pain of moderate severity was considered related to the study drug. The subject was treated with glyceryl trinitrate and acetylsalicylic acid, and the event resolved after about 4 h.
Discussion
The results of this head-to-head PK study show that active moiety concentrations were considerably more variable following administration of tolterodine ER compared with those following fesoterodine. There was considerably less variability in the PK of 5-HMT after fesoterodine administration compared with the PK of tolterodine + 5-HMT, the active moieties of tolterodine ER. This contrast is best illustrated through the ratios maximum : minimum exposure values observed across the EMs and PMs of CYP2D6 (up to seven- and 40-fold, following treatment with fesoterodine and tolterodine ER respectively).
Tolterodine, and not 5-HMT, was identified as the principal source of variability after tolterodine ER administration. The differential variability was attributable to formation of 5-HMT via CYP2D6 from tolterodine ER vs. via esterases from fesoterodine. The active moiety exposure in tolterodine ER-treated subjects involves two moieties and a genetically variant enzyme, whereas in fesoterodine-treated subjects it involves a singular active moiety that is formed by ubiquitous and genetically invariant esterases. It is known that genetic polymorphisms of the CYP2D6 enzyme can alter the metabolism of drugs requiring hepatic metabolism [12]. In this study, consistent with the prominent role of the CYP2D6 enzyme in the metabolism of tolterodine [15], the administration of the agent in PMs resulted in substantially higher concentrations of tolterodine and 5-HMT compared with EMs, resulting in higher drug exposure in PMs vs. EMs. Because CYP2D6 is not required for the formation of 5-HMT from fesoterodine [14, 16], there was reduced variability of active moiety exposures in EMs and PMs after administration of fesoterodine vs. tolterodine ER. This is particularly desirable for the pharmacologic management of the OAB patient population who may have additional contributors of exposure and/or response variability (i.e. concomitant illnesses and medications).
Once formed, the active moiety 5-HMT had substantial excretion in the urine and it appeared to be similar for both drugs. Mean urinary excretion of 5-HMT following fesoterodine 4 and 8 mg, respectively, was 0.44 and 0.89 mg in EMs and 0.60 and 1.32 mg in PMs. Following tolterodine ER 4 and 8 mg, it was 0.38 and 0.71 mg respectively (EMs only). Renal clearance was similar regardless of administered drug, dose or genotype. Geometric mean renal clearance of 5-HMT was 199 to 259 ml min−1 and was similar regardless of administered drug, dose or genotype.
In tolterodine ER treated subjects, the high pharmacokinetic variability and the attainment of excessively high exposures are specifically related to the tolterodine active moiety and CYP2D6 PM genotype (Figure 2B). Exposures may be even higher in patients with genetic polymorphisms taking concomitant medications considering that patients with high and unpredictable exposures to medications because of genetic polymorphisms can experience greater adverse effects. The relationship between pharmacogenomics and adverse drug reactions has been evaluated by Phillips et al. [17]. Through a systematic review, 27 drugs that are often cited in ADR studies were identified; 59% of these drugs were metabolized by at least one enzyme causing poor metabolism. Alternatively, only 7% to 22% of randomly selected drugs are known to be metabolized by enzymes with this genetic variability. Although the tolerability following tolterodine ER and fesoterodine treatments was not markedly different in this study in healthy volunteers, there is a potential for a genetics and high exposure related differential between the clinical effects of these two drugs. Clinical studies with tolterodine have been conducted with doses up to 12.8 mg in healthy volunteers in phase 1. During phase 2 development, tolterodine doses up to 8 mg daily (4 mg twice daily) were evaluated in subjects with OAB. Increased incidence of residual urinary volume and urinary retention at the highest dose limited the development of tolterodine in phase 3 to doses of 4 mg daily [18]. Fesoterodine has been evaluated at single and multiple once daily doses up to 28 mg in healthy volunteers in phase 1 studies and up to 12 mg once daily in subjects with OAB in phase 2 studies, leading to the selection of 4- and 8-mg once daily doses for phase 3 development [5]. Selection of the phase 3 doses of fesoterodine was based on benefit–risk evaluation of the added efficacy relative to the increased incidence of dry mouth at the 12-mg dose. Unlike tolterodine, urinary retention and residual volume were not dose-limiting [5]. As a result, it was possible to gain marketing authorization of 4-mg and the higher 8-mg daily doses of fesoterodine, whereas tolterodine could only be developed up to 4 mg daily.
The development of fesoterodine at two doses allows for individualization of therapy in OAB patients based on response. Higher active moiety exposures with fesoterodine 8 mg, combined with lower PK variability, will likely improve treatment response and its consistency and predictability when compared with tolterodine ER 4 mg. In a phase 3 study, treatment with fesoterodine 8 mg was shown to provide greater symptom improvement compared with tolterodine ER 4 mg. Although the incidence of dry mouth and constipation was somewhat higher with fesoterodine 8 mg, the related discontinuation rate was low and similar compared with tolterodine ER 4 mg [19]. As further evidence of the superior efficacy of fesoterodine 8 mg, results of two recent head-to-head clinical trials in OAB patients demonstrated that treatment with fesoterodine 8 mg provided greater symptom improvement vs. tolterodine ER 4 mg [20, 21].
In conclusion, the coefficient of variability and the span of the active moiety exposures were considerably lower following administration of fesoterodine (up to 48% and sevenfold respectively) compared with tolterodine ER (up to 87% and 40-fold). Tolterodine, and not 5-HMT, was identified as the principal source of variability after tolterodine ER administration. Fesoterodine delivers 5-HMT with less variability than tolterodine, regardless of CYP2D6 status, with up to 40% higher bioavailability compared with tolterodine ER. Unchanged 5-HMT was excreted in urine after administration of fesoterodine (about 12–13% of administered dose across EMs and PMs) and tolterodine ER (about 9–10% of administered dose in EMs only). The renal clearance of 5-HMT was similar, regardless of the administered drug, dose level or genotype. Both 4- and 8-mg doses of tolterodine ER and fesoterodine were well tolerated.
Acknowledgments
Funding for this study was provided by Schwarz BioSciences GmbH and Pfizer Inc. Editorial assistance was provided by Nancy Sheridan from Complete Healthcare Communications, Inc. and was funded by Pfizer Inc.
Competing Interests
BM, ED, PC and JF are Pfizer employees. PG was an employee of Pfizer Inc. at the time this research was performed. PC, JF and PG have shares in Pfizer.
REFERENCES
- 1.Andersson KE, Chapple CR, Cardozo L, Cruz F, Hashim H, Michel MC, Tannenbaum C, Wein AJ. Pharmacological treatment of overactive bladder: report from the International Consultation on Incontinence. Curr Opin Urol. 2009;19:380–94. doi: 10.1097/MOU.0b013e32832ce8a4. [DOI] [PubMed] [Google Scholar]
- 2.Chapple CR, Khullar V, Gabriel Z, Muston D, Bitoun CE, Weinstein D. The effects of antimuscarinic treatments in overactive bladder: an update of a systematic review and meta-analysis. Eur Urol. 2008;54:543–62. doi: 10.1016/j.eururo.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 3.Nilvebrant L. The mechanism of action of tolterodine. Rev Contemp Pharmacother. 2000;11:13–27. [Google Scholar]
- 4.Tubaro A, De Nunzio C. Comparison of peripherally active substance for treatment of detrusor overactivity: what is new; what is in the pipeline. EAU Update Series. 2004;2:161–9. [Google Scholar]
- 5.Cole P. Fesoterodine, an advanced antimuscarinic for the treatment of overactive bladder: a safety update. Drugs Future. 2004;29:715–20. [Google Scholar]
- 6.Zanger U, Raimundo S, Eichelbaum S. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:23–37. doi: 10.1007/s00210-003-0832-2. [DOI] [PubMed] [Google Scholar]
- 7.Toviaz (fesoterodine). Full Prescribing Information. New York: Pfizer Inc.; 2008. [Google Scholar]
- 8.Bock D, Forster A, Griese EU, Morike K, Brockmeier D, Eichelbaum M. The influence of environmental and genetic factors on CYP2D6, CYP1A2 and UDP-glucuronosyltransferases in man using sparteine, caffeine, and paracetamol as probes. Pharmacogenet Genomics. 1994;4:209–18. doi: 10.1097/00008571-199408000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5:6–13. doi: 10.1038/sj.tpj.6500285. [DOI] [PubMed] [Google Scholar]
- 10.Detrol LA® (tolterodine tartrate). Full Prescribing Information. Kalamazoo, MI: Pharmacia & Upjohn Company; 2005. [Google Scholar]
- 11.Olsson B, Szamosi J. Multiple dose pharmacokinetics of a new once daily extended release tolterodine formulation versus immediate release tolterodine. Clin Pharmacokinet. 2001;40:227–35. doi: 10.2165/00003088-200140030-00006. [DOI] [PubMed] [Google Scholar]
- 12.Xie HG, Kim RB, Wood AJ, Stein CM. Molecular basis of ethnic differences in drug disposition and response. Annu Rev Pharmacol Toxicol. 2001;41:815–50. doi: 10.1146/annurev.pharmtox.41.1.815. [DOI] [PubMed] [Google Scholar]
- 13.Malhotra B, Guan Z, Wood N, Gandelman K. Pharmacokinetic profile of fesoterodine. Int J Clin Pharmacol Ther. 2008;46:556–63. doi: 10.5414/cpp46556. [DOI] [PubMed] [Google Scholar]
- 14.Malhotra B, Gandelman K, Sachse R, Wood N, Michel MC. The design and development of fesoterodine as a prodrug of 5- hydroxymethyl tolterodine (5-HMT), the active metabolite of tolterodine. Curr Med Chem. 2009;16:481–9. doi: 10.2174/092986709789712835. [DOI] [PubMed] [Google Scholar]
- 15.Brynne N, Dalen P, Alvan G, Bertilsson L, Gabrielsson J. Influence of CYP2D6 polymorphism on the pharmacokinetics and pharmacodynamics of tolterodine. Clin Pharmacol Ther. 1998;63:529–39. doi: 10.1016/S0009-9236(98)90104-7. [DOI] [PubMed] [Google Scholar]
- 16.Michel MC. Fesoterodine: a novel muscarinic receptor antagonist for the treatment of overactive bladder syndrome. Expert Opin Pharmacother. 2008;9:1787–96. doi: 10.1517/14656566.9.10.1787. [DOI] [PubMed] [Google Scholar]
- 17.Phillips KA, Veenstra DL, Oren E, Lee JK, Sadee W. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA. 2001;286:2270–9. doi: 10.1001/jama.286.18.2270. [DOI] [PubMed] [Google Scholar]
- 18.Nilvebrant L. Clinical experiences with tolterodine. Life Sci. 2001;68:2549–56. doi: 10.1016/s0024-3205(01)01051-7. [DOI] [PubMed] [Google Scholar]
- 19.Chapple C, Van Kerrebroeck P, Tubaro A, Haag-Molkenteller C, Forst HT, Massow U, Wang J, Brodsky M. Clinical efficacy, safety, and tolerability of once-daily fesoterodine in subjects with overactive bladder. Eur Urol. 2007;52:1204–12. doi: 10.1016/j.eururo.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 20.Herschorn S, Swift S, Guan Z, Carlsson M, Morrow JD, Brodsky M, Gong J. Comparison of fesoterodine and tolterodine extended release for the treatment of overactive bladder: a head-to-head placebo-controlled trial. BJU Int. 2010;105:58–66. doi: 10.1111/j.1464-410X.2009.09086.x. [DOI] [PubMed] [Google Scholar]
- 21.Kaplan S, Schneider T, Foote J, Guan Z, Carlsson M, Gong J, on behalf of the 2nd Fesoterodine Assessment and Comparison Versus Tolterodine (FACT 2) Study Group Superior efficacy of fesoterodine over tolterodine extended release with rapid onset: a prospective, head-to-head, placebo-controlled trial. BJU Int. 2010 doi: 10.1111/j.1464-410X.2010.09640.x. doi: 10.1111/j.1464-410X.2010.09640.x. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]