

Abstract
AIMS
To assess and compare the mechanisms of central nervous system (CNS) penetration of antimuscarinic overactive bladder (OAB) agents.
METHODS
Physical properties were computed or compiled from the literature. Rats were administered 5-hydroxymethyl tolterodine (HMT), darifenacin, oxybutynin, solifenacin, tolterodine or trospium subcutaneously. At 1 h postdose, plasma, brain and cerebrospinal fluid (CSF) concentrations were determined using LC-MS/MS assays. Brain and plasma protein binding were determined in vitro. Permeability in the presence and absence of the efflux transporter P-glycoprotein (P-gp) was assessed in RRCK and MDCK-MDR1 transwell assays.
RESULTS
Oxybutynin displayed extensive CNS penetration, with brain : plasma ratios (B : P), unbound brain : unbound plasma ratios (Kp,free) and CSF : free plasma ratios each >1. Tolterodine (B : P = 2.95, Kp,free = 0.23 and CSF : free plasma = 0.16) and solifenacin (B : P = 3.04, Kp,free = 0.28 and CSF : free plasma = 1.41) showed significant CNS penetration but with some restriction from CNS as indicated by Kp,free values significantly <1. 5-HMT, darifenacin and trospium displayed much lower B : P (0.03–0.16), Kp,free (0.01–0.04) and CSF : free plasma (0.004–0.06), consistent with poor CNS penetration. Permeability in RRCK cells was low for trospium (0.63 × 10−6 cm s−1), moderate for 5-HMT (11.7 × 10−6 cm s−1) and high for darifenacin, solifenacin, tolterodine and oxybutynin (21.5–38.2 × 10−6 cm s−1). In MDCK-MDR1 cells 5-HMT, darifenacin and trospium, were P-gp substrates, whereas oxybutynin, solifenacin and tolterodine were not P-gp substrates.
CONCLUSIONS
Brain penetration was low for antimuscarinics that are P-gp substrates (5-HMT, darifenacin and trospium), and significant for those that are not P-gp substrates (oxybutynin, solifenacin and tolterodine). CNS adverse events reported in randomized controlled clinical trials show general alignment with the preclinical data described in this study.
Keywords: antimuscarinic, blood−brain barrier, CNS, fesoterodine, overactive bladder, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
This study provides antimuscarinic agents for overactive bladder (OAB) display variable association with side effects mediated by the central nervous system (CNS), which may be of particular concern in the elderly.
Adverse effects on CNS functioning are related to muscarinic receptor subtype selectivity and the ability of the agent to cross the blood—brain barrier, where P-gp plays a role in limiting permeability.
WHAT THIS STUDY ADDS
This study provides a parallel investigation of CNS penetration of antimuscarinic OAB agents in vivo and assessment of physical properties and permeability in cell monolayers in vitro.
It adds further understanding of the roles of passive transcellular permeability and P-gp in determining CNS penetration of antimuscarinic OAB agents.
It also enables a comparison of CNS side-effect profiles of OAB agents with preclinical CNS penetration data.
Introduction
Overactive bladder (OAB) is a syndrome defined by the International Continence Society as urgency, with or without urgency incontinence, usually with increased daytime frequency and nocturia [1]. OAB affects at least 10% of the adult population [2] and the prevalence increases with age [3]. Postulated aetiologies for this condition include increased afferent activity, decreased inhibitory control and increased sensitivity of the detrusor muscle to efferent stimulations [4, 5]. Muscarinic receptors are thought to mediate the detrusor contractions of normal voiding, but in OAB the muscarinic receptors are associated with bladder contraction leading to urinary frequency, urgency and urgency incontinence. Antimuscarinic agents, such as darifenacin, oxybutynin, solifenacin, trospium, tolterodine and fesoterodine have consistently demonstrated significant efficacy for the treatment of OAB symptoms [6, 7]. However, treatment is associated with typical anticholinergic side effects such as dry mouth, constipation, somnolence and blurred vision. The adverse effects (AEs) of antimuscarinic drugs may occur because muscarinic receptors are located throughout the body [8] where inhibition of specific receptor subtypes is associated with side effects. For example, in the central nervous system (CNS), muscarinic receptors, particularly the M1 subtype, are thought to play an important role in the consolidation of long-term memory. Cognitive impairment is associated with anticholinergic therapy in the elderly [9] and the side-effect profiles seen with some antimuscarinic OAB drugs are consistent with inhibition of muscarinic receptors in the CNS [10]. Although the incidence of CNS AEs of antimuscarinic agents is generally much lower than that of dry mouth [6, 7], CNS AEs can be of great concern, particularly in the elderly [11]. The incidence of CNS AEs among the available antimuscarinic agents for OAB seems to differ. For instance, while darifenacin has been shown to have no significant effects on memory vs. placebo, oxybutynin ER caused significant memory deterioration which was deemed comparable with brain ageing of 10 years [12]. The ability of certain antimuscarinic OAB drugs to exert CNS-related pharmacological effects at therapeutic doses for OAB treatment depends on their ability to penetrate the CNS as well as relative affinity for relevant muscarinic receptor subtypes in the CNS, particularly M1[13–15]. CNS penetration of drugs depends on the permeability properties of the blood−brain barrier (BBB) [16, 17] and the influence of active efflux transporters present in brain tissue, such as P-glycoprotein (P-gp) [18, 19]. Therefore, the relative permeability and affinity of OAB agents for P-gp is an important consideration in understanding their potential to exert AEs manifested in the CNS.
The purpose of the present paper was to present a comprehensive set of non-clinical in vitro and animal studies that investigated in parallel the CNS penetration potential of antimuscarinic OAB drugs. The following studies were conducted: (i) physicochemical characterization, including lipophilicity; (ii) in vitro RRCK cell passive permeability assessment; (iii) in vitro P-gp mediated efflux in MDCK-MDR1 transcellular flux assay; and (iv) in vivo brain, plasma and CSF concentrations following a single subcutaneous dose in rats. Strategies for assessment of brain penetration of compounds have focused on determination of unbound brain : unbound plasma concentration ratios (Kp,free), and consideration of the involvement of transporter proteins at the BBB, in particular P-gp [19]. Therefore, to understand further CNS disposition, the unbound brain : unbound plasma concentration ratios were estimated using brain and plasma binding experiments in vitro[19, 20]. The overall aim of this package of data was to enable an understanding of the brain penetration potential of antimuscarinic OAB drugs in relation to their physicochemical and permeability properties. The antimuscarinic agents included in these studies were 5-hydroxymethyl tolterodine (5-HMT, the active metabolite of tolterodine as well as fesoterodine), darifenacin, oxybutynin, solifenacin, tolterodine and trospium. Fesoterodine is a pro-drug that is rapidly and extensively converted to 5-HMT by esterases in vivo, and is not detectable after oral administration [21, 22]. Therefore, 5-HMT was evaluated as the relevant active moiety of fesoterodine in this study.
Methods
Materials
Oxybutynin, N-methylscopolamine HBr and atropine (internal standard) were purchased from Sigma-Aldrich. 5-hydroxymethyl tolterodine (5-HMT), solifenacin and trospium chloride were purchased from Toronto Research Chemicals Inc. (Ontario, Canada).
Fesoterodine and scopolamine were obtained from Pfizer Global Research and Development central compound stores (Milwaukee, WI, USA). Tolterodine was purchased from Sequoia Research Products (Pangbourne, UK). Darifenacin was purchased from Toronto Research Chemicals Inc. (Ontario, Canada) and Sequoia Research Products (Pangbourne, UK). Hanks's balanced salt solution (HBSS), cell culture media and supplements were purchased from Invitrogen (Paisley, UK). All other reagents were obtained from Sigma-Aldrich (Poole, UK) or J.T. Baker (Phillipsburg, NJ, USA).
Assessment of physical properties of compounds
The definitions of physical properties of compounds were: log D, logarithm of the distribution coefficient between octanol and buffer at pH 7.4; clog P, logarithm of the calculated partition coefficient between octanol and water, polar surface area (PSA), the area of molecular surface belonging to polar atoms (units: Å2) [17], rotatable bond count, count of all non-terminal single bonds and hydrogen bond acceptor and donor counts, counts of all atoms in a molecule that are potentially involved in a hydrogen bond as acceptor or donor atoms, respectively. All calculated parameters were determined within the proprietary Pfizer database (RGate), where clog P was determined using the BioByte program clog P, version 4.3 and PSA using a published method [17]. Log D values were taken from published sources [23].
Monolayer efflux studies in MDCK (Madin-Darby canine kidney) and RRCK cell lines
MDCK-MDR1 expressing P-gp were originally obtained from Netherlands Cancer Institute (Amsterdam, the Netherlands). RRCK cells were generated in house (Pfizer Inc., Groton, MA, USA) as a subclone of MDCK wild-type (MDCK-WT) cells that displayed low expression of endogenous P-gp (approximately 1–2% of MDCK-WT cells, based on mRNA level). The rank order of permeability values for compounds whose transcellular flux was predominantly by passive diffusion were similar for RRCK and MDCK-WT (data not shown). Monolayer efflux studies were conducted as previously described in the literature for MDCK-MDR1 cells [24]. Cells were cultured in minimal essential medium α with supplements and passaged when 70–80% confluent. Cell monolayer flux studies were conducted 5 days after seeding in 24-well transwell inserts [MDCK-MDR1 in 0.4-µm pore size (Corning Costar) at 1.8 × 105 cells cm−2; RRCK in 1-µm pore size (Becton Dickinson, Cowley, UK) at 4.2 × 104 cells cm−2]. Donor and acceptor solutions were prepared from HBSS, containing HEPES at 20 mm, pH 7.4. Stock solutions of test compounds were prepared at 10 mm in dimethyl sulphoxide (DMSO) and used to prepare donor solutions of 2 µm compound in 0.05% (v/v) DMSO and also containing 2 µm nadolol used as monolayer integrity marker. Apparent permeability (Papp) of compounds was determined in apical to basolateral (A→B) and basolateral to apical (B→A) directions in triplicate by incubation with compound for 2 h at 37°C. Samples of medium (20 µl) from both donor and acceptor chambers were analysed by tandem liquid chromatography and mass spectrometry (LC/MS-MS). The LC/MS-MS system consisted of a 2.1 × 15 mm C18 optilynx column (Optimize Technologies Inc., Oregon city, OR, USA) in line with Onyx monolithic C18 column 50 × 4.6 mm (Phenomenex, Torrance, CA, USA) operating at a flow rate of 3 ml min−1. The aqueous mobile phase consisted of 90% 2 mm ammonium acetate, 10% methanol, 0.1% formic acid. The organic mobile phase was 10% 2 mm ammonium acetate, 90% methanol, 0.1% formic acid. Mass spectrometry was performed on a SCIEX API 4000 triple quadrupole mass spectrometer (Applied Biosystems, Ontario, Canada), with turbo ion spray source. Data were acquired in positive ion mode with an ion spray probe voltage of 5.5 kV. The following selected reaction monitoring transitions, given as mass : charge ratio (m/z) were used to measure the compounds: tolterodine m/z 310→201 and nadolol m/z 310→254 at collision energy of 25 eV; darifenacin m/z 427→147, oxybutynin m/z 358→124, 5-HMT m/z 342→223, fesoterodine m/z 412→223 and nadolol m/z 310→201 at a collision energy of 40 eV; solifenacin m/z 364→110, trospium m/z 392→182 and nadolol m/z 310→56 at a collision energy of 55 eV.
Papp values were calculated according to the equation Papp = (Q/t) × 1/C0× 1/A, where Q is the sampled concentration in the acceptor compartment, t incubation time; C0 is the initial concentration in the donor compartment and A is the area of the filter of the transwell plate. Monolayers with nadolol Papp values of less than 1 × 106 cm s−1 were deemed intact.
In vivo brain penetration study
All procedures performed on animals were in accordance with US federal regulations and established NIH guidelines and were reviewed and approved by the Institutional Animal Care and Use Committee. Male Sprague-Dawley rats weighing approximately 250 g (n = 3) received a single dose of compound subcutaneously (0.3 or 1 mg kg−1). Dosing solutions were prepared fresh in saline on the day of the study and were administered at 1 ml kg−1. At 1 h postdose, animals were euthanized using CO2 and approximately 3 ml of blood removed by cardiac puncture and plasma prepared by centrifugation. Cerebrospinal fluid (CSF) was drawn from the cisterna magna using a 25 gauge needle attached to polyethylene tubing (2 mm) and a syringe. Brains were removed, rinsed with saline and weighed. All samples were immediately frozen on dry ice after processing and stored at −20°C until analysis. Brain samples were homogenized in four volumes of phosphate buffered saline (1 in 5 tissue dilution). Plasma, CSF and brain samples were analysed using a LC/MS-MS method. Calibration standard and quality control (QC) samples were prepared in untreated rat plasma or brain homogenate. The lower limit of quantitation (LLOQ) for all compounds was 0.1 ng ml−1 while the upper limit of quantitation (ULOQ) ranged from 25 to 100 ng ml−1, depending on the dose received. Aliquots of plasma or brain (50 µl) were basified using 50 mm K2HPO4, pH 8.0 (200 µl) and mixed with internal standard (atropine, 10 µl of 50 ng ml−1) before loading onto a 96-well solid phase extraction plate (Waters Oasis HLB, 10 mg). Following several washes (400-µl water, followed by 400-µl methanol : water 10:90), compounds were eluted using methanol (400 µl) and eluant dried under nitrogen heated to 40°C. Samples were reconstituted in 50-µl 5 mm ammonium acetate buffer containing 0.1% formic acid (v/v) before analysis by LC/MS-MS. Extracted samples (10 µl) were injected onto a Phenomenex Luna C18 analytical column (5 µm, 2.1 × 50 mm). The liquid chromatography system consisted of a gradient mixture of acetonitrile and 5 mm ammonium acetate pH adjusted with formic acid (0.1%, v/v) maintained at a flow rate of 0.3 ml min−1. Samples containing concentrations above the ULOQ were diluted with control matrix and re-analysed. CSF samples (20 µl) were mixed with internal standard and ammonium acetate buffer [5 mm with 0.1% formic acid (v/v)] and were quantified using a direct-inject method onto the LC/MS-MS system described above with standard curves prepared in untreated rat plasma ultrafiltrate. Data were acquired in positive ion mode with an ion spray probe voltage of 5.5 kV using the same reaction monitoring transitions as in the transcellular flux experiments, with the internal standard atropine monitored at m/z 290→124.
Plasma and brain binding
Binding of compounds to rat plasma or rat brain was determined by a previously described equilibrium dialysis method [20]. Briefly, untreated plasma or brain homogenate (5× diluted with PBS) was fortified with test compound to yield a final concentration of 1 µm. Samples were placed in a 96-well equilibrium dialysis block (HTDialysis, Gales Ferry, CT, USA) fitted with Spectra-Por 2 membranes (Spectrum Laboratories, Rancho Dominguez, CA, USA) and incubated at a temperature of 37°C, humidity of 95% and CO2 concentration of 5%. After 6 h, aliquots (10 µl) of buffer and matrix were removed and added directly to a 96-well polypropylene block containing internal standard in acetonitrile (atropine, 50 ng ml−1). An equal volume of the opposite matrix was added to each sample to yield uniform sample composition. All samples were analysed using LC/MS-MS with conditions similar to those described above. The unbound fraction in plasma (fup) was calculated using the ratio of drug concentrations in buffer to matrix. Unbound fraction in brain (fub) was calculated in a similar fashion but tissue dilution factor (D) was taken into account according to the correction described by Kalvass & Maurer [20], whereby fub = 1/D/[(1/fu2) − 1]+ 1/D, where fu2 = unbound fraction measured in diluted tissue homogenate.
Analysis of in vivo brain penetration data
The treatment of data obtained from in vivo brain penetration studies has been discussed extensively in the literature and has underlined the value of applying knowledge of tissue binding in comparing tissue concentrations, and hence understanding brain penetration [19, 20, 25]. Furthermore, comparsion of results obtained using unbound brain tissue homogenate concentrations with in vivo microdialysis has supported the use of free concentrations in brain and CSF as surrogates for concentrations in brain interstitial fluid [26]. Therefore, the tissue concentration, plasma protein binding and brain binding data were used to calculate the following parameters:
 |
 |
 |
 |
 |
 |
Results
Physicochemical property assessment of OAB agents
The physicochemical properties that describe the lipophilicity, hydrogen bonding potential, polarity and flexibility of the range of antimuscarinics studied are summarized in Table 1. The compounds used for this investigation possess relatively low molecular weight (range 325–426 Da), typical of small drug molecules and, with the exception of trospium, can be classed as lipophilic based on their log D and calculated log P values (ranges log D 0.74–>3.3 and clog P 3.6–4.9). The rank order of lipophilicity determined by log D [23] was oxybutynin > darifenacin > tolterodine ≈ solifenacin ≈ fesoterodine > 5HMT > trospium. Trospium (log D −1.22, clog P −1.2) is a much more hydrophilic compound in comparison due to the presence of a quaternary amine group that is ionized at physiological pH. Hydrogen bonding potential evaluated as the sum of hydrogen bond donors and acceptors was broadly similar between compounds, with rank order darifenacin = 5-HMT (5) > oxybutynin = trospium (4) > tolterodine = solifenacin = fesoterodine (3). All the compounds possessed relatively low PSA values ranging from 23.5Å2 for tolterodine to 55.6 Å2 for darifenacin. The number of rotatable bonds in a molecule is a measure of the flexibility of the compound. The range in the number of rotatable bonds in this series of compounds is relatively large with solifenacin having the lowest value (4) and 5-HMT and darifenacin having the highest value (8).
Table 1.
Physicochemical properties of various antimuscarinic agents
| Antimuscarinic agent | Clog P | Log D | Hydrogen bond acceptors | Hydrogen bond donors | Rotatable bonds | Molecular weight (Da) | PSA (Å2) |
|---|---|---|---|---|---|---|---|
| Trospium | −1.2 | −1.22 | 3 | 1 | 5 | 393 | 46.5 |
| 5-HMT | 3.7 | 0.74 | 3 | 2 | 8 | 341 | 43.7 |
| Darifenacin | 3.6 | 2.7 | 3 | 2 | 7 | 427 | 55.6 |
| Solifenacin | 4.7 | 1.69 | 3 | 0 | 4 | 362 | 32.8 |
| Tolterodine | 5.2 | 1.83 | 2 | 1 | 7 | 325 | 23.5 |
| Oxybutynin | 4.9 | >3.3 | 3 | 1 | 8 | 357 | 49.8 |
| Fesoterodine | 4.4 | ND | 2 | 1 | 11 | 412 | 49.8 |
For definition of parameters in the table, refer to Methods.
Transcellular flux across RRCK and MDR1- MDCK cell monolayers
In vitro cell membrane permeability and the influence of P-gp were assessed in cell lines designed to measure transcellular flux in the presence and absence of P-gp. A stable population of MDCK cells was selected by flow cytometry to have little or no functional P-gp that would contribute to efflux of substrates of this protein. In this cell line, designated RRCK, compounds displayed a range of flux values in the apical to basolateral direction (A→B) (Figure 1), and compounds could be classed as possessing low (<5 × 10−6 cm s−1), moderate (5–15 × 10−6 cm s−1) or high (>15 × 10−6 cm s−1) transcellular flux. Thus, trospium (Papp = 0.63 × 10−6 cm s−1) possessed low flux, 5-HMT moderate and solifenacin, tolterodine, darifenacin and oxybutynin high flux values.
Figure 1.
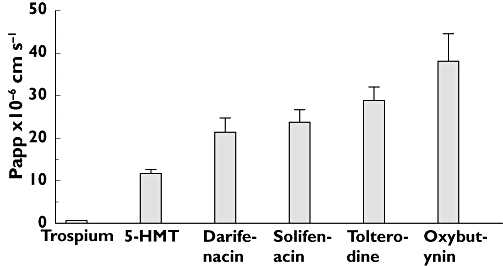
Passive transport of antimuscarinic agents in RRCK cells. Fluxes of compounds across RRCK cell monolayers in the apical to basolateral (A→B) direction are shown. Incubations were performed in triplicate and error bars represent SDs
The ability of compounds to act as P-gp substrates was assessed in MDCK cells transfected with the human mdr1 gene that expresses P-gp (MDCK-MDR-1). The ratio of apical to basolateral : basolateral to apical fluxes (A→B : B→A efflux ratio) was used as an index of P-gp-mediated efflux across MDCK-MDR-1 monolayers (Figure 2). The efflux ratios shown by the range of compounds varied between 0.6 and 13.7. Using an efflux ratio of >2 to indicate significant P-gp-mediated efflux [27], it was found that trospium, 5-HMT and darifenacin were likely to be substrates of the P-gp transporter, whereas solifenacin, tolterodine and oxybutynin were unlikely to act as substrates.
Figure 2.
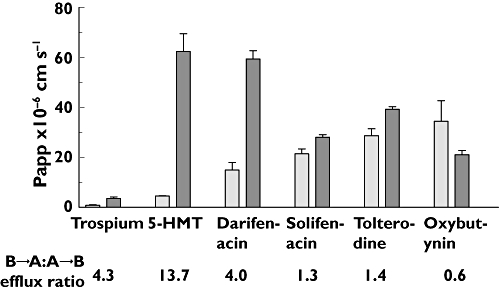
Transcellular flux of antimuscarinic agents in MDCK-MDR1 cells. Fluxes of compounds across MDCK-MDR1 cell monolayers were measured in apical to basolateral (A→B) and basolateral to apical (B→A) directions. The ratio of A→B/B→A fluxes (efflux ratio) is shown under each compound. Incubations were performed in triplicate and error bars represent SDs. A→B flux ( ); B→A (
); B→A ( )
)
In vivo CNS penetration of OAB agents in rats
Determination of plasma, brain and CSF drug concentrations by specific LC/MS-MS afforded the required sensitivity and selectivity for accurate determination of drug concentration in tissues and ensured that metabolites did not contribute to apparent drug concentrations (the tissue concentration data used to determine brain penetration are shown in Table 2). CNS penetration was initially assessed by calculating the total brain : plasma concentration ratios (B : P) and CSF : free plasma concentration ratios (CSF : free plasma). During collection of the brain in these experiments, blood remains in the tissue and following homogenization and LC/MS-MS analysis the residual blood can contribute an observed B : P of up to 0.04, reflecting presence of drug in the vasculature of the brain rather than true brain penetration [28]. Hence, values of B : P below 0.04 and CSF : free plasma approaching 0 are consistent with no significant brain penetration and no equilibrium between CSF and free plasma. The results of CNS penetration in vivo are summarized in Table 3 and displayed graphically in increasing order of their CNS penetration in Figure 3.
Table 2.
Mean concentrations and unbound fractions of antimuscarinic agents in rat tissues following subcutaneous dosing
| Compound | Dose (mg kg−1) | Plasma concentration (ng ml−1) | Brain concentration (ng g−1) | CSF concentration (ng ml−1) | fup | fub |
|---|---|---|---|---|---|---|
| Trospium | 1.0 | 106 ± 85 | 2.68 ± 1.31 | 0.33 ± 0.34 | 0.72 ± 0.02 | 0.19 ± 0.02 |
| 5-HMT | 0.3 | 27.3 ± 1.3 | 4.38 ± 0.58 | 0.74 ± 0.07 | 0.72 ± 0.03 | 0.16 ± 0.01 |
| Darifenacin | 0.3 | 39.0 ± 5.8 | 3.04 ± 0.10 | 0.17 ± 0.04 | 0.074 ± 0.006 | 0.019 ± 0 |
| Solifenacin | 1.0 | 59.3 ± 4.6 | 180 ± 55 | 10.9 ± 1.7 | 0.13 ± 0.02 | 0.012 ± 0.001 |
| Tolterodine | 0.3 | 20.8 ± 2.7 | 61.3 ± 6.7 | 1.07 ± 0.17 | 0.32 ± 0.02 | 0.025 ± 0.005 |
| Oxybutynin | 0.3 | 22.5 ± 5.2 | 141 ± 27 | 0.71 ± 0.26 | 0.019 ± 0.001 | 0.01 ± 0 |
| N-methylscopolamine | 0.3 | 45.9 ± 6.3 | 2.33 ± 0.92 | 0.38 ± 0.25 | 0.96 ± 0.06 | 0.40 ± 0.06 |
| Scopolamine | 0.3 | 29.8 ± 2.0 | 102 ± 6 | 27.9 ± NA | 0.83 ± 0.04 | 0.46 ± 0.02 |
Values in the table are mean ± SD for three separate determinations. fub, unbound fraction in brain; fup, unbound fraction in plasma.
Table 3.
In vivo CNS penetration of various antimuscarinic agents in rats following subcutaneous administration
| Compound | fup : fub | Brain : plasma concentration ratio | Kp,free | CSF : free plasma concentration ratio |
|---|---|---|---|---|
| Trospium | 3.8 | 0.03 | 0.01 | 0.004 |
| 5-HMT | 4.5 | 0.16 | 0.04 | 0.04 |
| Darifenacin | 3.9 | 0.08 | 0.02 | 0.06 |
| Solifenacin | 11 | 3.04 | 0.28 | 1.41 |
| Tolterodine | 13 | 2.95 | 0.23 | 0.16 |
| Oxybutynin | 1.9 | 6.27 | 3.30 | 1.66 |
| N-methylscopolamine | 2.4 | 0.05 | 0.02 | 0.01 |
| Scopolamine | 1.8 | 3.42 | 1.90 | 1.13 |
Definition of terms: fub, unbound fraction in brain; fup, unbound fraction in plasma; Kp,free, ratio of unbound concentrations in brain : plasma.
Figure 3.
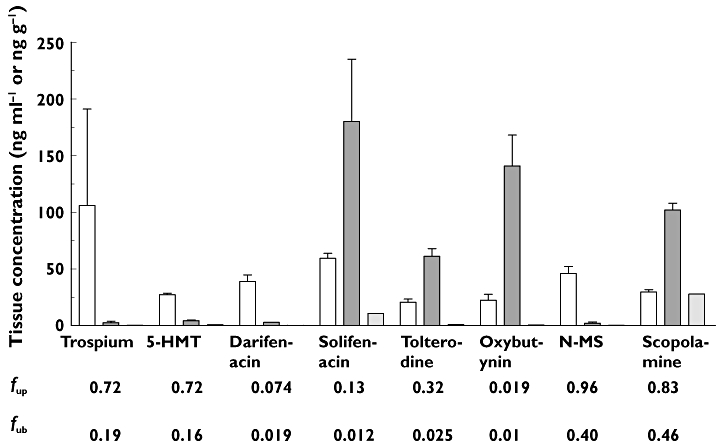
Tissue concentrations of antimuscarinic agents following subcutaneous administration in rats. Mean concentrations of compounds in plasma, brain and CSF determined following subcutaneous dosing to three animals are shown, where error bars represent SDs. Unbound fractions in plasma (fup) and brain (fub) are shown underneath each compound. plasma (ng ml−1) ( ); brain (ng g−1) (
); brain (ng g−1) ( ); CSF (ng ml−1) (
); CSF (ng ml−1) ( )
)
In parallel experiments, scopolamine (0.3 mg kg−1) and N-methylscopolamine (0.3 mg kg−1) were administered as comparator compounds that display significant (positive control) or marginal (negative control) CNS effects, respectively, at similar doses in mice [29] and in humans [30]. Scopolamine, oxybutynin and solifenacin displayed B : P and CSF : free plasma concentration ratios greater than 1, consistent with significant CNS penetration and equilibrium between CSF and free plasma. Tolterodine had a B : P >1 (2.95) and CSF : free plasma of 0.16, consistent with significant CNS penetration but with CSF not in equilibrium with free plasma under these experimental conditions. 5-HMT, darifenacin and trospium showed results that were similar to N-methylscoplamine, with B : P in the range 0.03–0.16 and CSF : free plasma of 0.004–0.06, consistent with no significant CNS penetration.
In vitro plasma and brain binding data and estimation of Kp,free
The binding of compounds in rat brain tissue homogenate and plasma were determined in vitro and data expressed as unbound fraction in brain (fub) and plasma (fup). The ratio of fup : fub was calculated in order to estimate the distribution of compounds between brain and plasma assuming CNS penetration is due to passive diffusion [31]. 5-HMT, darifenacin, oxybutynin and trospium displayed slightly higher unbound fractions in plasma than in brain (fup : fub ratios 1.9–4.5), whereas tolterodine and solifenacin showed even higher fup : fub ratios of 13 and 11, respectively. Scopolamine and N-methylscopolamine were found to have similar fup : fub ratios of 1.8 and 2.4, respectively. Assuming CNS distribution is due to passive permeability, the in vitro binding results would predict that all the antimuscarinic agents have significant CNS penetration (fup : fub >1).
The in vitro brain binding data and observed B : P ratio were used to calculate the unbound brain : unbound plasma ratio (Kp,free). Kp,free ratios were compared with the observed CSF : free plasma ratios to assess whether free brain and CSF concentrations were in equilibrium with free peripheral concentrations in the plasma (Table 3). 5-HMT, trospium and darifenacin could be categorized as having low Kp,free with values ranging from 0.01 to 0.04 and low CSF : free plasma with values between 0.004 and 0.06. The low ratios were similar to Kp,free and CSF : free plasma of N-methylscopolamine, 0.02 and 0.01, respectively. Oxybutynin and scopolamine afforded a high Kp,free and CSF : free plasma approaching unity. Tolterodine provided intermediate Kp,free and CSF : free plasma (0.23 and 0.16) while solifenacin had a similar Kp,free (0.28) but higher CSF : free plasma of approximately 1.41. The results are consistent with low CNS penetration of 5-HMT, darifenacin, N-methylscopolamine and trospium, and significant CNS penetration of oxybutynin, scopolamine, solifenacin and tolterodine. However, Kp,free values for solifenacin and tolterodine were significantly lower than unity, suggestive of some possible restriction in CNS penetration.
Discussion
The main objective of the present study was to determine the CNS penetration of antimuscarinic agents used for the treatment of OAB. CNS penetration of drugs may be limited by the passive permeability properties and efflux transporters, such as P-gp, in the endothelial cells of the cerebrovascular capillaries that comprise the BBB. Hence, a secondary objective was to provide a mechanistic explanation of any apparent differences in the ability of OAB drugs to cross the BBB. The drugs were assessed in a rat model that determined their CNS penetration in vivo by measuring tissue concentration and binding of compounds. The in vivo results were then put into context with physical property assessment and results from permeability and P-gp substrate assays to provide a mechanistic interpretation of the CNS penetration results.
CNS penetration of OAB agents in vivo
In the present study, rats were administered subcutaneous doses of compounds and brain, plasma and CSF were sampled 1 h after dosing. As a result of theoretical pharmacokinetic considerations combined with results of in vivo microdialysis experiments, it has been suggested that equilibration of drugs between brain and blood occurs very rapidly [32]. Other work that examined the time to reach brain equilibrium in rats dosed subcutaneously with a range of compounds found that compounds with a combination of low brain binding and high blood brain barrier permeability reached equilibrium (B : P at plateau) within 10 min to 2 h [33]. However, tissue sampling at a single time point remains a potential limitation in experimental design because it is possible that compounds may not have attained steady-state distribution between brain and plasma within that time. The most rigorous protocol would employ tissue sampling over a time course, allowing tissue concentration ratios to be determined at a time point shown to represent steady state, and potentially over a range of doses. However, such a protocol is extremely time consuming and considerably increases the number of animals used to generate the data. The single dose and time point design employed in our study aimed to minimize animal use and provide plasma and tissue concentrations high enough to permit their accurate measurement by specific LC/MS-MS assay, while avoiding saturation of transporter proteins that could affect tissue distribution.
The ratio of total concentration in brain and plasma (B : P) is most commonly used to assess CNS penetration. However, if CNS penetration is solely determined by passive permeability, steady-state B : P is dependent on the relative non-specific binding to plasma proteins (fup) and brain tissue (fub) suggesting that the binding ratio can be a good surrogate for in vivo steady state B : P [20, 31]. The fup : fub values obtained for the OAB agents were found to be in the range of 1.9 to 13. Therefore, if these compounds have high passive permeability and are not substrates for efflux transporters, these results taken alone would predict that all OAB agents will have significant brain penetration, as suggested by their free fractions in plasma relative to brain. However, based on tissue concentration data in rats, the OAB agents may be broadly categorized into two groups: one consisting of trospium, 5-HMT and darifenacin with no significant CNS penetration and another consisting of oxybutynin, solifenacin and tolterodine with significant CNS penetration. Within each group, the positive and the negative controls, scopolamine and N-methylscopolamine helped confirm the categorization as CNS-penetrant and non-penetrant, respectively. The observed B : P ratios for solifenacin, tolterodine and oxybutynin were >1 and similar to scopolamine, consistent with significant CNS penetration. For these compounds the in vitro fup : fub was consistent with the in vivo results since predictions were within ∼fourfold of observed. Tissue binding results were also used to calculate unbound tissue concentrations and enable comparison of free concentrations in plasma, brain and CSF, to indicate whether equilibrium between the compartments was likely to exist. As the concentration of protein in CSF is very low, it was assumed that free concentrations of drug are measured in CSF. The Kp,free and CSF : free plasma ratios for oxybutynin and scopolamine afforded ratios of approximately 1, indicating that equilibrium between free concentrations in the brain, CSF and plasma had been achieved. However, for solifenacin the CSF : free plasma was approximately 1, consistent with equilibrium between the CSF and free plasma compartment, but Kp,free was 0.28 indicating that free brain and free plasma concentrations were not at equilibrium. Tolterodine also demonstrated significant CNS penetration, yet Kp,free (0.23) and CSF : free plasma (0.16) did not reach unity indicating that equilibrium between CNS compartments had not been achieved. These results with solifenacin and tolterodine indicate some limitation in CNS penetration or may reflect slow attainment of equilibrium across the BBB relative to the other OAB agents. However, it cannot be ruled out that they act as substrates for a transporter protein other than P-gp that limits free brain and CSF concentrations. It is also possible that differences in the physiology of the CSF/plasma barrier and BBB play a role in the difference in Kp,free and CSF : free plasma ratio for solifenacin. CSF is produced at the choroid plexus, a fenestrated endothelial cell layer lacking the tight junctions of the BBB. Additionally, there have been reports of different expression and localization of transporter proteins in the choroid plexus relative to the BBB. These differences in permeability and transporter expression may lead to different brain tissue and CSF free concentrations and different rates in reaching equilibrium across the OAB class [34].
The B : P, Kp,free and CSF : free plasma ratios for trospium, 5-HMT and darifenacin were similar to those of N-methylscopolamine, which is consistent with no significant CNS penetration. For these compounds, the P : B ratios of unbound fractions (fup : fub) significantly over-predicted their respective B : P total drug concentration ratios by 28- to 127-fold, suggesting that passive permeability alone does not determine CNS penetration of these compounds. While it is recognized that further studies under confirmed steady-state conditions would be needed to define fully the brain penetration of these compounds, the single-dose results were well corroborated by the results of efflux transport experiments (discussed below). Furthermore, the single-dose results are relevant because of the known cognitive function effects of anticholinergic drugs like scopolamine and oxybutynin upon acute, as well as, chronic dosing [35]. Previous studies of receptor occupancy by OAB agents in rodents are broadly consistent with the above findings. Comparison of muscarinic receptor occupancy in the cerebral cortex of mice ex vivo demonstrated higher binding of oxybutynin than tolterodine and darifenacin following oral administration [36]. In rats, comparison of the muscarinic receptor occupancy of darifenacin, oxybutynin, solifenacin and tolterodine in regions of the brain suggested that oxybutynin crossed the BBB more easily than the other agents [37].
Physical properties and passive permeability as predictors of CNS penetration in vivo
Tight junctions between the vascular endothelial cells forming the BBB present a physical barrier that limits paracellular movement of molecules. Hence, CNS penetration requires small molecules to pass through the vascular endothelial cells. Passive permeability of cell membranes is directly related to the physical properties of a compound [16, 17]. For the prediction of BBB penetration, van de Waterbeemd et al. [16] identified a relatively simple set of rules based on the analysis of the properties associated with CNS drugs and those associated with non-CNS drugs, proposing that CNS-penetrant compounds require positive lipophilicity (log D/clog P) values, molecular weights of less than 450 Da and polar surface area (PSA) of below 90 Å2. Another investigation [38] suggested a similar molecular weight cut-off but indicated that the PSA should be <70 Å2, that the clog P should be in the range 0–6 and additionally proposed <9 rotatable bonds, <3 H bond donor and <5 H bond acceptors. Based on these physicochemical property criteria alone, trospium (quaternary ammonium with clog P −1.2), would not be expected to display good CNS penetration. Accordingly, trospium displayed low CNS penetration in rats and it is likely that this is a result of its low passive membrane permeability, as displayed in RRCK cells. The other OAB agents would be expected to possess high passive membrane permeability and to this extent have properties compatible with BBB penetration. In RRCK cells, 5-HMT, darifenacin oxybutynin, solifenacin and tolterodine demonstrated moderate to high permeability, consistent with the physicochemical property assessment that their passive permeability is unlikely to limit their CNS penetration. However, results in the rat model in vivo showed that 5-HMT and darifenacin did not show significant CNS penetration, based on Kp,free and CSF : free plasma values between 0.02 and 0.06, indicating the presence of another mechanism that restricts CNS penetration of these agents.
Role of P-gp efflux transporter in CNS penetration in vivo
The best studied efflux transporter limiting BBB penetration is P-glycoprotein (P-gp), which resides on the basolateral membrane of the capillary endothelial cells [39] and has been shown to limit the CNS penetration of many small molecules such as digoxin, loperamide, indinavir, ritonavir and non-sedating antihistamines [40–42]. In transfected MDR1-MDCK cells expressing P-gp, basal to apical transcellular fluxes of trospium, 5-HMT and darifenacin significantly exceeded those in the apical to basolateral direction (ratio >2), indicating that these compounds behaved as substrates for the P-gp efflux transporter. Under the conditions of the assay, 5-HMT displayed the highest efflux ratio of all the compounds examined, followed by trospium and darifenacin. Solifenacin, tolterodine and oxybutynin did not appear to be P-gp substrates in this assay. Therefore, an interaction with P-gp may further limit CNS penetration of trospium, as suggested elsewhere in studies of mdr1 knockout mice [43]. Indeed, the low passive permeability of trospium may provide a background against which P-gp-mediated active transport is able to effectively exclude this compound from the CNS. Although the RRCK data indicate that 5-HMT and darifenacin possess high intrinsic transcellular permeability, the in vivo results suggest that they are effectively excluded from the CNS. The results of the MDCK-MDR1 assay point to a role for P-gp-catalysed efflux in restricting brain penetration of these two compounds. In the case of darifenacin, this is consistent with its previously reported activity as a P-gp substrate [44]. The remaining compounds in this study, solifenacin, tolterodine and oxybutynin, were found to penetrate significantly brain in the rat and were also found to possess high transcellular permeability without evidence of a significant interaction with P-gp in the MDR1-MDCK system. In the case of solifenacin, this concurs with a previous report showing a lack of transport by MDR1 expressed in LLC-PK1 cells [45].
CNS penetration in relation to clinical CNS-related effects
The cognitive effects of anticholinergic medications have been assessed in older adults [9, 35]. While some medications with anticholinergic activity appear to affect negatively the cognitive performance in older adults, it remains to be systematically evaluated whether a reduction in the total anticholinergic burden in geriatric patients by substituting their medication with equally effective alternatives with lower anticholinergic activity will result in a favourable clinical impact on cognitive deficit. Analogous to this reasoning, it may be hypothesized that in patients taking medication with intended anticholinergic effects, such as those for treatment of OAB, choice of one with lower CNS penetration and/or lower affinity for M1 receptors may provide a more CNS-sparing clinical outcome. Several examples of drug classes exist where distribution into the CNS correlates with CNS side effects due to inhibition of centrally located receptors, including H1-receptor antagonists [42] and opiates [46, 47]. In these examples, restricted CNS penetration, where it occurs, is thought to result from an affinity for the efflux transporter, P-gp [47]. The CNS effects of some of the antimuscarinics for the treatment of OAB have been evaluated in clinical studies. In a study in older subjects, it was shown that mild cognitive deterioration occurs when patients take non-selective and CNS-penetrant antimuscarinic agents such as oxybutynin compared with darifenacin which has poor CNS penetration, as confirmed in this paper, and is M3-selective [12]. No significant CNS electrophysiological effects, assessed by EEG, were observed with trospium or tolterodine [48, 49], for which we have demonstrated poor and limited (although significant) CNS penetration, respectively. In contrast, cognitive impairment, sleep disorders and EEG effects on the CNS have been reported with oxybutynin [48, 50, 51] which showed the highest CNS penetration among the antimuscarinic agents tested. In a study in cognitively intact older adults [52], following administration of trospium extended release 60 mg once daily to steady state, trospium was undetectable in the CSF and also did not have a meaningful effect on learning or memory, assessed by the Hopkins Verbal Learning Test and Brief Visiospatial Memory Test. These clinical findings are consistent with the physicochemical properties, lack of CNS penetration and affinity for P-gp in our nonclinical models.
The CNS penetration properties of the various antimuscarinics, determined in the nonclinical models in this study, are summarized in Table 4 and are compared with reported clinical CNS side effects as documented in product labels. Across all the OAB drugs, CNS-related adverse event reports from post-marketing surveillance and clinical trials, as reported in the approved product labels, included headache, hallucinations, delirium, insomnia, nervousness, disorientation, memory impairment and confusion. Somnolence and dizziness were the principal CNS-related adverse events reported in excess of placebo rates and by ≥1% of patients receiving active antimuscarinic OAB treatment in the controlled phase 3 clinical trials. Consistent with the classification based on preclinical CNS penetration models, CNS AEs reported in clinical trials are the lowest for fesoterodine (pro-drug of 5-HMT) and trospium, intermediate for darifenacin, solifenacin and tolterodine and highest for oxybutynin. While dedicated clinical studies on cognitive function effects have not been conducted with all the current antimuscarinic drugs used for treatment for OAB, the CNS AE reports in randomized controlled clinical trials show general alignment with the preclinical data described in this study.
Table 4.
Summary evaluation of the CNS penetration potential of various antimuscarinic agents relative to reported clinical CNS side effects
| Antimuscarinic drug | Predicted CNS penetration potential, based on physicochemical properties | Measured permeability in vitro | Substrate of P-gp in vitro | Measured CNS penetration in rats in vivo | Observed clinical CNS side effects* |
|---|---|---|---|---|---|
| Trospium | Not significant | Low | Yes | Not significant | NR† |
| Fesoterodine (5-HMT) | Significant | Moderate | Yes | Not significant | NR‡ |
| Darifenacin | Significant | High | Yes | Not significant | 0.9–2.1% dizziness§ |
| Solifenacin | Significant | High | No | Significant | 1.8–1.9% dizziness¶ |
| Tolterodine | Significant | High | No | Significant | 3% somnolence** |
| 2% dizziness†† | |||||
| Oxybutynin | Significant | High | No | Significant | 2–12% somnolence‡‡ |
| 4–6% dizziness‡‡ | |||||
| 12.6% somnolence§§ | |||||
| 15.6% dizziness§§ |
Incidence of CNS adverse events in randomized controlled clinical trials, as reported in approved product labels.
Sanctura 20 mg twice daily and Sanctura XR 60 mg once daily.
Toviaz 4 and 8 mg once daily.
Enablex 7.5–15 mg once daily.
Vesicare 5 or 10 mg once daily.
Detrol LA 4 mg once daily.
Detrol 2 mg twice daily.
Ditropan XL 5–30 mg day−1 given once daily.
Ditropan 5–20 mg day−1 given three times daily.
NR: none reported by ≥1% (trospium) or ≥2% (trospium XR and fesoterodine) patients and exceeding placebo rates.
Competing Interests
EC, JP, BM, PB, RW, KF and SK are Pfizer employees and EC, JP, BM, PB, RW and KF are shareholders of Pfizer. Two of the compounds described in the manuscript, tolerodine and fesoterodine (pro-drug of 5-HmT) are Pfizer compounds. GK has served as a consultant to Pfizer, Novartis, Allergen and Watson on the CNS effects of antimuscarinic drugs. GK has also served as a speaker and has received research support from these companies. Within the last 5 years MCM has received research funds, consultancy and/or lecturer honoraria related to urogenital pharmacology from the following pharmaceutical companies: Allergan, Astellas, Bayer, Boehringer Ingelheim, Eli Lilly, GSK, Pfizer, Schwarz and Theravance.
REFERENCES
- 1.Haylen BT, de Ridder D, Freeman RM, Swift SE, Berghmans B, Lee J, Monga A, Petri E, Rizk DE, Sand PK, Gabriel N, Schaer GN. An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint report on the terminology for female pelvic floor dysfunction. Neurourol Urodyn. 2010;29:4–20. doi: 10.1002/nau.20798. [DOI] [PubMed] [Google Scholar]
- 2.Temml C, Haidinger G, Schmidbauer J, Schatzl G, Madersbacher S. Urinary incontinence in both sexes: prevalence rates and impact on quality of life and sexual life. Neurourol Urodyn. 2000;19:259–71. doi: 10.1002/(sici)1520-6777(2000)19:3<259::aid-nau7>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 3.Milsom I, Abrams P, Cardozo L, Roberts RG, Thüroff J, Wein AJ. How widespread are the symptoms of an overactive bladder and how are they managed? A population-based prevalence study. BJU Int. 2001;87:760–6. doi: 10.1046/j.1464-410x.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- 4.Michel M, Chapple CR. Basic mechanisms of urgency: preclinical and clinical evidence. Eur Urol. 2009;56:298–308. doi: 10.1016/j.eururo.2009.05.028. [DOI] [PubMed] [Google Scholar]
- 5.Mostwin JL. Pathophysiology: the varieties of bladder overactivity. Urology. 2002;5(Suppl. 1):22–7. doi: 10.1016/s0090-4295(02)01788-0. [DOI] [PubMed] [Google Scholar]
- 6.Chapple CR. Effects of antimuscarinic treatments in OAB: a systematic review and meta-analysis. Eur Urol. 2005;48:5–26. doi: 10.1016/j.eururo.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 7.Chapple CR, Khullar V, Gabriel Z, Muston D, Bitoun CE, Weinstein D. The effects of antimuscarinic treatments in overactive bladder: an update of a systematic review and meta-analysis. Eur Urol. 2008;54:543–62. doi: 10.1016/j.eururo.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 8.Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharrnacol Rev. 1998;50:279–90. [PubMed] [Google Scholar]
- 9.Ancelin ML, Artero S, Portet F, Dupuy A-M, Touchon J, Ritchie K. Non-degenerative mild cognitive impairment in elderly people and use of anticholinergic drugs: longitudinal cohort study. BMJ. 2006;332:455–8. doi: 10.1136/bmj.38740.439664.DE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kopelman MD. The cholinergic neurotransmitter system in human memory and dementia: a review. Q J Exp Psychol A. 1986;38:535–73. doi: 10.1080/14640748608401614. [DOI] [PubMed] [Google Scholar]
- 11.Scheife R, Takeda M. Central nervous system safety of anticholinergic drugs for the treatment of overactive bladder in the elderly. Clin Ther. 2005;27:144–53. doi: 10.1016/j.clinthera.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Kay GG, Crook T, Rekeda L, Lima R, Ebinger U, Arguinzoniz M, Steel M. Differential effects of the antimuscarinic agents darifenacin and oxybutynin ER on memory in older subjects. Eur Urol. 2006;50:317–26. doi: 10.1016/j.eururo.2006.03.057. [DOI] [PubMed] [Google Scholar]
- 13.Kay GG, Wesnes K. Pharmacodynamic effects of darifenacin, a muscarinic M3 selective receptor antagonist for the treatment of overactive bladder, in healthy volunteers. BJU Int. 2005;96:1055–62. doi: 10.1111/j.1464-410X.2005.05745.x. [DOI] [PubMed] [Google Scholar]
- 14.Kay GG, Granville L. Antimuscarinic agents: implications and concerns in the management of overactive bladder in the elderly. Clin Ther. 2005;27:127–38. doi: 10.1016/j.clinthera.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 15.Kay GG, Abou-Donia MB, Messer JWS, Murphy DG, Tsao JW, Ouslander JG. Antimuscarinic drugs for overactive bladder and their potential effects on cognitive function in older patients. J Am Geriatr Soc. 2005;53:2195–201. doi: 10.1111/j.1532-5415.2005.00537.x. [DOI] [PubMed] [Google Scholar]
- 16.Van de Waterbeemd H, Camenisch G, Folkers G, Chretien JR, Raevsky OA. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J Drug Target. 1998;6:151–65. doi: 10.3109/10611869808997889. [DOI] [PubMed] [Google Scholar]
- 17.Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43:3714–7. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 18.Eyal S, Hsiao P, Unadkat JD. Drug interactions at the blood-brain barrier: fact or fantasy? Pharmacol Ther. 2009;123:80–104. doi: 10.1016/j.pharmthera.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Che C, Smith BJ. Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther. 2008;325:349–56. doi: 10.1124/jpet.107.130294. [DOI] [PubMed] [Google Scholar]
- 20.Kalvass JC, Maurer TS. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos. 2002;23:327–38. doi: 10.1002/bdd.325. [DOI] [PubMed] [Google Scholar]
- 21.Michel MC. Fesoterodine: a novel muscarinic receptor antagonist for the treatment of overactive bladder syndrome. Expert Opin Pharmacother. 2008;9:1787–96. doi: 10.1517/14656566.9.10.1787. [DOI] [PubMed] [Google Scholar]
- 22.Malhotra BK, Guan Z, Wood N, Gandelman K. Pharmacokinetic profile of fesoterodine. Int J Clin Pharmacol Ther. 2008;46:556–63. doi: 10.5414/cpp46556. [DOI] [PubMed] [Google Scholar]
- 23.Malhotra BK, Gandelman K, Sachse R, Wood N, Michel MC. The design and development of fesoterodine as a prodrug of 5-hydroxymethyl tolterodine (5-HMT), the active metabolite of tolterodine. Curr Med Chem. 2009;16:4481–9. doi: 10.2174/092986709789712835. [DOI] [PubMed] [Google Scholar]
- 24.Feng B, Mills JB, Davidson RE, Mireles RJ, Janiszewski JS, Troutman MD, de Morais SM. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab Dispos. 2008;36:268–75. doi: 10.1124/dmd.107.017434. [DOI] [PubMed] [Google Scholar]
- 25.Kalvass JC, Maurer TS, Pollack GM. Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo P-glycoprotein efflux ratios. Drug Metab Dispos. 2007;35:660–6. doi: 10.1124/dmd.106.012294. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Van Natta K, Yeo H, Vilenski O, Weller PE, Worboys PD, Monshouwer M. Unbound drug concentration in brain homogenate and cerebral spinal fluid at steady state as a surrogate for unbound concentration in brain interstitial fluid. Drug Metab Dispos. 2009;37:7887–793. doi: 10.1124/dmd.108.024125. [DOI] [PubMed] [Google Scholar]
- 27.International Transporter Consortium. Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hitchcock SA, Pennington LD. Structure-brain exposure relationships. J Med Chem. 2006;49:7559–83. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- 29.Freedman SB, Harley EA, Patel S. Direct measurement of muscarinic agents in the central nervous system of mice using ex vivo binding. Eur J Pharmacol. 1989;174:253–60. doi: 10.1016/0014-2999(89)90317-8. [DOI] [PubMed] [Google Scholar]
- 30.Wesnes K, Warburton DM. Effects of scopolamine and nicotine on human rapid information processing performance. Psychopharmacology. 1984;82:147–50. doi: 10.1007/BF00427761. [DOI] [PubMed] [Google Scholar]
- 31.Maurer TS, DeBartolo DB, Tess DA, Scott DO. Relationship between exposure and non-specific binding of thirty-three central nervous system drugs in mice. Drug Metab Dispos. 2005;33:175–81. doi: 10.1124/dmd.104.001222. [DOI] [PubMed] [Google Scholar]
- 32.Hammarlund-Udenaes M, Paalow LK, de Lange CM. Drug equilibration across the blood-brain barrier – pharmacokinetic considerations based on the microdialysis method. Pharm Res. 1997;14:128–34. doi: 10.1023/a:1012080106490. [DOI] [PubMed] [Google Scholar]
- 33.Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, Cianfrogna J, Doran AC, Doran SD, Gibbs JP, Hosea N, Liu J, Nelsom FR, Szewc MA, Van Deusen J. Use of a physiologically based pharmacokinetic model to study the time to reach brain equilibrium: an experimental analysis of the role of blood-brain barrier permeability, plasma protein binding and brain tissue binding. J Pharmacol Exp Ther. 2005;313:1254–62. doi: 10.1124/jpet.104.079319. [DOI] [PubMed] [Google Scholar]
- 34.Rao VV, Dahlheimer LL, Bardgett ME, Snyder AZ, Finch RA, Sartorelli AC, Piwnica-Worms D. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood–cerebrospinal-fluid drug-permeability barrier. Proc Natl Acad Sci USA. 1999;96:3900–5. doi: 10.1073/pnas.96.7.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell N, Boustani M, Limbil T, Ott C, Fox C, Maidment I, Schubert CC, Munger S, Fick D, Miller D, Gulati R. The cognitive impact of anticholinergics: a clinical review. Clin Interv Aging. 2009;4:228–33. doi: 10.2147/cia.s5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oki T, Kageyama YT, Takagi Y, Uchida S, Yamada S. Comparative evaluation of central muscarinic receptor binding activity by oxybutynin, tolterodine and darifenacin used to treat overactive bladder. J Urol. 2007;177:766–770. doi: 10.1016/j.juro.2006.09.079. [DOI] [PubMed] [Google Scholar]
- 37.Maruyama S, Tsukada H, Nishiyama S, Kakiuchi T, Fukumoto D, Oku N, Yamada S. In vivo quantitative autoradiographic analysis of brain muscarinic receptor occupancy by antimuscarinic agents for overactive bladder treatment. J Pharmacol Exp Ther. 2008;325:774–81. doi: 10.1124/jpet.108.136390. [DOI] [PubMed] [Google Scholar]
- 38.Kelder J, Wagener M, Timmers M. Cheminformatics and drug design 2004. In: Noordik JH, editor. Chemoinformatics Development. History, Review and Current Research. Amsterdam: IOS Press; 2004. pp. 111–27. Chapter 5. [Google Scholar]
- 39.Roberts LM, Black DS, Raman C, Woodford K, Zhou M, Haggerty JE, Yan AT, Cwirla SE, Grindstaff KK. Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vitro biotinylation. Neuroscience. 2008;155:423–428. doi: 10.1016/j.neuroscience.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 40.Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. Absence of the mdrla P-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin. Am J Clin Invest. 1995;96:1698–705. doi: 10.1172/JCI118214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002;303:1029–37. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- 42.Chen C, Hanson E, Watson JW, Lee JS. P-glycoprotein limits the brain penetration of nonsedating but not sedating H1-antagonists. Drug Met Disp. 2003;31:312–8. doi: 10.1124/dmd.31.3.312. [DOI] [PubMed] [Google Scholar]
- 43.Geyer J, Gavrilova O, Petzinger E. The role of P-glycoprotein in limiting brain penetration of the peripherally acting anticholinergic overactive bladder drug trospium chloride. Drug Metab Dispos. 2009;37:1371–4. doi: 10.1124/dmd.109.027144. [DOI] [PubMed] [Google Scholar]
- 44.Skerjanec A. The clinical pharmacokinetics of darifenacin. Clin Pharmacokinet. 2006;45:325–50. doi: 10.2165/00003088-200645040-00001. [DOI] [PubMed] [Google Scholar]
- 45.Michel MC, Minematsu T, Hashimoto T, den Hoven WV, Swart PJ. In vitro studies on the potential of solifenacin for drug–drug-interactions: plasma protein binding and MDR1 transport. Br J Clin Pharmacol. 2005;59:647. [Google Scholar]
- 46.Mukwaya G, MacGregor T, Hoelscher D, Heming T, Legg D, Kavanaugh K, Johnson P, Sabo JP, McCallister S. Interaction of ritonavir-boosted tipranavir with loperamide does not result in loperamide-associated neurologic side effects in healthy volunteers. Antimicrob Agents Chemother. 2005;49:4903–10. doi: 10.1128/AAC.49.12.4903-4910.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schinkel AH, Wagenaar E, Mol C, van Deemter L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97:2517–24. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Todorova A, Vonderheid-Guth B, Dimpfel W. Effects of tolterodine, trospium chloride, and oxybutynin on the central nervous system. J Clin Pharmacol. 2001;41:636–44. doi: 10.1177/00912700122010528. [DOI] [PubMed] [Google Scholar]
- 49.Zinner NR, Mattiasson A, Stanton SL. Efficacy, safety, and tolerability of extended-release once-daily tolterodine treatment for overactive bladder in older versus younger patients. J Am Geriatr Soc. 2002;50:799–807. doi: 10.1046/j.1532-5415.2002.50203.x. [DOI] [PubMed] [Google Scholar]
- 50.Diefenbach K, Arold G, Wollny A, Schwantes U, Haselmann J, Roots I. Effects on sleep of anticholinergics used for overactive bladder treatment in healthy volunteers aged ≥50 years. BJU Int. 2005;95:346–9. doi: 10.1111/j.1464-410X.2005.05296.x. [DOI] [PubMed] [Google Scholar]
- 51.Katz IR, Sands LP, Bilker W, DiFilippo S, Boyce A, D'Angelo K. Identification of medications that cause cognitive impairment in older people: the case of oxybutynin chloride. J Am Geriatr Soc. 1998;46:8–13. doi: 10.1111/j.1532-5415.1998.tb01006.x. [DOI] [PubMed] [Google Scholar]
- 52.Staskin D, Kay G, Tannenbaum C, Goldman HB, Bhashi G, Ling J, Oefelein MG. Trospium chloride has no effect on memory testing and is assay undetectable in the central nervous system of older patients with overactive bladder. Int J Clin Pract. 2010;64:1294–300. doi: 10.1111/j.1742-1241.2010.02433.x. [DOI] [PubMed] [Google Scholar]