

Abstract
AIM
To determine the effect of the strong CYP2D6 inhibitor paroxetine and strong CYP3A4 inhibitor ketoconazole on the pharmacokinetics and safety (orthostatic challenge) of tamsulosin.
METHODS
Two open-label, randomized, two-way crossover studies were conducted in healthy male volunteers (extensive CYP2D6 metabolizers).
RESULTS
Co-administration of multiple oral doses of 20 mg paroxetine once daily with a single oral dose of the 0.4 mg tamsulosin HCl capsule increased the adjusted geometric mean (gMean) values of Cmax and AUC(0,∞) of tamsulosin by factors of 1.34 (90% CI 1.21, 1.49) and 1.64 (90% CI 1.44, 1.85), respectively, and increased the terminal half-life (t1/2) of tamsulosin HCl from 11.4 h to 15.3 h. Co-administration of multiple oral doses of 400 mg ketoconazole once dailywith a single oral dose of the 0.4 mg tamsulosin increased the gMean values of Cmax and AUC(0,∞) of tamsulosin by a factor of 2.20 (90% CI 1.96, 2.45) and 2.80 (90% CI 2.56, 3.07), respectively. The terminal half-life was slightly increased from 10.5 h to 11.8 h. These pharmacokinetic changes were not accompanied by clinically significant alterations of haemodynamic responses during orthostatic stress testing.
CONCLUSION
The exposure to tamsulosin is increased upon co-administration of strong CYP2D6 inhibitors and even more so of strong 3A4 inhibitors, but neither PK alteration was accompanied by clinically significant haemodynamic changes during orthostatic stress testing.
Keywords: CYP2D6, CYP3A4, ketoconazole, orthostatic challenge, paroxetine, tamsulosin
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Tamsulosin metabolism involves both CYP2D6 and 3A4. However, data on potential drug–drug interactions between tamsulosin and inhibitors of CYP2D6 and 3A4 are limited and information on potential pharmacodynamic consequences of such pharmacokinetic interactions is missing.
WHAT THIS STUDY ADDS
This study provides information on the drug–drug interactions of tamsulosin with strong CYP2D6 and strong CYP3A4 inhibitors after single dose administration in healthy subjects.
Introduction
Lower urinary tract symptoms (LUTS) suggestive of benign prostatic hyperplasia (BPH) are common among elderly males [1–3]. The medical treatment of LUTS/BPH mainly involves inhibition of the enzyme 5α-reductase to reduce prostate size [4] and α1-adrenoceptor antagonists. The latter are more frequently used as they reduce LUTS more effectively than the 5α-reductase inhibitors in most patients [5]. Globally, tamsulosin is the most frequently prescribed α1-adrenoceptor antagonist for the treatment of LUTS/BPH. It is generally well tolerated, and cardiovascular side effects occur only rarely in the standard therapeutic dose of 0.4 mg day−1[6, 7].
The average man receiving an α1-adrenoceptor antagonist prescription for the treatment of LUTS/BPH is in his mid-sixties and frequently has comorbidities and associated comedications [8, 9]. Such comedications may give rise to pharmacodynamic and/or pharmacokinetic drug–drug interactions which reduce the otherwise good tolerability. For example, among potential pharmacodynamic interactions it has been observed that the incidence of adverse events with tamsulosin quadruples when other α1-adrenoceptor antagonists or verapamil (which has α1-adrenoceptor antagonist effects in therapeutic doses) are used concomitantly [9]. In contrast, pharmacodynamic interactions with other blood pressure lowering drugs are rare [8, 10].
The pharmacokinetic profile of tamsulosin has been reviewed comprehensively recently [11] and it has also been studied in a paediatric population [12]. Its metabolism involves both CYP2D6 and 3A4 [13, 14] and yields at least some compounds with α1-adrenoceptor antagonist properties [15] but these metabolites have only low abundance in man. Nevertheless, only limited data are available on potential drug–drug interactions between tamsulosin and inhibitors of CYP2D6 and 3A4. Only one study with 400 mg of the weak CYP3A4 inhibitor cimetidine has been reported [16]. While that study indicated a limited potential for interaction with CYP 3A4 inhibitors, it lacked information on potential pharmacodynamic consequences of such pharmacokinetic interactions. Dedicated studies with strong inhibitors of CYP 2D6 or 3A4 are missing. Therefore, we have performed drug–drug interaction studies with paroxetine, a mechanism based inhibitor [17] and ketoconazole, which included not only pharmacokinetic but also pharmacodynamic safety assessments including forced orthostatic stress testing.
Preliminary results of these studies have been reported to the British Pharmacological Society [18, 19].
Methods
Study design
The study protocols were approved by the German authorities (BfArM) and the regional ethics committee of Rhineland-Palatinate. Healthy male volunteers (23–47 years, body mass index 19.2–29.9 kg m−2) were included after having given informed written consent. The trials were conducted in accordance with the guidelines on Good Clinical Practice and the Declaration of Helsinki at the Human Pharmacology Centre Ingelheim of Boehringer Ingelheim in Germany. Subjects were in good general health according to routine medical history, physical examination, vital signs, 12-lead ECG and laboratory data.
Two open-label randomized, two-way crossover studies of similar design were performed in 24 healthy male volunteers each, one assessing interactions with paroxetine and one with ketoconazole. Treatment A of both studies consisted of a single oral dose of 0.4 mg tamsulosin HCl as modified release capsule (Alna®; Source: BI Austria GmbH, Austria). Treatment B in the paroxetine study consisted of 10 mg paroxetine once daily (Seroxat®; Source: GlaxoSmithKline, Germany) for 3 days, then 20 mg paroxetine once daily for 9 days to achieve steady-state and full CYP2D6 inhibition and finally 10 mg paroxetine once daily for 3 days as taper out regimen. On day 8 of the 20 mg paroxetine treatment, subjects additionally received a single dose of 0.4 mg tamsulosin HCl. To maintain the full CYP2D6 inhibition over the pharmacokinetic (PK) sampling period of tamsulosin, paroxetine 20 mg was administered on day 9. Treatment B of the ketoconazole study consisted of 400 mg ketoconazole once daily (Source: Pliva, East Hanover, NJ, USA) for 5 days in order to achieve steady-state and full CYP3A4 inhibition and on day 4 additionally a single dose of 0.4 mg tamsulosin HCl. To maintain the full CYP3A4 inhibition over the PK sampling period of tamsulosin, ketoconazole 400 mg was administered up to day 5.
In both studies blood samples for PK were withdrawn before and at 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 14, 24, 30, 36 and 48 h after tamsulosin HCl administration. Paroxetine sparse sampling was performed before and at 3, 5 and 8 h after tamsulosin HCl administration. Ketoconazole sparse sampling was performed before and at 1, 2, 4, 7, and 24 h after tamsulosin HCl administration.
Safety and tolerability
Safety and tolerability were assessed by measurement of vital signs blood pressure and pulse rate including forced orthostatic stress testing as well as 12-lead ECGs. Orthostasis was assessed by measuring blood pressure 10 min after rest in the supine position and 2 min after rest in the standing position. Safety assessments were carried out before and at 2, 4, 6, 8 and 24 h after tamsulosin HCl administration, orthostasis was assessed before and at 6 and 24 h after tamsulosin HCl administration. Safety laboratory (haematology, serum chemistry and urinalysis) testing, performed at screening and end-of-trial visit, was assessed and adverse events were recorded.
Genotyping
Extensive metabolizers with regard to CYP2D6 were identified based on genotyping of subjects as carried out by an external provider (Epidauros Biotechnology AG, Am Neuland 1, 82347 Bernried, Germany) utilizing assays based on TaqMan® or sequencing methodology. For the participants of the paroxetine and ketoconazole studies the genotypes were *1/*1 in 11 and 10, *1/*2 in nine and 11, *1/*10 in two and two and *1/*41 in two and one subjects, respectively.
Bioanalytical methods
Tamsulosin
The bioanalytical method for the quantification of tamsulosin HCl in human plasma was based on high performance liquid chromatography with tandem mass spectrometry (HPLC-MS/MS). After addition of AB-289 (internal standard provided by Yamanouchi Europe B.V., Leiderdorp, the Netherlands) to the plasma, tamsulosin and the internal standard were extracted from plasma using liquid-liquid extraction [ethylacetate : cyclo-hexane (3:1, v : v)] under alkaline conditions. The organic phase was evaporated and the residue was reconstituted in ammonium acetate (20 mm) : acetonitrile (3:1, v : v). A sample was injected into a HPLC-MS/MS system to separate tamsulosin and the internal standard from matrix constituents using Symmetry C18 material (Waters, Saint-Quentin-en-Yvelines, France) with a mean size of 3.5 µm in a stainless steel column of 100 mm × 2.1 mm and acetonitrile : ammonium acetate (20 mm) (3:1, v : v) as mobile phase. A constant flow rate of 300 µl min−1 was employed.
Detection was performed using a triple stage quadrupole mass spectrometer (API 365, Applied Biosystems, Courtaboeuf Cedex, France) with a APCI (atmospheric pressure chemical ionization) interface in positive ion mode. Tamsulosin parent/daughter ions were detected with m/z = 409.2/228.0 and the internal standard AB-289 parent/daughter ions at m/z = 423.2/285.1. This method is suitable for the quantification of tamsulosin (as tamsulosin HCl) in human plasma at concentrations ranging between 0.100 and 50.0 ng ml−1. In-study inaccuracy and imprecision values for tamsulosin were within ± 7.5% and <8.1%, respectively [20].
Ketoconazole
The bioanalytical method for the quantification of ketoconazole in human plasma was based on HPLC-MS/MS and was validated at SGS Cephac Europe, France. After addition of itraconazole (internal standard, LGC-PROMOCHEM, Molsheim, France) to the plasma, ketoconazole and the internal standard were extracted from plasma using liquid-liquid extraction [heptane : 3-methylbutanol (95:5 v : v)] under alkaline conditions. The organic phase was evaporated and the residue was reconstituted in methanol : aqueous ammonium acetate (10 mm) (80:20%, v : v). The sample was injected into an HPLC-MS/MS system to separate ketoconazole and the internal standard from matrix constituents using BDS Hypersil C18 material with a mean size of 3 µm, in a column of 100 mm × 4 mm (supplied by Thermo Electron Corporation, Courtaboeuf, France) and methanol : aqueous ammonium acetate (10 mm) (80:20, v : v) as mobile phase. A constant flow rate of 1000 µl min−1 was employed.
Detection was performed using a triple stage quadrupole mass spectrometer (API 365, Applied Biosystems, Courtaboeuf Cedex, France) with a APCI interface in positive ion mode. Ketoconazole parent/daughter ions were detected with m/z = 531.1 >489.1 and the internal standard itraconazole parent/daughter ions at m/z = 705.3 >392.2. This method is suitable for the quantification of ketoconazole in human plasma at concentrations ranging between 10.0 and 10000 ng ml−1. In-study inaccuracy and imprecision values for ketoconazole were within ± 3.3% and <5.5%, respectively.
Paroxetine
The bioanalytical method for the quantification of paroxetine in human plasma was based on HPLC-MS/MS and was validated at SGS Cephac Europe, France. After addition of d4-paroxetine (internal standard, Toronto Research Chemicals Inc., North York, Ontario, Canada) to the plasma, paroxetine and the internal standard were extracted from plasma using Solid Phase Extraction on Oasis HLB 1 cc, 30 mg cartridges (Waters, Saint-Quentin-en-Yvelines, France). The cartridges were eluted with methanol containing 0.1% formic acid. The sample extract was evaporated to dryness and the residue was reconstituted in H2O containing 0.1% formic acid. The sample was injected into an HPLC-MS/MS system to separate paroxetine and the internal standard from matrix constituents using Zorbax SB-C18 material with a mean size of 3.5 µm, in a column of 2.1 × 50 mm (supplied by Agilent, Massy, France) and water : acetonitrile (65:35, v : v) containing 0.1% formic acid as mobile phase. A constant flow rate of 300 µl min−1 was employed.
Detection was performed using a triple stage quadrupole mass spectrometer (API 3000, Applied Biosystems, Courtaboeuf Cedex, France) with a TurboIonSpray interface in positive ion mode. Paroxetine parent/daughter ions were detected with m/z = 330.1 >192.2 and the internal standard itraconazole parent/daughter ions at m/z = 334.2 >196.1. This method is suitable for the quantification of paroxetine in human plasma at concentrations ranging between 0.050 and 50.0 ng ml−1. In-study inaccuracy and imprecision values for paroxetine were within ± 14.7% and <4.4%, respectively.
The lower limit of quantification was 0.1 ng ml−1 for tamsulosin, 0.05 ng ml−1 for paroxetine and 10 ng ml−1 for ketoconazole.
Pharmacokinetic data analysis
Pharmacokinetic parameters were calculated with non-compartmental analyses using WinNonlin professional version 5.2 (Pharsight Corporation, Mountain View, CA). Only concentrations within the validated concentration range and actual sampling times were used for the calculation of pharmacokinetic parameters. Individual Cmax and tmax values were directly determined from the plasma concentration time profiles of each subject. The apparent terminal rate constant (λz) was estimated from a regression of ln(C) vs. time over the terminal log-linear disposition portion of the concentration–time profiles. Terminal half-life (t1/2) was calculated as ln(2)/λz. Area under the concentration–time curve of tamsulosin HCl in plasma over the time interval from 0 to the last quantifiable data point (AUC(0,tlast)) was calculated using the linear up/log down algorithm. Area under the concentration–time curve over the time interval from 0 extrapolated to infinity (AUC(0,∞)) was calculated as 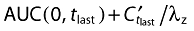 , where
, where 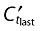 is the predicted concentration at the time tlast (last time point with a plasma concentration above the quantification limit). Apparent clearance (CL/F) and apparent volume of distribution were calculated as Dose/AUC(0,∞) and as (CL/F)/λz, respectively.
is the predicted concentration at the time tlast (last time point with a plasma concentration above the quantification limit). Apparent clearance (CL/F) and apparent volume of distribution were calculated as Dose/AUC(0,∞) and as (CL/F)/λz, respectively.
Statistical methods
AUC(0,∞), Cmax and AUC(0,tlast) were statistically analyzed by analysis of variance (anova) models on the logarithmic scale including effects accounting for the following sources of variation in the model: ‘sequence’, ‘subjects nested within sequences’, ‘period’ and ‘treatment’. The effect ‘subjects within sequences’ was considered as random, whereas the other effects were considered as fixed.
The pharmacokinetic parameters were log transformed (natural logarithm), prior to fitting the anova model. The difference between the expected means for log(test) − log(reference) was estimated by the difference in the corresponding Least Square Means (point estimate) and two-sided 90% confidence intervals based on the t-distribution were computed. These quantities were then back-transformed to the original scale to give the point estimator (geometric mean) and interval estimates for the median intra-subject ratio between response under test (tamsulosin + ketoconazole or tamsulosin + paroxetine) and response under reference (tamsulosin).
All subjects who provided at least one observation for at least one primary pharmacokinetic (PK) endpoint without important protocol violations relevant to the evaluation of pharmacokinetics were included in the analysis of primary PK endpoints.
All pharmacokinetic parameters (geometric mean, with geometric coefficient of variation, or median with range), adverse events (frequencies of subjects with adverse events), vital signs (mean and SD) and laboratory data were evaluated descriptively.
Results
Plasma concentrations of study drugs
The geometric mean (gMean) plasma concentration–time profile of paroxetine and ketoconazole reached the peak (at 5 and 4 h post dose at steady-state, respectively). The paroxetine and ketoconazole steady-state trough concentrations on the day of tamsulosin HCl administration ranged from 0.1 to 17.7 ng ml−1 and from 236 to 248 ng ml−1, respectively.
The gMean plasma concentration–time profiles of tamsulosin HCl after single oral administration of 0.4 mg tamsulosin HCl with and without co-administration of 20 mg paroxetine once daily or 400 mg ketoconazole once daily are illustrated in Figure 1. The gMean plasma concentrations of tamsulosin HCl reached its peak at around 7–8 h post dose and then declined gradually. The gMean plasma concentrations of tamsulosin HCl after co-administration with paroxetine or ketoconazole were generally higher than those without co-administration. There was no carry-over effect from the first period because all the pre-dose concentrations of tamsulosin HCl were below the limit of quantification before drug administration in the second period.
Figure 1.
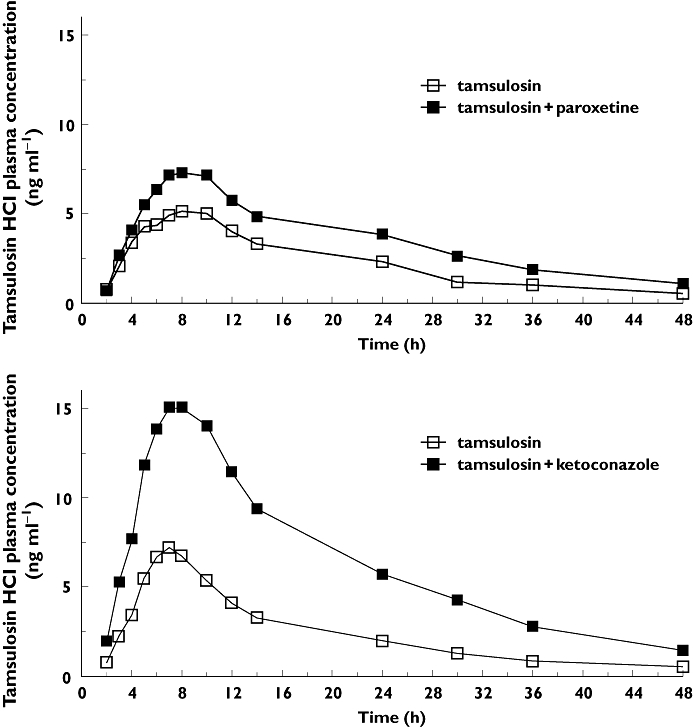
Plasma concentration–time profiles of tamsulosin HCl after single oral administration of 0.4 mg tamsulosin HCl (upper panel) with and without 20 mg day−1 paroxetine or 400 mg day−1 ketoconazole administration (lower panel). Data are gMean of 23 subjects for each combination treatment and 24 subjects for tamsulosin.
Pharmacokinetic parameters
During co-administration of paroxetine the gMean values of Cmax and AUC(0,∞) of tamsulosin HCl with paroxetine (8.83 ng ml−1 and 193 ng ml−1 h, respectively) were higher than those without paroxetine (6.57 ng ml−1 and 117 ng ml−1 h, respectively; Table 1). Paroxetine co-administration decreased the CL/F of tamsulosin HCl by approximately 39%. The gMean t1/2 of tamsulosin HCl with paroxetine (15.3 h) was longer than without paroxetine co-administration (11.4 h). The median values of tmax were similar for the two treatments (7 and 8 h). In two subjects, low paroxetine plasma concentrations (at trough) were measured. However, analysis of changes in Cmax and AUC(0,∞) of tamsulosin HCl did not indicate deviation from the overall increase shown in this study. Overall, paroxetine co-administration increased the Cmax and AUC(0,∞) of tamsulosin HCl in most of the subjects (21 out of 23 subjects for Cmax, 22 out of 23 subjects for AUC(0,∞)).
Table 1.
Comparison of pharmacokinetic parameters of tamsulosin HCl after single oral administration of 0.4 mg tamsulosin HCl with and without 20 mg paroxetine once daily co-administration
| Pharmacokinetic parameters of tamsulosin HCl | |||||||
|---|---|---|---|---|---|---|---|
| Tamsulosin alone | Tamsulosin + paroxetine | ||||||
| Parameter | n | gMean | gCV (%) | n | gMean | gCV (%) | |
| Cmax | (ng ml−1) | 24 | 6.57 | 37.2 | 23 | 8.83 | 31.6 |
| tmax* | (h) | 24 | 7.00 | (3.00–23.7) | 23 | 8.00 | (3.98–23.9) |
| AUC(0,tlast) | (ng ml−1 h) | 24 | 107 | 40.5 | 23 | 165 | 41.8 |
| AUC(0,∞) | (ng ml−1 h) | 24 | 117 | 42.8 | 23 | 193 | 45.8 |
| t1/2 | (h) | 24 | 11.4 | 26.4 | 23 | 15.3 | 24.8 |
| MRTpo | (h) | 24 | 20.5 | 24.5 | 23 | 25.6 | 21.9 |
| CL/F | (l h−1) | 24 | 3.41 | 42.8 | 23 | 2.07 | 45.8 |
| Vz/F | (l) | 24 | 56.4 | 35.0 | 23 | 45.7 | 37.4 |
Median (minimum–maximum)
Cmax, maximum plasma concentration; tmax, time to maximum plasma concentration; AUC(0,tlast), area under the concentration–time curve from 0 to the last quantifiable data point; AUC(0,∞), area under the concentration–time curve from 0 to ∞; t1/2, half-life; MRT, mean residence time; CL/F, apparent clearance; Vz/F, apparent volume of distribution.
During co-administration of ketoconazole the gMean values of Cmax (17.0 ng ml−1) and AUC(0,∞) (326 ng ml−1 h, Table 2) with ketoconazole were higher than those without ketoconazole (7.70 ng ml−1 and 115 ng ml−1 h, respectively). Co-administration of ketoconazole decreased the CL/F of tamsulosin HCl to approximately 35%. The gMean value of t1/2 (11.8 h) of tamsulosin HCl with ketoconazole was slightly longer than without ketoconazole co-administration (10.5 h). The median value of tmax was comparable between the two treatments (around 7 h).
Table 2.
Comparison of pharmacokinetic parameters of tamsulosin HCl after single oral administration of 0.4 mg tamsulosin HCl with and without 400 mg ketoconazole once daily co-administration
| Pharmacokinetic parameters of tamsulosin HCl | |||||||
|---|---|---|---|---|---|---|---|
| Tamsulosin alone | Tamsulosin + ketoconazole | ||||||
| Parameter | n | gMean | gCV (%) | n | gMean | gCV (%) | |
| Cmax | (ng·ml−1) | 24 | 7.70 | 43.3 | 23 | 17.0 | 34.6 |
| tmax* | (h) | 24 | 6.98 | (5.00–23.9) | 23 | 7.00 | (4.98–10.0) |
| AUC(0,tlast) | (ng ml−1 h) | 24 | 108 | 58.4 | 23 | 297 | 42.0 |
| AUC(0,∞) | (ng ml−1 h) | 24 | 115 | 60.8 | 23 | 326 | 46.2 |
| t1/2 | (h) | 24 | 10.5 | 25.4 | 23 | 11.8 | 29.3 |
| MRTpo | (h) | 24 | 18.2 | 21.8 | 23 | 21.0 | 25.5 |
| CL/F | (l·h−1) | 24 | 3.47 | 60.8 | 23 | 1.23 | 46.2 |
| Vz/F | (l) | 24 | 52.4 | 42.2 | 23 | 20.8 | 28.9 |
Median (minimum–maximum).
Cmax, maximum plasma concentration; tmax, time to maximum plasma concentration; AUC(0,tlast), area under the concentration–time curve from 0 to the last quantifiable data point; AUC(0,∞), area under the concentration–time curve from 0 to ∞; t1/2, half-life; MRT, mean residence time; CL/F, apparent clearance; Vz/F, apparent volume of distribution.
Statistical assessment of drug–drug interaction
Relative exposure was assessed by means of anova using the log-transformed pharmacokinetic parameters available from all periods. Relative exposure of test treatments (tamsulosin HCl co-administered with paroxetine or ketoconazole) vs. the reference treatment (tamsulosin HCl) based on the primary endpoints (Cmax and AUC(0,∞)) was evaluated. The adjusted gMean ratio (90% CI) of test treatment (tamsulosin HCl with paroxetine) to reference treatment was 134.1% (120.7, 149.0%) for Cmax and 163.5% (144.2, 185.3%) for AUC(0,∞). The degree of intra-individual variability of Cmax and AUC(0,∞) revealed a geometric coefficient of variation (gCV) of 21.0% and 25.2%, respectively. The adjusted gMean ratio (90% CI) of test treatment (tamsulosin HCl with ketoconazole) vs. reference treatment was 219.5% (196.4, 245.4%) for Cmax and 280.3% (256.2, 306.6%) for AUC(0,∞). The degree of intra-individual variability of Cmax and AUC(0,∞) revealed a gCV of 22.3% and 17.9%, respectively.
Vital signs and adverse events
The pharmacodynamic impact of concomitant treatment with CYP2D6 and 3A4 inhibitors was primarily assessed by forced orthostasis stress testing and investigated by descriptive statistics of the changes from supine (10 min at rest) to standing position before and at 6 and 24 h after tamsulosin administration (Figure 2). These data show that the increased tamsulosin exposure upon co-administration of paroxetine or ketoconazole did not result in greater haemodynamic changes upon standing at the group level. One case of postural dizziness was experienced which was mild in intensity after treatment with paroxetine and tamsulosin. However, when vital signs (blood pressure and pulse rate) were assessed immediately upon complaints, no findings of clinical significance were found. No orthostasis related AEs were reported for the tamsulosin/ketoconazole combination treatment. There was no clinically meaningful difference in systolic and diastolic blood pressure, as well as pulse rate, when tamsulosin was co-administered with ketoconazole or paroxetine.
Figure 2.
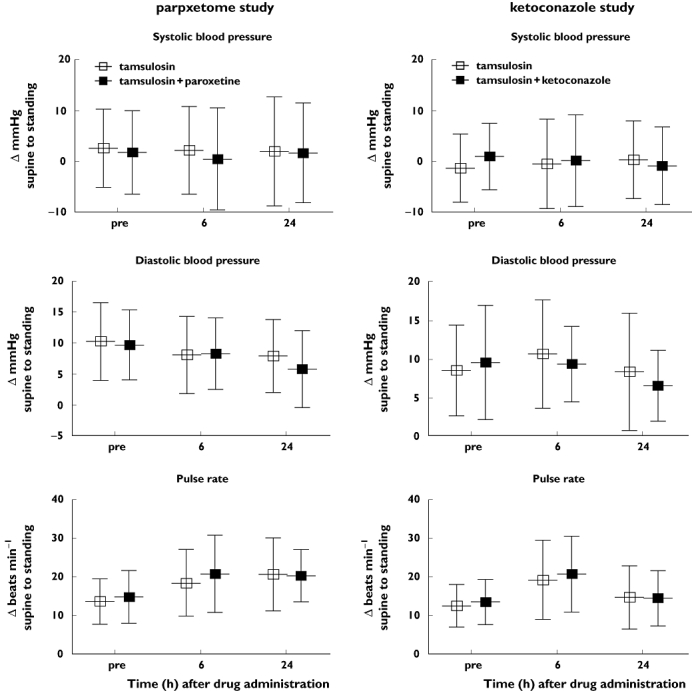
Effect of tamsulosin alone and tamsulosin plus paroxetine (left panels) or plus ketoconazole (right panels) on systolic blood pressure (upper panels), diastolic blood pressure (middle panels) and pulse rate (lower panels). Shown are mean differences and SD from supine to standing position in mmHg (blood pressure) and beats min−1 (pulse rate) as measured prior to tamsulosin administration (‘pre’) and 6 and 24 h thereafter.
There were no serious or severe adverse events reported. With regard to other significant adverse events, one subject discontinued treatment B on the second day of paroxetine administration due to nausea, i.e. prior to tamsulosin treatment. One further subject received treatment A completely but did not complete treatment B (ketoconazole) because of bad compliance.
Discussion
Tamsulosin is globally the most widely prescribed α1-adrenoceptor antagonist for the treatment of LUTS/BPH. It is metabolized primarily by CYP2D6 and 3A4 [13] but except for one drug–drug interaction study with cimetidine [16], no information was available on the effects of CYP2D6 or 3A4 inhibition on the PK and pharmacodynamics of tamsulosin. Therefore, we have performed two interaction studies with the CYP2D6 inhibitor paroxetine and the CYP3A4 inhibitor ketoconazole.
Critique of methods
Similar to previous studies, our trials were performed in male subjects because tamsulosin is used for an indication, LUTS/BPH, which only exists in men. PK parameters for tamsulosin in the absence of concomitant medications as determined in our two studies were in line with many previous PK studies of tamsulosin in healthy male populations [11]. Clinical studies have only rarely reported orthostasis as an adverse event [21–24]. Therefore, our study used forced orthostatic stress testing and alterations of systolic and diastolic blood pressure as well as pulse rate to explore pharmacodynamic consequences of increased tamsulosin exposure in the presence of co-medication. A time point of 6 h post dose was chosen for orthostatic stress testing, because the tmax was expected to be 6 h post-dose based on previous studies [11]. The actual observed tmax was 7 h post dose. Because gMean plasma concentrations of tamsulosin were 13.8 ng ml−1 at 6 h compared with 15.0 ng ml−1 at 7 h, this slight difference in the tamsulosin plasma concentration was not expected to cause a clinically significant different result for orthostatic stress testing. Furthermore, orthostasis tests for tamsulosin 0.4 mg vs. placebo (in elderly subjects) have been published for the 6 and 8 h time point, both yielding very similar results [25]. While age-related PK alterations seem not to exist to a relevant extent in the elderly [11], it is recognized that the elderly are inherently more sensitive to drug-induced orthostatic reactions [26] and extrapolation of safety data from healthy young subjects to diseased old patients is limited.
Overall, this approach has been shown to be sensitive even to detect small differences in previous studies, e.g. with regard to food intake or in comparison with other α1-adrenoceptor antagonists [21, 27–30]. On the other hand, those previous studies including a placebo arm had not reported major differences between tamsulosin and placebo. Therefore, our present study did not include a placebo arm for the hemodynamic measurements.
Both paroxetine [27] and ketoconazole [28, 29] were administered with regimens which are known to achieve effective inhibition of CYP2D6 and 3A4, respectively, and the measured plasma levels of both drugs confirm effective dosing. This study only included subjects with extensive CYP2D6 metabolizer status and therefore caution should be taken when tamsulosin is additionally combined with CYP3A4 inhibitors in these patients. As the CYP2D6 status is unknown in most patients, this may be more a theoretical concern. Tamsulosin has a broad therapeutic margin, as confirmed by the lack of increase in orthostatic reactions in our study despite an increased drug exposure.
It is suggested that accumulation should be accounted for in the study design by multiple dosing of a substrate, when drugs are known to have an elimination half-life of about 11 h and longer [30]. After multiple dosing, due to accumulation, higher tamsulosin plasma concentrations could produce a greater orthostatic response than observed after single dose. Because tamsulosin has dose linear PK ([11] and data on file), it can be expected that a very similar extent of interaction would have been observed after multiple dosing.
Drug–drug interaction data
In line with the role of CYP2D6 in the metabolism of tamsulosin [13, 14], co-administration of the strong CYP2D6 inhibitor paroxetine increased tamsulosin exposure. The increase was moderate and consisted of an elevated Cmax and AUC(0,∞) (134.1% and 163.5% of values of reference treatment, respectively). As expected for a co-medication interfering with drug metabolism, this was associated with a reduced CL/F and an increased t1/2 for tamsulosin (from 11.4 to 15.3 h). Co-administration of the strong CYP3A4 inhibitor ketoconazole increased tamsulosin exposure to a greater extent (219.5% and 280.3% of values of reference treatment for Cmax and AUC(0,∞), respectively). With co-administration of ketoconazole, CL/F of tamsulosin HCl was decreased to approximately 35% with a slight increase of t1/2 (from 10.5 to 11.8 h). Based on mechanistic considerations, inhibition of CYP3A4 by ketoconazole may result in an increased bioavailability and/or a decreased clearance of CYP3A4 substrates. Because the bioavailability of tamsulosin HCl is high (mean ± SD 100 ± 19%) for the oral dose of 0.4 mg [31], an increased bioavailability can only account for a small part of the observed increase in exposure. Furthermore, a decreased elimination would also result in an increased t1/2. However, the t1/2 of tamsulosin was not elevated to any extent (from 10.5 to 11.8 h) which would explain the increase in exposure by a decrease in clearance (from 3.47 to 1.23 l h−1) only. Therefore, the results of our study suggest that other unknown effects contributed to the increase in exposure in addition to the inhibitory effect of ketoconazole for which sufficient exposure to achieve CYP3A4 inhibition is proven. The observed effect on the AUC is compatible with the inhibition of CYP3A4 but additional effects, e.g. due to P-gp inhibition cannot be excluded at the present, because it is unknown whether tamsulosin is a P-gp substrate.
Of interest, while in most countries a maximum tamsulosin dose of 0.4 mg once daily is used [5], a dose of 0.8 mg once daily is also available in the US [32, 33]. The increase in tamsulosin exposure with co-administration of paroxetine is much smaller than would be expected from a doubling of dose. Dose-linearity of exposure has formally been established between 0.05 and 1 mg of tamsulosin HCl ([11] and data on file). On the other hand, the increase in exposure upon co-administration of ketoconazole is expected to be the approximate equivalent of a dose increase to 0.8 mg once daily. In line with the documented safety and tolerability of 0.8 mg once daily of tamsulosin [32, 33], our forced orthostasis stress tests did not reveal a notable increase in haemodynamic effects upon co-administration of paroxetine or ketoconazole despite the proven sensitivity of our approach [25, 34–37]. Moreover, the combination treatment was safe and well tolerated in both trials.
In conclusion, the co-administration of strong inhibitors of CYP2D6 has only limited effect on the exposure to tamsulosin, whereas that of strong inhibitors of CYP3A4 about doubles the tamsulosin exposure. However, neither PK alteration was accompanied by clinically significant haemodynamic changes during orthostatic stress testing. These findings should be taken into account when using tamsulosin in combination with strong inhibitors of CYP2D6 (e.g. paroxetine) or strong inhibitors of CYP3A4 (e.g. ketoconazole).
Acknowledgments
The authors acknowledge the assistance of Heike Wölfel, Kevin Knoche und Kerstin Saar during the conduct of this study and Laurette Millérioux and Nathalie Plaud (SGS Cephac Europe) for their involvement in the bioanalytical work.
Competing Interests
J.T., M.M., L.M. are employees of Boehringer Ingelheim Pharma GmbH & Co. KG, S.T and Y.T. are employees of Nippon Boehringer Ingelheim Co., Ltd. This study was funded by Boehringer Ingelheim Pharma GmbH & Co.KG. M.C.M. has received research support, consultancy and lecturer honoraria from Boehringer Ingelheim related to tamsulosin and from Recordati related to silodosin.
REFERENCES
- 1.Kok ET, Schouten BW, Bohnen AM, Groeneveld FPMW, Thomas S, Bosch JLHR. Risk factors for lower urinary tract symptoms suggestive of benign prostatic hyperplasia in a community based population of healthy aging men: the Krimpen study. J Urol. 2009;181:710–6. doi: 10.1016/j.juro.2008.10.025. [DOI] [PubMed] [Google Scholar]
- 2.Fowke JH, Murff HJ, Signorello LB, Lund L, Blot WJ. Race and socioeconomic status are independently associated with benign prostatic hyperplasia. J Urol. 2008;180:2091–6. doi: 10.1016/j.juro.2008.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarma AV, Jacobson DJ, McGree ME, Roberts RO, Lieber MM, Jacobsen SJ. A population based study of incidence and treatment of benign prostatic hyperplasia among residents of Olmsted County, Minnesota: 1987 to 1997. J Urol. 2005;173:2048–53. doi: 10.1097/01.ju.0000158443.13918.d6. [DOI] [PubMed] [Google Scholar]
- 4.Naslund MJ, Miner M. A review of the clinical efficacy and safety of 5α-reductase inhibitors for the enlarged prostate. Clin Ther. 2007;29:17–25. doi: 10.1016/j.clinthera.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Michel MC. The forefront of novel therapeutic agents based on the pathophysiology of lower urinary tract dysfunction: α-blockers in the treatment of male voiding dysfunction – How do they work and why do they differ in tolerability? J Pharmacol Sci. 2010;112:151–7. doi: 10.1254/jphs.09r15fm. [DOI] [PubMed] [Google Scholar]
- 6.Djavan B, Marberger M. A meta-analysis on the efficacy and tolerability of α1-adrenoceptor antagonists in patients with lower urinary tract symptoms suggestive of benign prostatic obstruction. Eur Urol. 1999;36:1–13. doi: 10.1159/000019919. [DOI] [PubMed] [Google Scholar]
- 7.Nickel JC, Sander S, Moon TD. A meta-analysis of the vascular-related safety profile and efficacy of α-adrenergic blockers for symptoms related to benign prostatic hyperplasia. Int J Clin Pract. 2008;62:1547–59. doi: 10.1111/j.1742-1241.2008.01880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michel MC, Mehlburger L, Bressel H-U, Schumacher H, Schäfers RF, Goepel M. Tamsulosin treatment of 19,365 patients with lower urinary tract symptoms: does comorbidity alter tolerability? J Urol. 1998;160:784–91. doi: 10.1016/S0022-5347(01)62787-3. [DOI] [PubMed] [Google Scholar]
- 9.Michel MC, Bressel H-U, Goepel M, Rübben H. A 6-months large-scale study into the safety of tamsulosin. Br J Clin Pharmacol. 2001;51:609–14. doi: 10.1046/j.0306-5251.2001.01388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowe FC. Coadministration of tamsulosin and three antihypertensive agents in patients with benign prostatic hyperplasia: pharmacodynamic effect. Clin Ther. 1997;19:730–42. doi: 10.1016/s0149-2918(97)80097-5. [DOI] [PubMed] [Google Scholar]
- 11.Franco-Salinas G, de la Rosette JJMCH, Michel MC. Pharmacokinetics and pharmacodynamics of tamsulosin in its modified-release and oral-controlled absorption system formulations. Clin Pharmacokin. 2010;49:177–88. doi: 10.2165/11317580-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 12.Tsuda Y, Tatami S, Yamamura N, Tadayasu Y, Sarashina A, Liesenfeld K-H, Staab A, Schäfer H-G, Ieiri I, Higuchi S. Population pharmacokinetics of tamsulosin hydrochloride in paediatric patients with neuropathic and non-neuropathic bladder. Br J Clin Pharmacol. 2010;70:88–101. doi: 10.1111/j.1365-2125.2010.03662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soeishi Y, Matsushima H, Watanabe T, Higuchi S, Cornelissen K, Ward J. Absorption, metabolism and excretion of tamsulosin hydrochloride in man. Xenobiotica. 1996;26:637–45. doi: 10.3109/00498259609046739. [DOI] [PubMed] [Google Scholar]
- 14.Kamimura H, Oishi S, Matsushima H, Watanabe T, Higuchi S, Hall M, Wood SG, Chasseaud LF. Identification of cytochrome P450 isozymes involved in metabolism of the α1-adrenoceptor blocker tamsulosin in human liver microsomes. Xenobiotica. 1998;28:909–22. doi: 10.1080/004982598238985. [DOI] [PubMed] [Google Scholar]
- 15.Taguchi K, Saitoh M, Sato S, Asano M, Michel MC. Effects of tamsulosin metabolites at alpha-1 adrenoceptor subtypes. J Pharmacol Exp Ther. 1997;280:1–5. [PubMed] [Google Scholar]
- 16.Miyazawa Y, Forrest A, Schentag JJ, Kamimura H, Swarz H, Ito Y. Effect of concomitant administration of cimetidine hydrochloride on the pharmacokinetic and safety profile of tamsulosin hydrochloride 0.4 mg in healthy subjects. Curr Ther Res. 2002;63:15–26. [Google Scholar]
- 17.Bertelsen KM, Vankatakrishnan K, von Moltke LL, Obach RS, Greenblatt DJ. Apparent mechanism-based inhibition of human CYP2D6 in vitro by paroxetine: comparison with fluoxetine and quinidine. Drug Metab Dispos. 2003;31:289–93. doi: 10.1124/dmd.31.3.289. [DOI] [PubMed] [Google Scholar]
- 18.Troost J, Tatami S, Tsuda Y, Mattheus M, Mehlburger L, Michel MC. Effects of the CYP3A4 inhibitor ketoconazole on the pharmacokinetics of a single oral dose of tamsulosin. Br J Clin Pharmacol. 2010;70:305. doi: 10.1111/j.1365-2125.2011.03988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Troost J, Tatami S, Tsuda Y, Mattheus M, Mehlburger L, Michel MC. Effects of the CYP2D6 inhibitor paroxetine on the pharmacokinetics of a single oral dose of tamsulosin. Br J Clin Pharmacol. 2010;70:305–6. doi: 10.1111/j.1365-2125.2011.03988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michel MC, Korstanje C, Krauwinkel W, Kuipers M. The pharmacokinetic profile of tamsulosin oral controlled absorption system (OCASR) Eur Urol Suppl. 2005;4:15–24. [Google Scholar]
- 21.Chapple CR, Wyndaele JJ, Nordling J, Boeminghaus F, Ypma AFGVM, Abrams P. Tamsulosin, the first prostate-selective a1A-adrenoceptor antagonist. A meta-analysis of two randomized, placebo-controlled multicentre studies in patients with benign prostatic obstruction (symptomatic BPH. Eur Urol. 1996;29:155–67. [PubMed] [Google Scholar]
- 22.Abrams P, Speakman M, Stott M, Arkell D, Pocock R. A dose-ranging study of the efficacy and safety of tamsulosin, the first prostate-selective a1A-adrenoceptor antagonist, in patients with benign prostatic obstruction (symptomatic benign prostatic hyperplasia. Br J Urol. 1997;80:587–96. doi: 10.1046/j.1464-410x.1997.00380.x. [DOI] [PubMed] [Google Scholar]
- 23.Lepor H. Phase III multicenter placebo-controlled study of tamsulosin in benign prostatic hyperplasia. Urology. 1998;51:892–900. doi: 10.1016/s0090-4295(98)00126-5. [DOI] [PubMed] [Google Scholar]
- 24.Narayan P, Tewari A, Members of United States 93-01 Study Group A second phase III multicenter placebo controlled study of 2 dosages of modified release tamsulosin in patients with symptoms of benign prostatic hyperplasia. J Urol. 1998;160:1701–6. [PubMed] [Google Scholar]
- 25.Michel MC, Korstanje C, Krauwinkel W. Cardiovascular safety of tamsulosin modified release in the fasted and fed state in elderly healthy subjects. Eur Urol Suppl. 2005;4:9–14. [Google Scholar]
- 26.Mets TF. Drug-induced orthostatic hypotension in older patients. Drugs Aging. 1995;6:219–28. doi: 10.2165/00002512-199506030-00005. [DOI] [PubMed] [Google Scholar]
- 27.Hemeryck A, Lefebvre RA, de Vriendt C, Belpaire FM. Paroxetine affects metoprolol pharmacokinetics and pharmacodynamics in healthy volunteers. Clin Pharmacol Ther. 2000;67:283–91. doi: 10.1067/mcp.2000.104788. [DOI] [PubMed] [Google Scholar]
- 28.Olkkola KT, Backman JT, Neuvonen PJ. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;55:481–5. doi: 10.1038/clpt.1994.60. [DOI] [PubMed] [Google Scholar]
- 29.Varhe A, Olkkola KT, Neuvonen PJ. Oral triazolam is potentially hazardous to patients receiving systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;56:601–7. doi: 10.1038/clpt.1994.184. [DOI] [PubMed] [Google Scholar]
- 30.Lewis LD. Drug-drug interactions: is there an optimal way to study them? Br J Clin Pharmacol. 2010;70:781–3. doi: 10.1111/j.1365-2125.2010.03829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Hoogdalem EJ, Soeishi Y, Matsushima H, Higuchi S. Disposition of the selective α1A-adrenoceptor antagonist tamsulosin in humans: comparison with data from interspecies scaling. J Pharm Sci. 1997;86:1156–61. doi: 10.1021/js960303k. [DOI] [PubMed] [Google Scholar]
- 32.Lepor H. Phase III multicenter placebo-controlled study of tamsulosin in benign prostatic hyperplasia. Urology. 1998;51:892–900. doi: 10.1016/s0090-4295(98)00126-5. [DOI] [PubMed] [Google Scholar]
- 33.Narayan P, Tewari A. Members of United States 93-01 Study Group. A second phase III multicenter placebo controlled study of 2 dosages of modified release tamsulosin in patients with symptoms of benign prostatic hyperplasia. J Urol. 1998;160:1701–6. [PubMed] [Google Scholar]
- 34.de Mey C, Michel MC, McEwen J, Moreland T. A double-blind comparison of terazosin and tamsulosin on their differential effects on ambulatory blood pressure and nocturnal orthostatic stress testing. Eur Urol. 1998;33:481–8. doi: 10.1159/000019639. [DOI] [PubMed] [Google Scholar]
- 35.de Mey C, Terpstra I. Orthostatic effects of alfuzosin twice daily vs. tamsulosin one daily in the morning. J Urol. 2000;163(Suppl.):220. [Google Scholar]
- 36.Michel MC, Korstanje C, Krauwinkel W, Shear M, Davies J, Quartel A. Cardiovascular safety of the oral controlled absorption system (OCAS) formulation of tamsulosin compared to the modified release (MR) formulation. Eur Urol Suppl. 2005;4:53–60. [Google Scholar]
- 37.Nieminen T, Ylitalo R, Kööbi T, Ylitalo P, Kähönen M. The vasodilatory effect of alfuzosin and tamsulosin in passive orthostasis: a randomised, double-blind, placebo-controlled study. Eur Urol. 2005;47:340–5. doi: 10.1016/j.eururo.2004.11.002. [DOI] [PubMed] [Google Scholar]