

Abstract
AIMS
To assess the effects of fluconazole, a moderate CYP3A4 inhibitor, on the pharmacokinetics (PK) and safety/tolerability of fesoterodine.
METHODS
In this open-label, randomized, two-way crossover study, 28 healthy subjects (18–55 years) received single doses of fesoterodine 8 mg alone or with fluconazole 200 mg. PK endpoints, including the area under the plasma concentration–time curve from 0 to infinity (AUC(0,∞)), maximum plasma concentration (Cmax), time to Cmax (tmax), and half-life (t1/2), were assessed for 5-hydroxymethyl tolterodine (5-HMT), the active moiety of fesoterodine.
RESULTS
Concomitant administration of fesoterodine with fluconazole increased AUC(0,∞) and Cmax of 5-HMT by approximately 27% and 19%, respectively, with corresponding 90% confidence intervals of (18%, 36%) and (11%, 28%). There was no apparent effect of fluconazole on 5-HMT tmax or t½. Fesoterodine was generally well tolerated regardless of fluconazole co-administration, with no reports of death, serious adverse events (AEs) or severe AEs. Following co-administration of fesoterodine with fluconazole, 13 subjects (48%) experienced a total of 40 AEs; following administration of fesoterodine alone, six subjects (22%) experienced a total of 19 AEs. The majority of AEs were of mild intensity. There were no clinically significant changes in laboratory or physical examination parameters.
CONCLUSION
Fesoterodine 8 mg single dose was well tolerated when administered alone or with fluconazole. Based on the observed increase in 5-HMT exposures being within the inherent variability of 5-HMT pharmacokinetics, adjustment of fesoterodine dose is not warranted when co-administered with a moderate CYP3A4 inhibitor provided they are not also inhibitors of transporters.
Keywords: drug–drug interactions, fesoterodine, fluconazole, overactive bladder, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Available data suggest that fesoterodine dosage should not exceed 4 mg once daily when taken concomitantly with potent CYP3A4 inhibitors, such as ketoconazole. Currently, no information is available on whether dose adjustment is necessary when fesoterodine is administered with a moderate CYP3A4 inhibitor.
WHAT THIS STUDY ADDS
This study shows that adjustment of fesoterodine dose is not warranted when co-administered with a moderate CYP3A4 inhibitor.
Introduction
Fesoterodine is an oral antimuscarinic drug that is approved for the treatment of overactive bladder (OAB) symptoms in 4 mg and 8 mg once daily doses [1, 2]. Fesoterodine is a prodrug that is rapidly and extensively converted to its active metabolite, 5-hydroxymethyl tolterodine (5-HMT) by nonspecific and ubiquitous esterases [3]. Accordingly, fesoterodine is not detectable in plasma after oral dosing and its antimuscarinic effects are attributable to 5-HMT [4]. Fesoterodine exhibits linear pharmacokinetics (PK) across a wide range of doses (4–28 mg) [4], with maximum plasma concentrations of 5-HMT achieved approximately 5 h after fesoterodine administration [4, 5]. The terminal half-life of 5-HMT following administration of fesoterodine extended release tablets is approximately 7 h and reflects the release rate of the formulation due to flip-flop pharmacokinetics [1, 5, 6]. After oral administration of fesoterodine, approximately 70% of the administered dose was recovered in urine as the active metabolite 5-HMT (16%), carboxy metabolite (34%), carboxy-N-desisopropylmetabolite (18%), or N-desisopropyl metabolite (1%), and a smaller amount (7%) was recovered in faeces [1, 3]. Study results show that the elimination of 5-HMT is mediated by multiple pathways [3, 6] including two equally prominent hepatic metabolism pathways involving cytochrome P450 (CYP) isozymes CYP2D6 and CYP3A4. Therefore, it is relevant to examine the effects of reduced elimination of 5-HMT that might result from interactions with drugs that modify CYP3A4 activity on 5-HMT exposure. The renal clearance of 5-HMT in healthy subjects is about 14.4 l h–1, which is considerably in excess of the nominal glomerular filtration rate of approximately 7.5 l h–1, suggesting the involvement of a secretory component in 5-HMT renal clearance [7].
Because of the availability of multiple pathways for the elimination of 5-HMT, hepatic impairment [8], renal impairment [7] and administration of potent CYP3A4 or CYP2D6 inhibitors [9] have only modest effects on 5-HMT exposure following fesoterodine administration. For example, co-administration of the potent CYP3A4 inhibitor ketoconazole with fesoterodine has been shown to cause an approximately 2-fold increase in 5-HMT exposure in both CYP2D6 poor metabolizers (PMs) and extensive metabolizers (EMs) [9]. According to regulatory guidelines [10, 11] the finding of a statistically significant interaction with a potent CYP3A4 inhibitor, such as ketoconazole, may provide cause for further studies with moderate CYP3A4 inhibitors in order to assess the relative degree of inhibition, to provide recommendations for dosage adjustment, and to predict interactions with other moderate CYP3A4 inhibitors that are likely to be co-administered with the drug under development.
The draft drug interaction regulatory guidelines, under review by both the CPMP and FDA, classify an agent that increases midazolam AUC between 2- and 5-fold as a moderate CYP3A4 inhibitor [10, 11]. Ketoconazole and fluconazole are among choices of potent [10, 11] and moderate [10] CYP3A inhibitors, respectively, that are recommended in regulatory guidelines. Acute (400 mg) and steady-state (200 mg once daily) administration of fluconazole has been shown to increase AUC of orally administered midazolam approximately 3.6-fold [12]. A 3.9-fold increase in oral midazolam AUC was observed when midazolam was administered 2 h following a single 200 mg dose of fluconazole [13]. Co-administration of midazolam with a single 400 mg dose of fluconazole resulted in mean midazolam AUC about 3.7-fold higher compared with that following midazolam administration alone [13, 14]. Based on these results, midazolam exposures are up to 3.9-fold higher upon single and multiple dose fluconazole pretreatment, which meets the criterion for a moderate CYP3A4 inhibitor [10–12, 14].
In fasted normal volunteers, the terminal plasma elimination half-life of fluconazole is approximately 30 h (range 20–50 h) following oral administration of a single oral 400 mg dose. Steady state concentrations are reached within 5–10 days following oral doses of 50–400 mg given once daily. In general, a loading dose of twice the daily dose is recommended on the first day of therapy to result in plasma concentrations close to steady-state by the second day of therapy. In a study of fluconazole interaction after administration of fluconazole 400 mg on the first day and then 200 mg daily for 5 days, fluconazole concentrations were 6.6 ± 2.2, 9.0 ± 2.1 and 9.2 ± 0.8 µg ml–1, on days 1, 4 and 6, respectively, and appeared to be at steady-state by day 4 [12]. In this study, fluconazole increased AUC of orally administered midazolam to a similar extent (approximately 3.6-fold) on days 1 and 6.
Fluconazole is an inhibitor of CYP3A4, CYP2C9 and 2C19 enzymes but not an inhibitor of transporters [10, 12, 15]. The metabolism of 5-HMT is mediated by CYP3A4 and CYP2D6 enzymes, and does not involve CYP2C9 or 2C19 enzymes [3, 6, 9]. Therefore, the effects of fluconazole on the PK of 5-HMT would be considered representative of the interactions of moderate CYP3A4 inhibitors with fesoterodine. Furthermore, in the ketoconazole-fesoterodine interaction study, there was a similar increase in 5-HMT exposures in CYP2D6 EMs vs. PMs: AUC increased 2.3- and 2.5-fold and Cmax increased 2.0- and 2.1-fold in EMs and PMs, respectively [9]. Therefore, this CYP3A4 interaction study was performed in subjects without specific regard to CYP2D6 genotype.
Based on the 2-fold increase in 5-HMT exposures as a result of interaction with a potent CYP3A4 inhibitor (ketoconazole) and the availability of multiple pathways of elimination, co-administration of a moderate CYP3A4 inhibitor with fesoterodine is not expected to have a statistically significant effect on 5-HMT PK. However, this remains to be assessed in a clinical study. The objective of this study was to estimate the effects of fluconazole, a model moderate CYP3A4 inhibitor, on the PK of a single 8 mg oral dose of fesoterodine in healthy adult subjects. An 8 mg dose of fesoterodine was selected because it is the highest approved therapeutic dose of fesoterodine [1, 2].
Methods
Study design
This was an open-label, randomized, two-period, two-treatment, two-way crossover study. Subject screening assessments included medical history, physical examination with haemodynamics (including blood pressure and heart rate), 12-lead electrocardiogram (ECG), genotyping for CYP2D6 status (if not known) and laboratory screening. Eligible subjects were admitted to the Pfizer Clinical Research Unit (New Haven, CT) on the evening before each study period. Twenty-eight subjects aged 18 to 55 years received a single dose of fesoterodine 8 mg alone (treatment A) and a single dose of fesoterodine 8 mg co-administered with fluconazole 200 mg (treatment B). The crossover treatments were separated by a 9 day washout period. Specifically, in both treatment periods, single dose fesoterodine 8 mg was administered orally in a fasted state on day 1. During treatment B, fluconazole 200 mg was administered 1 h before and approximately 11 h after fesoterodine 8 mg administration on day 1, and also on day 2 approximately 24 and 36 h after fesoterodine 8 mg administration on day 1. Subjects completed the two treatment periods in a randomized order.
The study was conducted in compliance with the Declaration of Helsinki, the International Conference on Harmonization Good Clinical Practice Guidelines and all local regulatory requirements. The protocol was approved by the appropriate Institutional Review Board for the study centre. All subjects provided written informed consent before entering the study.
Subjects
Eligible subjects included healthy men or women 18 to 55 years of age with a body mass index (BMI) between 17.5 and 30.5 kg m–2 and with a total body weight >50 kg. Subjects were excluded if they had a severe acute or chronic medical or psychiatric condition or laboratory abnormality, had current or previous urinary retention, bladder obstruction, micturition disturbance, urinary tract infections, nocturia, prostatic hyperplasia, or urethral stricture, had uncontrolled narrow angle glaucoma, had regular alcohol consumption exceeding seven drinks per week for women or 14 drinks per week for men within 6 months of screening and had 12-lead ECG demonstrating corrected QT interval (QTc) >450 ms at screening.
Assessment of pharmacokinetics and safety
Blood samples for 5-HMT PK analysis were collected pre dose and at 1, 2, 3, 4, 5, 6, 8, 10, 12, 15, 24, 36 and 48 h after fesoterodine dosing. The total volume of blood samples collected from each subject was approximately 197 ml. Blood samples were placed immediately into an ice/water bath and centrifuged at 1700 g for 10 min at 4°C, and the resultant plasma samples (minimum of 2 ml) were stored at approximately −20°C within 1 h of collection. Plasma samples were analyzed for 5-HMT concentrations at Advion Bioservices, Inc (Ithaca, NY) using a validated liquid chromatography tandem mass spectrometry (LC-MS-MS) method, as previously described [5].
Plasma 5-HMT PK parameters, which included AUC from time zero to infinity (AUC(0,∞)), AUC from time 0 to the time of the last quantifiable concentration (AUC(0,tlast)), maximum plasma concentration (Cmax), time to Cmax (tmax) and half-life (t1/2) were determined from plasma concentration–time data using standard noncompartmental methods.
Safety was assessed via adverse event (AE) monitoring, laboratory evaluations and vital sign measurements. Safety data were analyzed descriptively and included all subjects who received at least one dose of study drug.
Statistics
A sample size of 24 subjects was required to provide 90% confidence intervals (CIs) for the difference between treatments of ± 0.085 and ± 0.1309 on a natural log-transformed scale, or for the ratio between treatments in the range of (0.919, 1.089) and (0.877, 1.140) on the non-transformed scale, for AUC(0,∞) and Cmax, respectively, with 90% coverage probability. This estimate was based on estimates of within-subject standard deviations of 0.1449 and 0.2232 for ln AUC(0,∞) and ln Cmax[or coefficient of variation (CV) of 0.1457 and 0.2260 for AUC(0,∞) and Cmax,], respectively, obtained from the results of previous studies. A total of 28 subjects were enrolled. The additional subjects were enrolled, anticipating discontinuation by up to four subjects (about 20%) based on prior experience for studies of similar design conducted at the clinical research unit.
Log transformed AUC(0,∞), AUC(0,last) and Cmax were analyzed using analysis of variance (anova), with sequence, period and treatment as fixed effects and subject within sequence as a random effect, to yield estimated adjusted means of PK parameters and corresponding 90% CIs for fesoterodine (reference) and fesoterodine plus fluconazole (test) groups. The adjusted mean differences and 90% CIs for the differences were exponentiated to provide point estimates of the ratio of adjusted geometric means (test : reference) and 90% CIs for the ratios. All subjects with at least one concentration measurement in at least one treatment period were included in concentration summaries. All subjects with at least one PK parameter of interest in at least one treatment were included in the statistical analysis of PK parameters.
Physiologically based pharmacokinetic (PBPK) methodology
A PBPK model (SimCYP V7.1) for 5-HMT was generated incorporating physico-chemical properties (log P = 4.3, base, pKa = 10.3), fraction unbound in plasma (0.49), blood : plasma ratio (1.0), absorption (0.81) and steady-state volume of distribution (1.7 l kg–1) together with intrinsic clearances for CYPs 3A4 and 2D6 (both 13.4 µl min−1mg−1 microsomal protein) and renal clearance (15 l h–1).
The contributions of CYP 3A4 and 2D6 to the clearance of 5-HMT were calculated, knowing the total clearance (43 l h–1) and renal clearance of 5-HMT (15 l h–1). The difference between total and renal clearance was assumed to be metabolic clearance. The drug–drug interaction (DDI) studies with the CYP3A4 inhibitor and CYP2D6 EMs and PMs indicated that CYPs 3A4 and 2D6 contributed equally to metabolic clearance [9]. Thus intrinsic clearance inputs for CYPs 3A4 and 2D6 were back calculated from the well-stirred model for hepatic clearance assuming equal contribution for the two enzymes and used in the PBPK model. The model was validated by comparing clinical PK data with PBPK simulations.
The trial design in PBPK for all simulations used a 10 × 10 trial with subjects aged 18 to 65 years, with 34% of the individuals being female. The population was selected as being composed of a standard Caucasian population with 8% CYP2D6 PM subjects. For the fluconazole and ketoconazole DDI simulations, a single dose of 8 mg fesoterodine was administered on day 4 following administration of fluconazole (200 mg twice daily) or ketoconazole (200 mg twice daily), which were the doses used in the clinical studies.
Results
Among 28 subjects assigned to study treatment, 26 subjects received both the assigned treatments and completed the study. All subjects were between 19 and 48 years old (mean age 34.8 years) with a BMI between 20 to 30 kg m–2 (mean BMI 25.6 kg m–2). The majority (22/28, 79%) of subjects were male and 13 subjects were Black.
Plasma concentration–time profiles of 5-HMT following administration of fesoterodine alone and concomitantly with fluconazole is shown in Figure 1. Compared with fesoterodine administration alone, concomitant administration of fesoterodine with fluconazole resulted in an increase in mean adjusted geometric mean 5-HMT AUC(0,∞) from 49.5 to 62.8 ng ml–1 h and an increase in adjusted geometric mean Cmax from 4.42 to 5.27 ng ml–1. Point estimates (90% CI) for the ratios of AUC(0,∞) and Cmax for concomitant administration of fesoterodine with fluconazole vs. fesoterodine alone were 126.8 (118.3, 135.9) and 119.2 (110.8, 128.3), respectively (Table 1). The within-subject CV% was 14.7, 14.5 and 15.6 for AUC(0,∞), AUC(0,tlast) and Cmax, respectively, and the between-subject CV% was 30.6, 30.9 and 32.4 for AUC(0,∞), AUC(0,tlast) and Cmax, respectively. There was no apparent effect of fluconazole on the median tmax or mean t1/2 for 5-HMT (Table 1).
Figure 1.
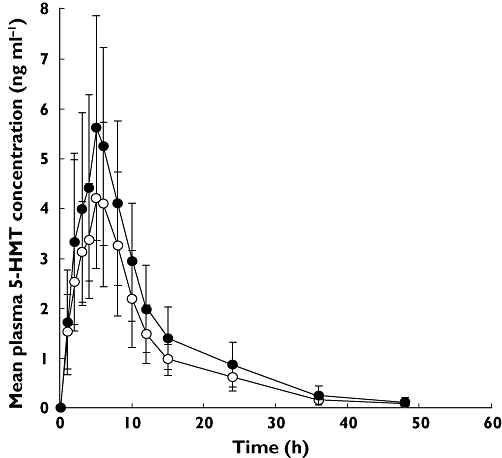
Plasma concentration–time profiles of 5-hydroxymethyl tolterodine (5-HMT) following administration of fesoterodine alone (○) and concomitantly with fluconazole 200 mg (•)
Table 1.
Pharmacokinetics of 5-HMT following administration of single dose fesoterodine 8 mg alone or co-administration of fesoterodine with fluconazole 200 mg*
| Fesoterodine + fluconazole (Test) | Fesoterodine alone (Reference) | Test : reference ratio† (90% CI) | |
|---|---|---|---|
| n | 27 | 27 | – |
| AUC(0,∞) (ng ml–1 h) | 62.8 | 49.5 | 126.8 (118.3, 135.9) |
| AUC(0,tlast) (ng ml–1 h) | 61.4 | 48.3 | 127.1 (118.7, 136.1) |
| Cmax (ng ml–1) | 5.27 | 4.42 | 119.2 (110.8, 128.3) |
| tmax (h) | 5.0 (2.1–6.0) | 5.0 (3.0–6.0) | ND |
| t1/2 (h) | 8.05 (35) | 7.83 (40) | ND |
AUC(0,∞), AUC(0,tlast) and Cmax presented as adjusted geometric means, tmax presented as median (range), t1/2 presented as arithmetic mean (% coefficient of variation).
Ratio of adjusted geometric means.
5-HMT = 5-hydroxymethyl tolterodine; CI = confidence interval; AUC(0,∞) = area under the plasma concentration–time curve from time zero to infinity; AUC(0,tlast) = area under the plasma concentration–time profile from time 0 to the time of the last quantifiable concentration (Clast); Cmax = maximum plasma concentration; ND = not determined; tmax = time to Cmax; t1/2 = half-life.
Fesoterodine was generally well tolerated regardless of fluconazole co-administration, with no reports of death, serious AEs or severe AEs. Two subjects were discontinued due to AEs (one treatment-related and one non treatment-related). The subject discontinued due to a treatment-emergent AE experienced moderate vomiting following administration of fesoterodine plus fluconazole (test) on day 1. However, the AE had resolved by the end of study. Following co-administration of fesoterodine plus fluconazole, 13 subjects (48%) experienced a total of 40 all causality AEs (38 treatment-related); following administration of fesoterodine, six subjects (22%) experienced a total of 19 all causality AEs (all treatment-related), with the majority of AEs of mild intensity (Table 2). There were no clinically significant changes in laboratory parameters or physical examination findings for either regimen.
Table 2.
Incidence of treatment-emergent adverse events*
| Fesoterodine + fluconazole (n = 27) | Fesoterodine (n = 27) | |||
|---|---|---|---|---|
| All-causality adverse events | Treatment-related adverse events | All-causality adverse events | Treatment-related adverse events | |
| Nausea | 5 | 5 | 2 | 2 |
| Dizziness | 5 | 5 | 0 | 0 |
| Vision blurred | 3 | 3 | 1 | 1 |
| Headache | 2 | 2 | 2 | 2 |
| Abdominal distension | 3 | 3 | 0 | 0 |
| Vomiting | 2 | 2 | 1 | 1 |
| Pollakiuria | 2 | 2 | 1 | 1 |
| Abdominal pain | 2 | 2 | 0 | 0 |
| Flatulence | 2 | 1 | 0 | 0 |
Data indicate number of subjects experiencing adverse event; only all-causality, treatment-emergent adverse events occurring in ≥ two subjects during either crossover treatment are shown.
PBPK results
In a comparison with the clinical data on co-administration of fesoterodine 8 mg with 200 mg twice daily of ketoconazole, the results show the ketoconazole DDI simulation slightly underpredicted the magnitude of clinical DDI. The mean (90% CI) increase in 5-HMT AUC for CYP2D6 EMs in the ketoconazole simulation was 1.53 (1.17, 2.19) vs. 2.30 (1.99, 2.72) in the corresponding clinical DDI study [9]. However, the extent of the interaction between fluconazole and 5-HMT predicted by the fluconazole DDI simulation was more consistent with the results of the clinical DDI study reported here. The mean (90% CI) increase in 5-HMT AUC in CYP2D6 EMs was 1.38 (1.15, 1.72) for fluconazole simulation vs. 1.27 (1.18, 1.36) for the corresponding clinical DDI study. When the simulation using a population of CYP2D6 92% EM + 8% PM subjects was compared with 100% EM subjects, similar AUC results of 1.40 (1.15, 1.79) vs. 1.38 (1.15, 1.72), respectively, were predicted. These results suggested a similar extent of CYP3A4 inhibition in CYP2D6 EMs and PMs, and that the equal contributions of CYP3A4 and CYP2D6 to 5-HMT metabolism in humans were accurately represented by the PBPK model.
Discussion
The results of this study showed increases of approximately 27% and 19% in the AUC(0,∞) and Cmax of 5-HMT, respectively, following the co-administration of fluconazole with fesoterodine compared with fesoterodine alone. This magnitude of increase in 5-HMT exposure is considerably smaller than the over 100% increase observed after co-administration with a potent CYP3A4 inhibitor, ketoconazole [9]. Fesoterodine 8 mg was generally well tolerated in the present study, both in the presence or absence of fluconazole. The frequency of AEs in this study are consistent with the results of other phase 1 studies [5, 8], and phase 2 and 3 controlled studies [16, 17]. The increased incidence of AEs following concomitant administration of fesoterodine and fluconazole in this study is consistent with the somewhat increased 5-HMT exposure and/or treatment with fluconazole. The increase in nausea observed with co-administration of fluconazole and fesoterodine is expected as it is a common adverse event associated with fluconazole and can be attributed to its concomitant administration [18].
Fluconazole at 400 mg once daily (multiple dose) is typically cited as giving a maximal inhibitory effect for this compound [10]. Thus, fluconazole at 200 mg twice daily (multiple dose) requires several days to reach steady-state. The PBPK simulations achieved steady-state fluconazole concentrations but the clinical protocol used would not reach maximum concentrations of fluconazole. However, given that the proportion of CYP3A4 mediated clearance is about a third of the total (i.v.) clearance of 5-HMT in CYP2D6 EM individuals, then a moderate CYP3A4 inhibitor (assuming no other DDI mechanisms) would still not be expected to show a clinically relevant effect on 5-HMT PK.
In the present study, all subjects except one were genotyped as CYP2D6 EMs, and the one CYP2D6 PM discontinued. Thus, our results do not provide information on whether the effect of CYP3A4 inhibition on 5-HMT PK differs based on CYP2D6 function. However, it has been shown that the magnitude of the effects of co-administration of ketoconazole with fesoterodine on 5-HMT AUC and Cmax were similar in CYP2D6 EMs and PMs (i.e. AUC increased 2.3- and 2.5-fold and Cmax increased 2.0- and 2.1-fold in EMs and PMs, respectively), with no effect on t1/2 or tmax in either group, compared with fesoterodine alone [9]. Similar to ketoconazole co-administration with fesoterodine, co-administration of fluconazole with fesoterodine in the present study had no apparent effect on the mean t1/2 or median tmax of 5-HMT. These data suggest that the effects of a potent CYP3A4 inhibitor on 5-HMT exposure are independent of CYP2D6 activity. Similarly, the effect of moderate or weak CYP3A4 inhibitors with 5-HMT PK are not expected to be different in CYP2D6 EMs compared with PMs. Therefore, the present study was designed to assess the effect of fluconazole on 5-HMT PK in the entire study population, regardless of the subjects' CYP2D6 genotype.
PBPK modelling has been used prior to the design of this study. The PBPK model for 5-HMT following fesoterodine administration incorporated clearance inputs for CYP3A4 and 2D6 together with renal clearance so that approximately equal fractions of the dose were cleared by the three respective mechanisms, and was validated by comparing clinical plasma concentration–time curves following administration of either intravenous 5-HMT or oral fesoterodine with PBPK simulated data. The change in 5-HMT AUC produced by ketoconazole administration was due to inhibition of CYP3A4 together with a reduction of renal clearance, possibly due to an inhibitory effect of an active component of renal clearance. Ketoconazole is a known inhibitor of some active transporters such as P-gp and 5-HMT is a substrate for the P-gp transporter. CYP3A4 inhibition but not renal clearance inhibition can be visualized in PBPK, thus offering some explanation for the under prediction of the ketoconazole DDI. In contrast, the fluconazole DDI was well predicted by PBPK, because the DDI was expected to be primarily at the level of CYP3A4. Fluconazole is not known to be an inhibitor of transporters including P-gp. 5-HMT is not known to be a substrate of other transporters. Further investigation of other transporters is in progress.
The effect of fluconazole on 5-HMT PK observed in the present study is relatively small compared with its effect on the PK of other CYP3A4 substrates [19]. For example, fluconazole has been shown to increase the AUC of diazepam by 2.5-fold and to prolong t1/2 by approximately 2-fold [20]. The effect of fluconazole on diazepam exposure is pronounced as the elimination of diazepam depends predominantly on CYP2C19 and CYP3A4 and its inhibition by fluconazole is reflected by increased AUC as well as prolonged terminal elimination half-life following immediate release oral administration of diazepam [20, 21]. As described previously, the elimination of 5-HMT is mediated by multiple pathways [3, 6], including two equally prominent hepatic metabolism pathways, CYP2D6 and CYP3A4 enzymes. In addition, 5-HMT is excreted unchanged in the urine, accounting for approximately 16% of the fesoterodine dose. The marginal increase in 5-HMT exposures by a moderate CYP3A4 inhibitor relative to the overall variability of 5-HMT's PK in this study is consistent with the relatively small contribution of the CYP3A4 pathway to the total elimination of 5-HMT [6, 8, 9].
Overall, the 90% CIs for the test : reference 5-HMT exposure ratios (fluconazole + fesoterodine : fesoterodine alone) of 5-HMT are relatively narrow (<18% interval widths with the intervals of (118%, 136%) and (111%, 128%) for AUC(0,∞) and Cmax, respectively) supporting the precision of the estimated magnitude of the interaction. The upper 90% CI limits for AUC and Cmax were contained within 1.4-fold. The effect of a high fat meal on the PK of 5-HMT is of a similar magnitude as that of a moderate CYP3A4 inhibitor (approximately 19% and 18% increase in 5-HMT AUC and Cmax, respectively, tmax and t1/2 unchanged) [22]. Fesoterodine is indicated for administration without regard to food intake [1, 2]. Similarly, no dose adjustment is recommended for the patients with mild to moderate renal impairment in whom exposure is increased 1.5–1.8-fold compared with patients with no renal impairment [1, 2]. Considering the demonstrated safety and tolerability at doses up to 12 mg and the highest approved dose of 8 mg, the less than 30% increase in 5-HMT exposure observed from fluconazole co-adminstration in the present study is not deemed clinically meaningful. Therefore, co-administration of fesoterodine with moderate CYP3A4 inhibitors that are not also inhibitors of other transporters that influence the PK of 5-HMT does not warrant specific dosing recommendations or additional consideration for dose escalation.
Acknowledgments
Funding for this study was provided by Pfizer Inc. Editorial assistance was provided by Nancy Sheridan and Colin P. Mitchell, PhD, from Complete Healthcare Communications, Inc., and was funded by Pfizer Inc.
Competing Interests
BM, CA, ZJ, GD and KG are Pfizer employees and own Pfizer stock. There are no other competing interests to declare.
REFERENCES
- 1.Pfizer Inc. Toviaz® (Fesoterodine Fumarate). Full Prescribing Information. New York: Pfizer Inc; 2010. [Google Scholar]
- 2.European Medicines Agency (EMEA) EPARs for authorized medicinal products for human use. European Public Assessment Report for Toviaz (Product Information). Updated August 2009. Available at http://www.emea.europa.eu/humandocs/Humans/EPAR/toviaz/toviaz.htm (last accessed 20 October 2009)
- 3.Michel MC. Fesoterodine: a novel muscarinic receptor antagonist for the treatment of overactive bladder syndrome. Expert Opin Pharmacother. 2008;9:1787–96. doi: 10.1517/14656566.9.10.1787. [DOI] [PubMed] [Google Scholar]
- 4.Cole P. Fesoterodine, an advanced antimuscarinic for the treatment of overactive bladder: a safety update. Drugs Future. 2004;29:715–20. [Google Scholar]
- 5.Malhotra B, Guan Z, Wood N, Gandelman K. Pharmacokinetic profile of fesoterodine. Int J Clin Pharmacol Ther. 2008;46:556–63. doi: 10.5414/cpp46556. [DOI] [PubMed] [Google Scholar]
- 6.Malhotra B, Gandelman K, Sachse R, Wood N, Michel MC. The design and development of fesoterodine as a prodrug of 5- hydroxymethyl tolterodine (5-HMT), the active metabolite of tolterodine. Curr Med Chem. 2009;16:4481–9. doi: 10.2174/092986709789712835. [DOI] [PubMed] [Google Scholar]
- 7.Malhotra B, Gandelman K, Sachse R, Wood N. Assessment of the effects of renal impairment on the pharmacokinetic profile of fesoterodine. J Clin Pharmacol. 2009;49:477–82. doi: 10.1177/0091270009332434. [DOI] [PubMed] [Google Scholar]
- 8.de Mey C, Mateva L, Krastev Z, Sachse R, Wood N, Malhotra B. Effects of hepatic dysfunction on the single-dose pharmacokinetics of fesoterodine. J Clin Pharmacol. 2011;51:397–405. doi: 10.1177/0091270010365547. [DOI] [PubMed] [Google Scholar]
- 9.Malhotra B, Sachse R, Wood N. Evaluation of drug-drug interactions with fesoterodine. Eur J Clin Pharmacol. 2009;65:551–60. doi: 10.1007/s00228-009-0648-1. [DOI] [PubMed] [Google Scholar]
- 10.Food and Drug Administration. Guidance for Industry: Drug Interaction Studies-Study Design, Data Analysis, and Implications for Dosing and Labeling. Draft Guidance. September 2006. Available at http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm064982.htm (last accessed 20 October 2009)
- 11.European Agency for the Evaluation of Medicinal Products, Committee for Propietary Medicinal Products. Note for guidance on the clinical investigation of drug interactions. June 2008. Report No.: CPMP/EWP/560.95.
- 12.Olkkola KT, Ahonen J, Neuvonen PJ. The effects of the systemic antimycotics, itraconazole and fluconazole, on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Anesth Analg. 1996;82:511–6. doi: 10.1097/00000539-199603000-00015. [DOI] [PubMed] [Google Scholar]
- 13.Kharasch ED, Walker A, Hoffer C, Sheffels P. Sensitivity of intravenous and oral alfentanil and pupillary miosis as minimally invasive and noninvasive probes for hepatic and first-pass CYP3A activity. J Clin Pharmacol. 2005;45:1187–97. doi: 10.1177/0091270005280077. [DOI] [PubMed] [Google Scholar]
- 14.Ahonen J, Olkkola KT, Neuvonen P. Effect of route of administration of fluconazole on the interaction between fluconazole and midazolam. Eur J Clin Pharmacol. 1997;51:415–9. doi: 10.1007/s002280050223. [DOI] [PubMed] [Google Scholar]
- 15.Kunze KL, Wienkers LC, Thummel KE, Trager WF, Warfarin-fluconazole I. Inhibition of the human cytochrome P450-dependent metabolism of warfarin by fluconazole: in vitro studies. Drug Metab Dispos. 1996;24:414–21. [PubMed] [Google Scholar]
- 16.Chapple C, Van Kerrebroeck P, Tubaro A, Haag-Molkenteller C, Forst HT, Massow U, Wang J, Brodsky M. Clinical efficacy, safety, and tolerability of once-daily fesoterodine in subjects with overactive bladder. Eur Urol. 2007;52:1204–12. doi: 10.1016/j.eururo.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 17.Nitti V, Dmochowski R, Sand P, Forst H-T, Haag-Molkenteller C, Massow U, Wang J, Brodsky M, Bavendam T. Efficacy, safety, and tolerability of fesoterodine in subjects with overactive bladder. J Urol. 2007;178:2488–94. doi: 10.1016/j.juro.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 18.Pfizer Inc. Diflucan (Fluconazole). Full Prescribing Information. New York: Pfizer Inc; 1990. [Google Scholar]
- 19.Nivoix Y, Leveque D, Herbrecht R, Koffel JC, Beretz L, Ubeaud-Sequier G. The enzymatic basis of drug-drug interactions with systemic triazole antifungals. Clin Pharmacokinet. 2008;47:779–92. doi: 10.2165/0003088-200847120-00003. [DOI] [PubMed] [Google Scholar]
- 20.Saari TI, Laine K, Bertilsson L, Neuvonen PJ, Olkkola KT. Voriconazole and fluconazole increase the exposure to oral diazepam. Eur J Clin Pharmacol. 2007;63:941–9. doi: 10.1007/s00228-007-0350-0. [DOI] [PubMed] [Google Scholar]
- 21.Saari TI, Olkkola KT. Azole antimycotics and drug interactions in the perioperative period. Curr Opin Anaesthesiol. 2010;23:441–8. doi: 10.1097/ACO.0b013e32833a254d. [DOI] [PubMed] [Google Scholar]
- 22.Malhotra B, Sachse R, Wood N. Influence of food on the pharmacokinetic profile of fesoterodine. Int J Clin Pharmacol Ther. 2009;47:384–90. doi: 10.5414/cpp47384. [DOI] [PubMed] [Google Scholar]