

Abstract
The androgen receptor (AR) signaling axis plays a critical role in the development, function and homeostasis of the prostate. The classical action of AR is to regulate gene transcriptional processes via AR nuclear translocation, binding to androgen response elements on target genes and recruitment of, or crosstalk with, transcription factors. Prostate cancer initiation and progression is also uniquely dependent on AR. Androgen deprivation therapy remains the standard of care for treatment of advanced prostate cancer. Despite an initial favorable response, almost all patients invariably progress to a more aggressive, castrate-resistant phenotype. Considerable evidence now supports the concept that development of castrate-resistant prostate cancer (CRPC) is causally related to continued transactivation of AR. Understanding the critical events and complexities of AR signaling in the progression to CRPC is essential in developing successful future therapies. This review provides a synopsis of AR structure and signaling in prostate cancer progression, with a special focus on recent findings on the role of AR in CRPC. Clinical implications of these findings and potential directions for future research are also outlined.
Keywords: Androgen receptor, castrate-resistant prostate cancer, signaling
BACKGROUND
Prostate cancer is the most frequently diagnosed non-cutaneous malignancy in men, and the second leading cause of male cancer-related mortality in the United States.[1] Clinically localized prostate cancer is managed primarily through surgery or radiation therapy.[2] For patients who recur systemically following definitive treatment or who present with locally advanced or metastatic disease, the mainstay of treatment is androgen-deprivation therapy (ADT), typically with a luteinizing-hormone-releasing hormone (LRHR) agonist.[3] However, this effect is temporary and after a median of 18-24 months, there is disease progression, heralded by rising serum prostate-specific antigen (PSA), increasing tumor size, new metastatic spread and disease-related symptoms.[4] This represents the lethal phenotype of the disease, and is referred to as castration-resistant prostate cancer (CRPC).
The androgen receptor (AR) plays a pivotal role in the normal growth and development of the prostate gland, and also in prostate carcinogenesis and progression to androgen-independent disease. The AR is expressed to some degree in nearly all primary prostate cancers.[5–7] Studies in both humans and animal models suggest a relationship between the cellular AR level in both primary and metastatic lesions; and, in subsequent disease progression to CRPC.[8–10] This evolution from a clinically localized hormone-naïve state to a castrate-resistant phenotype involves a complex interplay of a network of signaling molecules and is attributed to aberrant AR signaling.[10–13]
Rising serum PSA, indicates that AR activity is inappropriately restored in CRPC,[14] a hypothesis that has been solidified though intensive investigation into the mechanisms of therapeutic failure. These mechanisms include (a) AR amplification/overexpression;[15] (b) gain-of-function AR mutations (mostly in the ligand-binding domain, conferring ligand promiscuity);[16] (c) intracrine androgen production;[17] (d) overexpression of AR cofactors (sensitizing cells to low levels of androgens);[18] (e) ligand independent AR activation by cytokines or growth factors;[19] and (f) constitutively active messenger ribonucleic acid (mRNA) splice variants of AR.[20] Thus, AR remains a critical factor in the progression to CRPC. In the following sections, we will give an overview of androgen signaling in prostate cancer development and progression, with a special focus on recent findings on the role of AR in CRPC. Clinical implications of these findings and potential directions for future research are also outlined.
ANDROGEN RECEPTOR STRUCTURE
The human AR gene is a nuclear transcription factor and a member of the steroid hormone receptor superfamily of genes. It is located on the X chromosome (q11-12) and consists of 8 exons. It codes for a protein of 919 amino acids with a mass of 110 kDa. The AR consists of four structurally and functionally distinct domains [Figure 1], a poorly conserved N-terminal domain (NTD), a highly conserved deoxyribonucleic acid (DNA) -binding domain (DBD) and a moderately conserved ligand-binding domain (LBD). A short amino acid sequence called the ‘hinge region’ separates the LBD from the DBD and also contains part of a bipartite ligand-dependent nuclear localization signal (NLS) for AR nuclear transport. AR domain structure and function has been extensively reviewed elsewhere.[21–23]
Figure 1.
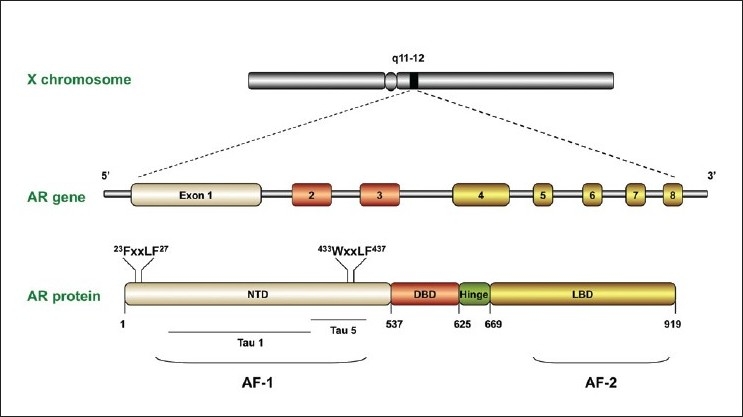
Schematic representation of the androgen receptor gene and protein, with indications of its specific motifs and domains
N-terminal domain
The NTD (amino acids 1-537 coded by exon 1) is considered to be constitutively active, and can activate transcription independently of androgenic stimulus in LBD-deletion mutants.[24,25] The NTD also harbors transcriptional activation function (AF)-1, which encompasses two transcriptional activation units (TAU): TAU-1 and TAU-5.[26] The core domain mediating TAU1 transcriptional activity has been mapped to a discreet 178LKDIL182 motif within the NTD.[27,28] However, TAU5 is responsible for the majority of constitutive transcriptional activity within the NTD, and is mediated through the core sequence 435WHTLF439, accounting for approximately 50% aberrant AR activity in CRPC cells.[29]
Deoxyribonucleic acid-binding domain
The DBD (68 amino acids coded by exons 2 and 3) consists primarily of two zinc finger domains. The first zinc finger contains a conserved P-box motif that co-ordinates gene specific nucleotide contacts within the DNA groove. The second zinc finger contains a conserved D-box motif, which functions as a DBD/DBD binding site for receptor homodimer formation.[30]
Hinge region
A short sequence of approximately 50 amino acids (625-669) called the hinge region separates the LBD from the DBD. This region also contains part of a bipartite ligand-dependent nuclear localization signal (NLS) for AR nuclear import. A cytoskeletal protein Filamin-A (FlnA) interacts with the hinge, DBD and LBD of AR, facilitating AR translocation to the nucleus. FlnA negative cell lines do not show nuclear translocation of AR, and AR remains cytoplasmic even after prolonged androgen exposure.[31] FlnA cytoplasmic localization in clinical specimens also correlates with increased metastatic potential and a hormone-refractory phenotype.[32] Two additional NLS exist in the NTD and LBD with distinct pathways for nuclear import: the NLS of DBD is Ran and importin /β-dependent, whereas the NLSs of NTD and LBD are Ran dependent but importin /β-independent.[33] This suggests that the nuclear import of AR is regulated by interplay between each domain of the AR.
Ligand-binding domain
The AR LBD (amino acids 669-919) facilitates binding of the AR ligands, testosterone and dihydrotestosterone (DHT), which represents the primary control mechanism of the androgen-signaling axis. Similar to the AF1 region in the NTD, AF2 interacts with LxxLL-containing co-regulators (the steroid receptor coactivator [SRC] /p160 family) and 23FQNLF27 and 433WHTLF437 motifs in the NTD.[34,35] Most of the AR point mutations in prostate cancer have been mapped to regions of the LBD including: amino acids 670-676, 701-730 and 874-919.[36] Although the association of point mutations in the AR with resistance to anti-androgens such as bicalutamide is strong, the overall frequency of AR mutations cannot account for most cases of CRPC.[37–39]
CLASSICAL ANDROGEN RECEPTOR SIGNALING
Androgenic steroids are 19-carbon steroids of which, testosterone is the prototype. It is produced primarily by the testes in males with a small contribution from the adrenal glands. Androgens play a role in a wide range of developmental and physiological responses. The cytochrome P450 enzyme, 5α-reductase, which converts testosterone to DHT, is highly expressed within the prostate and genital tissues.[40,41] Both testosterone and DHT can bind to and activate AR under physiological conditions, with DHT having a significantly greater affinity for AR, therefore activating target genes at lower concentrations than testosterone.[42,43].
In the absence of ligand, the AR is located primarily in the cytoplasm [Figure 2], where it associates with heat shock proteins (HSP)-90, -70, -56, cytoskeletal proteins and other chaperones (reviewed in[44]). HSPs are believed to be tethered to cytoskeletal proteins, such as FlnA. FlnA interacts directly with the hinge-region of AR, thereby modulating nuclear translocation and transcriptional action of AR as well as androgen dependence of LNCaP cells.[31,45,46] Androgens enhance association and co-localization of AR with FlnA. This complex also recruits integrin beta 1 and induces activation of Rac1 and focal adhesion kinase (FAK), which both coordinate cell migration.[47] This complex appears to act as a link between androgen signaling and actin cytoskeleton, thereby driving cell migration and may affect prostate cancer progression and metastasis.
Figure 2.
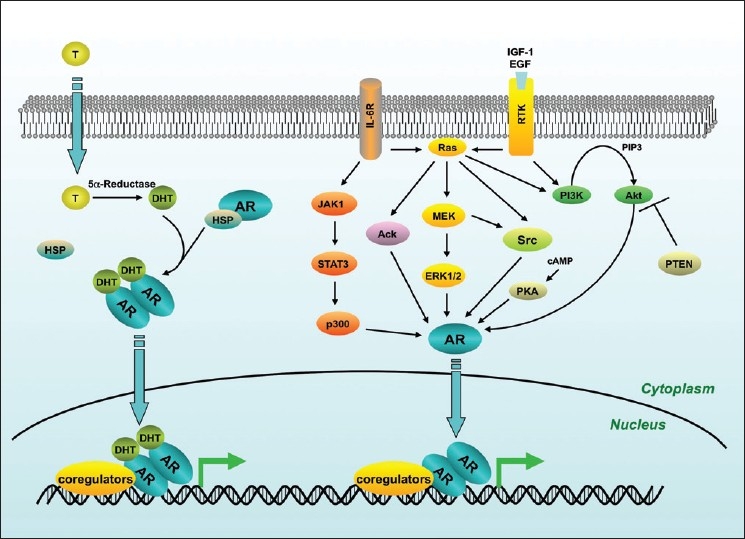
Summary of the major androgen receptor signaling pathways in prostate cancer. Upon binding to dihydrotestosterone, androgen receptor translocates to the nucleus, binds to its target genes and regulates their expression. Androgen receptor can also be transactivated in the absence, or in very low levels of dihydrotestosterone. Activating signals arise from several, non-mutually-exclusive mechanisms including extracellular peptides such as Insulin-like growth factor, Epidermal growth factor and Interleukin-6
Binding of ligand to the AR ligand-binding pocket induces a conformational change in AR whereby helices 3, 4 and 12 within the LBD, form the AF-2 binding surface. AF-2 is the principal protein-protein interaction surface used by nuclear receptors to recruit LxxLL-motif containing coactivators.[34] However, AR differs from other nuclear receptors in this respect and interacts with coactivators in a unique manner. This pocket in the LBD binds preferentially to FxxLF motifs found in the NTD, and interacts poorly with LxxLL motifs commonly found in coactivators.[48,49]
As a result, the hydrophobic pocket within the AR LBD facilitates intramolecular and intermolecular interaction between the AR NTD and its carboxy-terminal domain (CTD ), resulting in the dimerization of AR. This NTD/CTD interaction occurs predominately when AR is not bound to DNA.[50] Several AR-associated coactivators that contain FxxLF motifs have been isolated,[51] suggesting that competition exists between these regulatory proteins and the NTD for binding to the AF-2. The significance of this is unclear, but suggests that additional binding sites outside this well-defined coactivator pocket enable AR to interact with its coactivators, and that different classes of coactivators may interact with different AR surfaces.[18] These interactions facilitate the nuclear targeting of AR and AR homodimer formation. Once inside the nucleus, AR binds to specific recognition sequences known as androgen response elements (AREs) in the promoter and enhancer regions of target genes. The AR transcriptional complex is completed by recruitment of coregulators, which ultimately results in modulation of gene expression.[52]
Androgen receptor coregulatory proteins
Almost 200 AR coregulators have now been identified (reviewed by Heemers and Tindall.[18] These proteins have specific and distinct functions, either enhancing (coactivators) or repressing (corepressors) AR activity, depending on the target gene.[53] However, unlike general and specific transcription factors, they do not significantly alter the basal transcription rate and do not typically possess DNA binding capabilities. Instead, coregulators act at AR target gene promoter and/or enhancer regions to facilitate DNA occupancy, induce chromatin remodeling, and/or recruit general transcription factors associated with RNA polymerase activity.[18] These proteins are broadly divided into 4 main types: (i) molecular chaperones that coordinate AR maturation and movement, (ii) histone modifiers, (iii) coordinators of transcription and (iv) DNA structural modifiers.
The formation of an active AR-directed transcription pre-initiation complex occurs via the sequential recruitment of coregulators with distinct activities on the ligand-bound nuclear AR. The first identified and most widely understood of these coregulatory proteins is the p160 coactivator family, which consists of three 160 kDa proteins: SRC1, transcription intermediary factor 2 (TIF2; and its mouse homologue GRIP1) and SRC3. Immunohistochemical studies have shown that SRC1 expression is increased in 50% of androgen-dependent prostate cancer samples, compared to benign or normal prostate tissues. Moreover, SRC1 and TIF2 expression are increased in 63% of CRPC samples.[54] A correlation between increased levels of SRC3 and prostate tumor grade and stage has been identified in clinically localized disease.[55,56] The p160 coactivators interact with the AR NTD[57] and also the LBD, thereby enhancing ligand-dependent, AR-mediated transcription of target genes.[58] Their recruitment directly influences AR transactivation capacity via intrinsic histone acetyltransferase activity,[59] and indirectly by acting as platforms for the recruitment of secondary coactivators possessing chromatin remodeling and protein acetyltransferase capabilities such as p300.[60] In tissue samples from patients with biopsy-proven prostate cancer who underwent prostatectomy, p300 levels not only correlated with proliferation in vivo, but also predicted larger tumor volumes, the likelihood of extra-prostatic extension, seminal vesicle involvement at surgery as well as progression after surgery.[61] Further evidence in vitro has shown that an increase in p300 expression, fostered by androgen deprivation, offers a growth advantage to androgen-insensitive prostate cancer cells.[62]
The role of the non-AR specific coregulator, ARA70, is less clear. ARA70 protein has been found to be overexpressed in high grade prostate carcinomas, prostate cancer cell lines and xenografts,[63] whereas ARA70 mRNA expression was found to be decreased in prostate tumor tissue,[64] increased in response to hormone deprivation[65] or unchanged between normal and prostate cancer in the same tissue.[66] As a result of ARA70's ability to interact with other nuclear receptors, the significance of ARA70-AR interaction remains to be fully elucidated. ARA24/Ran interacts with the TAU-1 region of the NTD, leading to polyglutamine repeat expansion in AR.[67] The role of ARA24 in prostate cancer progression remains inconclusive. In one study, ARA24 mRNA expression was found to be increased in early primary prostate cancer specimens,[64] whereas another study found ARA24 expression to be similar between benign prostate hypertrophy, primary prostate tumors and CRPC tumors.[68]
Overall, these studies providence evidence that prostate cancer is associated with overexpression of certain AR coregulators, which may contribute to disease progression. However, the simultaneous involvement of multiple coregulators and their overlapping interaction, suggest that additional studies are required to determine the full contribution of AR coregulators in prostate carcinogenesis.
Androgen receptor target genes
Defining the androgen-regulated gene expression program in normal and malignant prostate cancer cells has been an area of intense investigation over the last number of years. This has been facilitated by advances in high throughput gene expression analysis. The majority of these studies have been performed in LNCaP cells, with other models systems including rat ventral prostate, rat ventral prostate epithelial cells (rVPECs) and a variety of other human prostate cancer cell lines such as 22Rv1, MDACaP2a, MDACaP2b and LAPC4 (reviewed by Dehm and Tindall[52]). Studies have estimated the LNCaP transcriptome to be anywhere from 10,570[69] to 23,448[70] polyadenylated RNAs, with 1.5-4.3% of the transcriptome either directly or indirectly regulated by androgens.[52]
More recently, ChIP-on-chip analysis, a technique that combines chromatin immunoprecipitation (“ChIP”) with microarray technology (“chip”), has been used to screen for novel androgen responsive genes.[71] Wang and colleagues mapped the AR-binding sites on chromosomes 21 and 22 in LNCaP cells by combining ChIP with tiled oligonucleotide microarrays[72] and expanded on this with comparisons between LNCaP and castrate-resistant LNCaP-abl cells, in an attempt to identify direct AR-dependent target genes in both androgen-dependent disease as well as in CRPC.[73] Ultimately, they determined that the role of the AR in CRPC is to execute a distinct program resulting in androgen-independent growth, involving mitotic phase (M-phase) regulatory genes and in particular, UBE2C which is overexpressed in CRPC tissues.[73] Significantly, silencing of UBE2C significantly decreases growth in CRPC cells by arresting Gap 2 (G2)/M and synthesis phases (S phases), providing an exciting potential therapeutic target.
One of the most significant findings with respect to prostate cancer development and progression was the identification of chromosomal rearrangements leading to novel fusions between the androgen-regulated promoter of the TMPRSS2 gene to the 3’ end of the oncogenic ETS transcription factor family members, ERG or ETV1.[74] TMPRSS2 was identified using complementary DNA (cDNA) microarrays derived from human prostate tissues to examine the transcript expression profiles of androgen-responsive LNCaP cells under conditions of androgen deprivation or androgen supplementation. TMPRSS2 mRNA is induced within 2 hours of androgen stimulation and reaches a maximum level within 24 hours.[75] Subsequent studies have reported that TMPRSS2 is highly specific to prostatic tissue and localized to prostate luminal epithelial cells.[76,77] Fusions between TMPRSS2 and ERG or ETV1 were identified using a novel bioinformatics approach called cancer outlier profile analysis (COPA)[74] . COPA was applied to databases of microarray data to identify overexpressed oncogenes in subsets of malignant versus normal tissues. Strong outlier profiles were identified for ERG and ETV1, and overexpression of these genes was confirmed for a subset of malignant prostate tissue. Using a combination of genomic analysis techniques, ERG or ETV1 overexpression was attributed to chromosomal translocations, which result in various fusions between the 5′ end of the TMPRSS2 gene and the 3’ end of either ERG or ETV1. In microarray datasets, ERG1 or ETV1 was overexpressed in 57% of prostate cancer cases but not in benign tissues, and the fusion with TMPRSS2 was found in 20 of 22 cases that overexpressed ERG or ETV.[74] Data from larger cohorts support these findings, and indicate that TMPRSS2 fusion with ETS members is the most frequent rearrangement in prostate cancer.[78,79] Recently, it has been reported that androgen treatment can induce these fusion events.[80,81] Interestingly, TMPRSS2:ERG fusion has been detected in non-malignant prostate cancer epithelial cells following long-term exposure to DHT,[82] suggesting that these fusions may be an early event in prostate carcinogenesis and play a pivotal role in prostate cancer progression. These landmark findings have positioned chromosomal rearrangements as critical initiating events in prostate cancer, elaborating our understanding the mechanisms of carcinogenesis and potentially opening new avenues for therapeutic intervention.[83,84]
ALTERNATIVE ANDROGEN RECEPTOR SIGNALING PATHWAYS
Considerable evidence now exists that AR remains transcriptionally active in CRPC. Numerous immunohis-tochemical studies have shown that AR protein is expressed at high levels (compared to levels in untreated tumors), in most cases of CRPC.[5,85,86] Consistent with this finding, real-time quantitative reverse transcription-polymerase chain reaction assay studies have shown that expression of AR mRNA in CRPC is higher compared to primary untreated tumors.[87,88] At least one mechanism for the increased AR mRNA expression is AR gene amplification, which occurs in 20-33% of CRPC cases.[37,87,89–90] In addition to AR, multiple AR regulated genes (such as PSA) are expressed, indicating that AR transcriptional activity is active in CRPC.[37] AR mutations that occur in CRPC is a possible mechanism for this AR transcriptional activity, however the overall frequency of CRPC that harbor somatic mutations in the AR gene is approximately 10%,[39] and unlikely to account for most cases of CRPC.
Interleukins
A considerable body of evidence now exists that implicates extracellular peptide signals in the form of growth factors and cytokines in the maintenance of the transcriptional activity of AR in CRPC. The role of interleukins (IL), and in particular IL-6 and IL-8, in the regulation of cellular events in different cancers has been extensively investigated (reviewed by Culig[91]). IL-6 is a multifunctional cytokine produced by many cells including prostate, immune cells and osteoblasts. IL-6 binds to the IL-6 receptor, which is composed of the ligand binding subunit gp80 and the ubiquitously expressed signal-transducing subunit gp130. This leads to phosphorylation of Janus kinases (JAK), after which signal transducer and activator of transcription (STAT) factor-3 is phosphorylated and translocated to the nucleus. However, activation of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3 kinase (PI3K) pathways can also occur depending on the cell type.[92] The rationale for studies on AR regulation by IL-6 is that elevated circulating IL-6 levels have been associated with advanced stage,[93,94] distant metastases,[95] metastasis-related morbidity[93,96] and decreased survival.[95] IL-6 can transactivate AR in prostate cancer cells; however, the effect of IL-6 on ligand-independent AR activation, tumor formation and castrate-resistant growth is variable depending on the status of AR, as well as crosstalk with other signaling pathways.[97,98] IL-6 enhances proliferation in LAPC-4 cells (which expresses wild-type AR) and PCa-2b (which expresses two AR substitution mutants at T877A and L701H), however IL-6 inhibits ligand independent AR activity in LNCaP cells (which express the AR T877A mutant in the LBD)[98] . PSA transcription is inhibited by IL-6 treatment in the presence of androgen in LNCaP cells, by preventing recruitment of p300 to the PSA promoter.[99] However, prolonged IL-6 treatment of LNCaP cells leads to activation of AR and PSA expression, via STAT signaling, even in the absence of androgen.[100] STAT3 interacts directly with amino acids 234-558 in the NTD of AR,[101] and AR transcriptional activation is dependent on phosphorylation of STAT3 at Serine-772.[102] As expression of IL-6 is increased in tissues and sera of CRPC, it has been explored as a target for therapy. Siltuximab (CNTO 328) is a chimeric murine-human monoclonal IL-6 antibody. Promising effects were observed in PC-3 cells and LuCap 35 xenografts where the antibody delayed tumor progression to CRPC.[103,104] Recently, a phase II clinical study found that the antibody has possible biological activity in patients previously treated with docetaxel, as evidenced by reduced C-reactive protein. However, a clinical response was not observed, and only 3.8% of patients had a PSA response.[105] Given the heterogeneous nature of CRPC, it may be reasonable to combine anti-IL-6 therapy with other established or experimental treatments.
IL-8 is another cytokine that has been implicated in prostate cancer progression. Serum levels of IL-8 correlate with prostate cancer clinical stage, and allow differentiation between benign and malignant disease.[106] IL-8 contributes to angiogenesis and metastasis by inducing metalloproteinase-9.[107] .Similar to IL-6, IL-8 upregulates AR transcriptional activity leading to enhanced tumor growth in vitro and in vivo.[108] Overexpression of IL-8 decreases the efficacy of the antiandrogen, bicalutamide, and promotes androgen independent tumor growth.[109] Interestingly, depletion of endogenous IL-8 in prostate cancer cells by small interfering RNA, induced apoptosis and increased sensitivity to the chemotherapeutic agent docetaxel.[110] However, the application of anti-IL-8 therapy to prostate cancer in the clinic has been limited so far.
Growth Factors
The ability of AR to cross-talk with key growth factor signaling events in the regulation of cell cycle, apoptosis and differentiation in prostate cancer cells is an active area of research (reviewed by Zhu and Kyprianou[19]). Epidermal growth factor (EGF) and its membrane receptor, epidermal growth factor-1 receptor (EGFR) have been implicated in the pathogenesis of several cancers, including prostate cancer.[111] There is increased expression of both EGF and EGFR in patients with metastatic disease, and this correlates with disease progression to CRPC.[112] EGF and EGFR activation in prostate cancer cells leads to activation of the MAPK pathway.[113] The first evidence of cross-talk between EGF and AR was shown over a decade ago, where EGF treatment was found to induce an AR-mediated reporter gene transcription in DU145 cells (a prostate cancer cell line that expresses neither AR nor PSA).[114] Growth factor-induced reporter gene expression was dependent on cotransfection of the AR expression construct, and was blocked by the AR antagonist, bicalutamide. These experiments demonstrated that growth factor signaling can regulate androgen responsive genes by a mechanism that is AR dependent and androgen independent. In prostate cancer cells that express endogenous AR such as LNCaP cells, treatment with EGF in the absence of androgen induces phosphorylation of AR at Tryrosine-267 and -534 by Src and Ack1 kinases.[115] Androgen-activated AR activates the MAPK pathway[116] and conversely, EGF-activated MAPK signaling cascade interferes with AR function, modulating the androgen response. MAPK extracellular kinase (MEK) inhibition reverses the EGF-mediated AR down regulation in differentiated cells, thus suggesting the existence of an inverse correlation between EGF and androgen signaling in non-tumor epithelial cells.[117] EGF can also induce IL-6 upregulation in prostate cancer cells.
Increased levels of STAT3 leads to STAT3-AR complex formation in response to EGF and IL-6. Moreover, STAT3 increases the EGF-induced transcriptional activation of AR, while androgen pre-treatment increases STAT3 levels in an IL-6 autocrine/paracrine-dependent manner, suggesting an intracellular feedback loop.[102] With increased expression of EGFR in metastatic disease, and correlation with disease progression, EGFR has potential as a therapeutic target. Gefitinib, an EGFR-selective tyrosine kinase inhibitor, exhibits antitumor activity in xenograft models of both androgen-dependent and androgen-independent human prostate cancer.[118] However, as a single agent, gefitinib, failed to demonstrate any objective or PSA responses in phase II studies in patients with either metastatic or non-metastatic CRPC.[119] A Phase II study in CRPC of lapatinib, an inhibitor of EGFR and human epidermal growth factor receptor 2 (HER2), showed a PSA response only in a very small number of patients.[120]
Insulin-like growth factor (IGF)-1 signaling is of significant biological importance. Large-scale epidemiological studies have suggested a correlation between elevated serum levels of IGF-1 and low levels of IGFBP-3 (Insulin-like growth factor-binding protein 3 , a serum protein that regulates the binding of free IGF-1 to the IGF receptor), and increased risk of developing prostate cancer.[121] However, several subsequent studies have failed to show this effect.[122,123] None-the-less, the IFG pathway has been implicated in the modulation of AR signaling. Potential mechanisms include AR phosphorylation,[124] AR translocation into the nucleus[124] or simulation of expression/activity of AR cofactors,[54] thereby affecting AR transcriptional activation both in the presence and absence of androgens. Activation of androgen-responsive promoters with IGF-1 treatment is equivalent to androgen treatment in ectopic AR-expressing DU-145 cells.[114] However, the cross-talk between IGF-1 and AR is apparent in other cell types, including non-transformed cells.[19] Therefore, the action of IGF-1 on AR activity appears to be non-specific. As a result, the IGF-1 signaling pathway is not an attractive pathway as a therapeutic target.
Intracellular Kinase Signaling
The extracellular signals discussed in the previous section represent just a small part of the complex mechanism of AR transactivation in the absence of androgens. These factors work in isolation or, more likely, in concert with each other to activate intracellular kinase signaling cascades that target fundamental cellular processes such as proliferation and transcription initiation. Kinase signal cascades are rapidly transduced and mediated through protein-protein interactions. The deregulated cell growth observed in cancer occurs as a result of perturbed signal transduction, with alterations in the activities of certain kinases driving the development and progression of many cancers, including those of the prostate.[125] Therapeutically, targeting these complex protein-protein interactions or phosphorylation events is difficult and requires vigorous validation and confirmation of specificity. However, an understanding of the principal intracellular signaling cascades responsible for prostate cancer progression provides important insights into the role of AR during progression to CRPC.
The MAPK signaling pathway has been implicated in many cancers, including prostate cancer. As discussed in the previous section, a variety of extracellular stimuli can activate the MAPK signaling pathway. The many downstream targets of the MAPK pathway provide a huge number of potential regulators of AR activity, only a few of which have been characterized, including sarcoma-related kinase (Src) and p42/44 extracellular-signal-regulated kinases (ERK).
Src is a powerful oncogene that is activated in a variety of tumors. In LNCaP and LAPC-4 cells, Src phosphorylates AR at Tyrosine-534, which induces transcriptional activity by promoting nuclear translocation and DNA binding in the absence of androgen.[126,127] Activation of Src occurs as a result of a number of stimuli, including EGF, described earlier. The activity of Src on AR transactivation is modulated, at least in part through its binding partner and associated scaffold protein, receptor for activated protein kinase C-1 (RACK1)[127]. Treatment of C4-2 cells with the Src inhibitor, protein phosphatase-2 in androgen free conditions results in decreased AR activation of reporter activities and reduced recruitment of AR to the PSA enhancer region.[128]
p42/44 (ERK1 and ERK2, respectively) is another key effector of the MAPK pathway. Immunohistochemical studies of prostate tumors have shown the level of activated MAP K increases with increasing Gleason score and tumor stage.[129] High levels of activated MAP kinase were also detected in CRPC, suggesting an increase in the activation of the MAP kinase signal pathway during prostate cancer progression.[129] Treatment of C4-2 cells with the MAP kinase inhibitor, UO126 in steroid depleted conditions, substantially decreases AR stability and protein expression, suggesting that enhanced autocrine growth factor signaling contributes to the maintenance of AR expression and facilitates AR activity in the absence of androgens.[130] G protein-related factors have also been shown to regulate AR activity through stimulation of the MAP kinase pathway. The expression of Vav3, a Rho GTPase guanine nucleotide exchange factor, is upregulated in the progression of LNCaP cells to the androgen independent phenotype of LNCaP-R1 cells.[131] Vav3 has also been shown to enhance EGF activation of AR in the absence of androgens.[132] This signaling pathway induces nuclear localization of AR via the activation of Rho GTPase, Rac1, whose downstream signaling includes members of the MAP kinase family, including ERK[132].
The phosphatidylinositol 3-kinase (PI3K) pathway has been implicated in prostate carcinogenesis to CRPC, however its precise function remains to be fully elucidated (reviewed by Sarker and colleagues[133]). Activation of PI3K leads to the generation of second messenger phosphatidylinositol 3,5-triphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate (PIP2).This results in recruitment of pleckstrin homology domains of a number of signaling kinases including Akt (Akt/PKB is a serine/threonine protein kinase), driving their conformational change and phosphorylation by the constitutively active phosphoinositide-dependent kinase 1 and 2. Activated Akt translocates to the nucleus and activates downstream targets involved in fundamental cellular processes such as proliferation, cell cycle progression, growth and angiogenesis. Akt is negatively regulated by the tumor suppressor, phosphatase and tensin homolog deleted on chromosome 10 (PTEN).[133]
Immunohistochemical studies indicate that loss of PTEN expression is found in 20-27% of primary tumors[134] and in 79% of CRPC specimens.[135] PTEN loss correlates with advanced stage and Gleason score.[136] Functional loss of PTEN is associated with increased activity of Akt-1 and correlates with prognosis,[137] predicting disease recurrence after primary treatment.[138] Targeted Cre-mediated PTEN homozygous deletion in mice leads to a significant shortened latency of prostate intraepithelial neoplasia (PIN) formation and results in prostate cancer progression to a metastatic stage, despite androgen deprivation therapy, mimicking the disease progression seen in humans.[139] These data support the concept that PI3K signaling induces continued AR gain of function despite reduced androgen levels, possibly though activation by posttranslational modification or reduced corepressor activity.[133]
As discussed previously, the role of the TMPRSS2:ERG fusion in prostate carcinogenesis has gained considerable interest. Recent data has shown that loss of PTEN cooperates with TMPRSS2:ERG in prostate cancer. Transgenic mice expressing TMPRSS2:ERG in the prostate failed to develop PIN or invasive cancer. However, when they were crossed with either PTEN+/- mice or prostate-specific Akt transgenic mice, PIN but not invasive cancer developed.[140] Other studies have found transgenic overexpression of ERG in mouse prostate tissue promotes marked acceleration and progression of high-grade PIN to adenocarcinoma when crossed with PTEN+/- mice.[141] This study also showed prostate cancer specimens containing the TMPRSS2:ERG rearrangement are significantly enriched for PTEN loss.[141] Together these data suggest cooperativity between PI3K-pathway activation and ERG aberration in induction of PIN, but suggest that additional events are likely required for invasive malignancy.
Nuclear factor-κB (NF-κB) transcription factor members are important mediators of oncogenesis in many cancers, including prostate cancer. The activity of NF-κB is higher in androgen-independent cell lines, and androgen-independent xenografts compared with androgen-dependent grafts,[142] as well as in metastatic prostate cancer compared to localized disease.[143] Elevation of NF-κB activity in primary prostate cancer correlates with poor prognosis and predicts biochemical relapse.[144] Activation of NF-κB in transgenic mouse models leads to continued tumor growth despite surgical castration,[145] indicating that activation of the NF-κB pathway may play a critical role in progression of the tumor to androgen independence. The mechanism of this remains largely unknown; however, recent data suggests that NF-κB/p52 can activate AR, leading to increased transcription of AR-responsive genes in a ligand independent manner.[146] NF-κB/p52 enhances nuclear translocation of AR by binding to the AR NTD and enhancing recruitment of AR coactivators such as p300 to the promoter regions of AR target genes.[146]
Protein kinase A, which is regulated by intracellular cyclic adenosine monophosphate (cAMP) levels, can also modulate AR activity in the absence of androgens. This was first demonstrated over a decade ago.[147] AR can be activated in the absence of androgens through an alternate signaling pathway involving forskolin, which activates adenyl cyclase, thus increasing intracellular levels of cAMP and consequently protein kinase A (PKA) .[148] This activation is blocked by bicalutamide, confirming that this ligand-independent pathway is AR dependent.[147] Aurora-A is a serine-threonine protein kinase frequently overexpressed in several cancers, including prostate. It is overexpressed in high-grade PIN lesions,[149] primary prostate cancer specimens and also several prostate cancer cell lines.[150] In addition, Aurora-A expression correlates with tumorigenicity and invasive potential as well as the clinical staging, surgical margin status, and seminal vesicle invasion in radical prostatectomy specimens.[150] Aurora-A interacts with AR, phosphorylates AR at Threonine 282 and Serine 293 and induces AR transactivation in a phosphorylation dependent-manner. Overexpression of Aurora-A in LNCaP cells induces PSA and cell survival. However, inhibition of Aurora-A sensitizes LNCaP-RF cells (an androgen independent subline) to apoptosis and cell growth arrest.[151] This data suggests that AR is a substrate of Aurora-A and that elevated Aurora-A may contribute to androgen-independent growth by phosphorylation and activation of AR. The Aurora kinase inhibitor, VX680, attenuates phosphorylation of histone H3 and reduces survival in PC3 and LNCaP cells[150]
Intracellular kinase signaling represents an important mediator of extra-cellular signals that promote cancer cell proliferation and survival. The ability of kinases such as those discussed here, to directly modify AR and promote AR activity in CRPC provides attractive targets for therapeutic intervention. The redundancy between many of these pathways makes achieving specific and durable inhibition difficult. This is further complicated by the uniquely heterogeneous nature of CRPC. Further investigation into the interplay between these pathways and AR [Figure 2], may lead to the identification of distinct subsets of CRPC patients who may benefit from a multi-drug approach.
CONCLUSIONS AND FUTURE DIRECTIONS
Ever since the seminal observation by Huggins and Hodges in 1941 that castration was beneficial in CRPC, abrogation of AR action has remained the therapeutic objective in clinical management of the disease. There is now incontrovertible evidence that the onset of CRPC coincides with renewed AR signaling. Given that metastatic prostate cancer is a molecularly heterogeneous disease even within a single patient,[152] multiple mechanisms may simultaneously and ultimately give rise to a molecularly diverse group of CRPCs. Despite these challenges, a new generation of AR antagonists such as MDV3100 and EPI-001 has shown promise. MDV3100 which binds to the AR LBD, impairs nuclear translocation, DNA binding, and coactivator recruitment,[153] and has shown encouraging antitumor activity in phase 1-2 studies in patients.[154] In contrast to MDV3100, EPI-001 binds to the AR NTD. Data from preclinical studies has shown that EPI-001 treatment leads to cytoreduction of CRPC in xenografts dependent on AR for growth and survival, without causing toxicity.[155] These emerging next-generation AR antagonists, used either alone or in combination with existing treatments have huge potential to significantly change treatment options in metastatic disease in the future.
DISCLOSURES
The authors declare no conflict of interests.
AUTHOR’S PROFILE
Peter E Lonergan, MB, BCh, MRCSI is a Research Fellow in the Department of Urology, Mayo Clinic, Rochester, Minnesota, USA
Donald J Tindall, PhD is the Carl Rosen Professor of Urology, Director and Vice Chair of Urologic Research at Mayo Clinic, Rochester, Minnesota, USA.
ACKNOWLEDGEMENTS
This work is supported by the NIH (CA125747, CA091956 and CA121277) and the T.J. Martell Foundation (DJT). PEL is supported by a Fulbright Scholarship and the Postgraduate Traveling Scholarship in Medicine from Trinity College, Dublin.
REFERENCES
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Klein EA, Ciezki J, Kupelian PA, Mahadevan A. Outcomes for intermediate risk prostate cancer: are there advantages for surgery, external radiation, or brachytherapy? Urol Oncol. 2009;27:67–71. doi: 10.1016/j.urolonc.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Loblaw DA, Virgo KS, Nam R, Somerfield MR, Ben-Josef E, Mendelson DS, et al. Initial hormonal management of androgen-sensitive metastatic, recurrent, or progressive prostate cancer: 2006 update of an American Society of Clinical Oncology practice guideline. J Clin Oncol. 2007;25:1596–605. doi: 10.1200/JCO.2006.10.1949. [DOI] [PubMed] [Google Scholar]
- 4.Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006;12:1665–71. doi: 10.1158/1078-0432.CCR-06-0067. [DOI] [PubMed] [Google Scholar]
- 5.Ruizeveld de Winter JA, Janssen PJ, Sleddens HM, Verleun-Mooijman MC, Trapman J, Brinkmann AO, et al. Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am J Pathol. 1994;144:735–46. [PMC free article] [PubMed] [Google Scholar]
- 6.Chodak GW, Kranc DM, Puy LA, Takeda H, Johnson K, Chang C. Nuclear localization of androgen receptor in heterogeneous samples of normal, hyperplastic and neoplastic human prostate. J Urol. 1992;147:798–803. doi: 10.1016/s0022-5347(17)37389-5. [DOI] [PubMed] [Google Scholar]
- 7.Sadi MV, Walsh PC, Barrack ER. Immunohistochemical study of androgen receptors in metastatic prostate cancer. Comparison of receptor content and response to hormonal therapy. Cancer. 1991;67:3057–64. doi: 10.1002/1097-0142(19910615)67:12<3057::aid-cncr2820671221>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 8.Henshall SM, Quinn DI, Lee CS, Head DR, Golovsky D, Brenner PC, et al. Altered expression of androgen receptor in the malignant epithelium and adjacent stroma is associated with early relapse in prostate cancer. Cancer Res. 2001;61:423–7. [PubMed] [Google Scholar]
- 9.Ricciardelli C, Choong CS, Buchanan G, Vivekanandan S, Neufing P, Stahl J, et al. Androgen receptor levels in prostate cancer epithelial and peritumoral stromal cells identify non-organ confined disease. Prostate. 2005;63:19–28. doi: 10.1002/pros.20154. [DOI] [PubMed] [Google Scholar]
- 10.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 11.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 12.Scher HI, Buchanan G, Gerald W, Butler LM, Tilley WD. Targeting the androgen receptor: improving outcomes for castration-resistant prostate cancer. Endocr Relat Cancer. 2004;11:459–76. doi: 10.1677/erc.1.00525. [DOI] [PubMed] [Google Scholar]
- 13.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 14.Ryan CJ, Smith A, Lal P, Satagopan J, Reuter V, Scardino P, et al. Persistent prostate-specific antigen expression after neoadjuvant androgen depletion: an early predictor of relapse or incomplete androgen suppression. Urology. 2006;68:834–9. doi: 10.1016/j.urology.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 15.Buchanan G, Irvine RA, Coetzee GA, Tilley WD. Contribution of the androgen receptor to prostate cancer predisposition and progression. Cancer Metastasis Rev. 2001;20:207–23. doi: 10.1023/a:1015531326689. [DOI] [PubMed] [Google Scholar]
- 16.Steinkamp MP, O’Mahony OA, Brogley M, Rehman H, Lapensee EW, Dhanasekaran S, et al. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. 2009;69:4434–42. doi: 10.1158/0008-5472.CAN-08-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–15. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 18.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 19.Zhu ML, Kyprianou N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer. 2008;15:841–9. doi: 10.1677/ERC-08-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergerat JP, Ceraline J. Pleiotropic functional properties of androgen receptor mutants in prostate cancer. Hum Mutat. 2009;30:145–57. doi: 10.1002/humu.20848. [DOI] [PubMed] [Google Scholar]
- 21.Claessens F, Denayer S, Van Tilborgh N, Kerkhofs S, Helsen C, Haelens A. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl Recept Signal. 2008;6:e008. doi: 10.1621/nrs.06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–15. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- 23.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 24.Jenster G, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol. 1991;5:1396–404. doi: 10.1210/mend-5-10-1396. [DOI] [PubMed] [Google Scholar]
- 25.Simental JA, Sar M, Lane MV, French FS, Wilson EM. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem. 1991;266:510–8. [PubMed] [Google Scholar]
- 26.Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995;270:7341–6. doi: 10.1074/jbc.270.13.7341. [DOI] [PubMed] [Google Scholar]
- 27.Callewaert L, Van Tilborgh N, Claessens F. Interplay between two hormone-independent activation domains in the androgen receptor. Cancer Res. 2006;66:543–53. doi: 10.1158/0008-5472.CAN-05-2389. [DOI] [PubMed] [Google Scholar]
- 28.Chamberlain NL, Whitacre DC, Miesfeld RL. Delineation of two distinct type 1 activation functions in the androgen receptor amino-terminal domain. J Biol Chem. 1996;271:26772–8. doi: 10.1074/jbc.271.43.26772. [DOI] [PubMed] [Google Scholar]
- 29.Dehm SM, Regan KM, Schmidt LJ, Tindall DJ. Selective role of an NH2-terminal WxxLF motif for aberrant androgen receptor activation in androgen depletion-independent prostate cancer cells. Cancer Res. 2007;67:10067–77. doi: 10.1158/0008-5472.CAN-07-1267. [DOI] [PubMed] [Google Scholar]
- 30.Umesono K, Evans RM. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell. 1989;57:1139–46. doi: 10.1016/0092-8674(89)90051-2. [DOI] [PubMed] [Google Scholar]
- 31.Ozanne DM, Brady ME, Cook S, Gaughan L, Neal DE, Robson CN. Androgen receptor nuclear translocation is facilitated by the f-actin cross-linking protein filamin. Mol Endocrinol. 2000;14:1618–26. doi: 10.1210/mend.14.10.0541. [DOI] [PubMed] [Google Scholar]
- 32.Bedolla RG, Wang Y, Asuncion A, Chamie K, Siddiqui S, Mudryj MM, et al. Nuclear versus cytoplasmic localization of filamin A in prostate cancer: immunohistochemical correlation with metastases. Clin Cancer Res. 2009;15:788–96. doi: 10.1158/1078-0432.CCR-08-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaku N, Matsuda K, Tsujimura A, Kawata M. Characterization of nuclear import of the domain-specific androgen receptor in association with the importin alpha/beta and Ran-guanosine 5’-triphosphate systems. Endocrinology. 2008;149:3960–9. doi: 10.1210/en.2008-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–6. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 35.Duff J, Davies P, Watt K, McEwan IJ. Structural dynamics of the human androgen receptor: implications for prostate cancer and neurodegenerative disease. Biochem Soc Trans. 2006;34:1098–102. doi: 10.1042/BST0341098. [DOI] [PubMed] [Google Scholar]
- 36.Buchanan G, Greenberg NM, Scher HI, Harris JM, Marshall VR, Tilley WD. Collocation of androgen receptor gene mutations in prostate cancer. Clin Cancer Res. 2001;7:1273–81. [PubMed] [Google Scholar]
- 37.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 38.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 39.Taplin ME, Rajeshkumar B, Halabi S, Werner CP, Woda BA, Picus J, et al. Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol. 2003;21:2673–8. doi: 10.1200/JCO.2003.11.102. [DOI] [PubMed] [Google Scholar]
- 40.Wilson JD. The role of 5 alpha-reduction in steroid hormone physiology. Reprod Fertil Dev. 2001;13:673–8. doi: 10.1071/rd01074. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt LJ, Tindall DJ. Steroid 5 alpha-reductase inhibitors targeting BPH and prostate cancer. J Steroid Biochem Mol Biol. 2011;125:32–8. doi: 10.1016/j.jsbmb.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Zhou ZX, Lane MV, Kemppainen JA, French FS, Wilson EM. Specificity of ligand-dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Mol Endocrinol. 1995;9:208–18. doi: 10.1210/mend.9.2.7776971. [DOI] [PubMed] [Google Scholar]
- 43.Wright AS, Thomas LN, Douglas RC, Lazier CB, Rittmaster RS. Relative potency of testosterone and dihydrotestosterone in preventing atrophy and apoptosis in the prostate of the castrated rat. J Clin Invest. 1996;98:2558–63. doi: 10.1172/JCI119074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith DF, Toft DO. Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endocrinol. 2008;22:2229–40. doi: 10.1210/me.2008-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loy CJ, Sim KS, Yong EL. Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. Proc Natl Acad Sci U S A. 2003;100:4562–7. doi: 10.1073/pnas.0736237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Kreisberg JI, Bedolla RG, Mikhailova M, deVere White RW, Ghosh PM. A 90 kDa fragment of filamin A promotes Casodex-induced growth inhibition in Casodex-resistant androgen receptor positive C4-2 prostate cancer cells. Oncogene. 2007;26:6061–70. doi: 10.1038/sj.onc.1210435. [DOI] [PubMed] [Google Scholar]
- 47.Castoria G, D’Amato L, Ciociola A, Giovannelli P, Giraldi T, Sepe L, et al. Androgen-induced cell migration: role of androgen receptor/filamin A association. PLoS One. 2011;6:e17218. doi: 10.1371/journal.pone.0017218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem. 2000;275:22986–94. doi: 10.1074/jbc.M002807200. [DOI] [PubMed] [Google Scholar]
- 49.Chang CY, McDonnell DP. Evaluation of ligand-dependent changes in AR structure using peptide probes. Mol Endocrinol. 2002;16:647–60. doi: 10.1210/mend.16.4.0818. [DOI] [PubMed] [Google Scholar]
- 50.van Royen ME, Cunha SM, Brink MC, Mattern KA, Nigg AL, Dubbink HJ, et al. Compartmentalization of androgen receptor protein-protein interactions in living cells. J Cell Biol. 2007;177:63–72. doi: 10.1083/jcb.200609178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He B, Minges JT, Lee LW, Wilson EM. The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J Biol Chem. 2002;277:10226–35. doi: 10.1074/jbc.M111975200. [DOI] [PubMed] [Google Scholar]
- 52.Dehm SM, Tindall DJ. Molecular regulation of androgen action in prostate cancer. J Cell Biochem. 2006;99:333–44. doi: 10.1002/jcb.20794. [DOI] [PubMed] [Google Scholar]
- 53.Agoulnik IU, Weigel NL. Coactivator selective regulation of androgen receptor activity. Steroids. 2009;74:669–74. doi: 10.1016/j.steroids.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, et al. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–9. [PubMed] [Google Scholar]
- 55.Zhou HJ, Yan J, Luo W, Ayala G, Lin SH, Erdem H, et al. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 2005;65:7976–83. doi: 10.1158/0008-5472.CAN-04-4076. [DOI] [PubMed] [Google Scholar]
- 56.Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN. Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Brit J Cancer. 2001;85:1928–36. doi: 10.1054/bjoc.2001.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma H, Hong H, Huang SM, Irvine RA, Webb P, Kushner PJ, et al. Multiple signal input and output domains of the 160-kilodalton nuclear receptor coactivator proteins. Mol Cell Biol. 1999;19:6164–73. doi: 10.1128/mcb.19.9.6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alen P, Claessens F, Verhoeven G, Rombauts W, Peeters B. The androgen receptor amino-terminal domain plays a key role in p160 coactivator-stimulated gene transcription. Mol Cell Biol. 1999;19:6085–97. doi: 10.1128/mcb.19.9.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389:194–8. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- 60.Chakravarti D, LaMorte VJ, Nelson MC, Nakajima T, Schulman IG, Juguilon H, et al. Role of CBP/P300 in nuclear receptor signalling. Nature. 1996;383:99–103. doi: 10.1038/383099a0. [DOI] [PubMed] [Google Scholar]
- 61.Debes JD, Sebo TJ, Lohse CM, Murphy LM, Haugen DA, Tindall DJ. p300 in prostate cancer proliferation and progression. Cancer Res. 2003;63:7638–40. [PubMed] [Google Scholar]
- 62.Heemers HV, Sebo TJ, Debes JD, Regan KM, Raclaw KA, Murphy LM, et al. Androgen deprivation increases p300 expression in prostate cancer cells. Cancer Res. 2007;67:3422–30. doi: 10.1158/0008-5472.CAN-06-2836. [DOI] [PubMed] [Google Scholar]
- 63.Hu YC, Yeh S, Yeh SD, Sampson ER, Huang J, Li P, et al. Functional domain and motif analyses of androgen receptor coregulator ARA70 and its differential expression in prostate cancer. J Biol Chem. 2004;279:33438–46. doi: 10.1074/jbc.M401781200. [DOI] [PubMed] [Google Scholar]
- 64.Li P, Yu X, Ge K, Melamed J, Roeder RG, Wang Z. Heterogeneous expression and functions of androgen receptor co-factors in primary prostate cancer. Am J Pathol. 2002;161:1467–74. doi: 10.1016/S0002-9440(10)64422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang HC, Chen SC, Chen J, Hsieh JT. In vitro gene expression changes of androgen receptor coactivators after hormone deprivation in an androgen-dependent prostate cancer cell line. J Formos Med Assoc. 2005;104:652–8. [PubMed] [Google Scholar]
- 66.Mestayer C, Blanchère M, Jaubert F, Dufour B, Mowszowicz I. Expression of androgen receptor coactivators in normal and cancer prostate tissues and cultured cell lines. Prostate. 2003;56:192–200. doi: 10.1002/pros.10229. [DOI] [PubMed] [Google Scholar]
- 67.Hsiao PW, Lin DL, Nakao R, Chang C. The linkage of Kennedy's neuron disease to ARA24, the first identified androgen receptor polyglutamine region-associated coactivator. J Biol Chem. 1999;274:20229–34. doi: 10.1074/jbc.274.29.20229. [DOI] [PubMed] [Google Scholar]
- 68.Linja MJ, Porkka KP, Kang Z, Savinainen KJ, Jänne OA, Tammela TL, et al. Expression of androgen receptor coregulators in prostate cancer. Clin Cancer Res. 2004;10:1032–40. doi: 10.1158/1078-0432.ccr-0990-3. [DOI] [PubMed] [Google Scholar]
- 69.Oudes AJ, Roach JC, Walashek LS, Eichner LJ, True LD, Vessella RL, et al. Application of Affymetrix array and Massively Parallel Signature Sequencing for identification of genes involved in prostate cancer progression. BMC Cancer. 2005;5:86. doi: 10.1186/1471-2407-5-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu LL, Su YP, Labiche R, Segawa T, Shanmugam N, McLeod DG, et al. Quantitative expression profile of androgen-regulated genes in prostate cancer cells and identification of prostate-specific genes. Int J Cancer. 2001;92:322–8. doi: 10.1002/ijc.1196. [DOI] [PubMed] [Google Scholar]
- 71.Takayama K, Kaneshiro K, Tsutsumi S, Horie-Inoue K, Ikeda K, Urano T, et al. Identification of novel androgen response genes in prostate cancer cells by coupling chromatin immunoprecipitation and genomic microarray analysis. Oncogene. 2007;26:4453–63. doi: 10.1038/sj.onc.1210229. [DOI] [PubMed] [Google Scholar]
- 72.Wang Q, Li W, Liu XS, Carroll JS, Jänne OA, Keeton EK, et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27:380–92. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–56. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 75.Lin B, Ferguson C, White JT, Wang S, Vessella R, True LD, et al. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res. 1999;59:4180–4. [PubMed] [Google Scholar]
- 76.Afar DE, Vivanco I, Hubert RS, Kuo J, Chen E, Saffran DC, et al. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res. 2001;61:1686–92. [PubMed] [Google Scholar]
- 77.Vaarala MH, Porvari KS, Kellokumpu S, Kyllönen AP, Vihko PT. Expression of transmembrane serine protease TMPRSS2 in mouse and human tissues. J Pathol. 2001;193:134–40. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH743>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 78.Mehra R, Tomlins SA, Shen R, Nadeem O, Wang L, Wei JT, et al. Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod Pathol. 2007;20:538–44. doi: 10.1038/modpathol.3800769. [DOI] [PubMed] [Google Scholar]
- 79.Rajput AB, Miller MA, De Luca A, Boyd N, Leung S, Hurtado-Coll A, et al. Frequency of the TMPRSS2:ERG gene fusion is increased in moderate to poorly differentiated prostate cancers. J Clin Pathol. 2007;60:1238–43. doi: 10.1136/jcp.2006.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–83. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, Varambally S, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bastus NC, Boyd LK, Mao X, Stankiewicz E, Kudahetti SC, Oliver RT, et al. Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate epithelial cells. Cancer Res. 2009;70:9544–8. doi: 10.1158/0008-5472.CAN-10-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–9. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 85.van der Kwast TH, Schalken J, Ruizeveld de Winter JA, van Vroonhoven CC, Mulder E, Boersma W, et al. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int J Cancer. 1991;48:189–93. doi: 10.1002/ijc.2910480206. [DOI] [PubMed] [Google Scholar]
- 86.Tilley WD, Lim-Tio SS, Horsfall DJ, Aspinall JO, Marshall VR, Skinner JM. Detection of discrete androgen receptor epitopes in prostate cancer by immunostaining: measurement by color video image analysis. Cancer Res. 1994;54:4096–102. [PubMed] [Google Scholar]
- 87.Linja MJ, Savinainen KJ, Saramäki OR, Tammela TL, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–5. [PubMed] [Google Scholar]
- 88.Latil A, Bièche I, Vidaud D, Lidereau R, Berthon P, Cussenot O, et al. Evaluation of androgen, estrogen (ER alpha and ER beta), and progesterone receptor expression in human prostate cancer by real-time quantitative reverse transcription-polymerase chain reaction assays. Cancer Res. 2001;61:1919–26. [PubMed] [Google Scholar]
- 89.Bubendorf L, Kononen J, Koivisto P, Schraml P, Moch H, Gasser TC, Willi N, Mihatsch MJ, Sauter G, Kallioniemi OP. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999;59(4):803–806. [PubMed] [Google Scholar]
- 90.Edwards J, Krishna NS, Grigor KM, Bartlett JM. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer. 2003;89:552–6. doi: 10.1038/sj.bjc.6601127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Culig Z. Cytokine disbalance in common human cancers. Biochim Biophys Acta. 2011;1813:308–14. doi: 10.1016/j.bbamcr.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 92.Culig Z, Steiner H, Bartsch G, Hobisch A. Interleukin-6 regulation of prostate cancer cell growth. J Cell Biochem. 2005;95:497–505. doi: 10.1002/jcb.20477. [DOI] [PubMed] [Google Scholar]
- 93.Adler HL, McCurdy MA, Kattan MW, Timme TL, Scardino PT, Thompson TC. Elevated levels of circulating interleukin-6 and transforming growth factor-beta1 in patients with metastatic prostatic carcinoma. J Urol. 1999;161:182–7. [PubMed] [Google Scholar]
- 94.Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, Slawin KM. Plasma levels of interleukin-6 and its soluble receptor are associated with prostate cancer progression and metastasis. Urology. 2001;58:1008–15. doi: 10.1016/s0090-4295(01)01405-4. [DOI] [PubMed] [Google Scholar]
- 95.Nakashima J, Tachibana M, Horiguchi Y, Oya M, Ohigashi T, Asakura H, et al. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin Cancer Res. 2000;6:2702–6. [PubMed] [Google Scholar]
- 96.Twillie DA, Eisenberger MA, Carducci MA, Hseih WS, Kim WY, Simons JW. Interleukin-6: a candidate mediator of human prostate cancer morbidity. Urology. 1995;45:542–9. doi: 10.1016/S0090-4295(99)80034-X. [DOI] [PubMed] [Google Scholar]
- 97.Hobisch A, Eder IE, Putz T, Horninger W, Bartsch G, Klocker H, et al. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998;58:4640–5. [PubMed] [Google Scholar]
- 98.Malinowska K, Neuwirt H, Cavarretta IT, Bektic J, Steiner H, Dietrich H, et al. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr Relat Cancer. 2009;16:155–69. doi: 10.1677/ERC-08-0174. [DOI] [PubMed] [Google Scholar]
- 99.Jia L, Choong CS, Ricciardelli C, Kim J, Tilley WD, Coetzee GA. Androgen receptor signaling: mechanism of interleukin-6 inhibition. Cancer Res. 2004;64:2619–26. doi: 10.1158/0008-5472.can-03-3486. [DOI] [PubMed] [Google Scholar]
- 100.Debes JD, Comuzzi B, Schmidt LJ, Dehm SM, Culig Z, Tindall DJ. p300 regulates androgen receptor-independent expression of Prostate-Specific antigen in prostate cancer cells treated chronically with interleukin-6. Cancer Res. 2005;65:5965–73. doi: 10.1158/0008-5472.CAN-04-2837. [DOI] [PubMed] [Google Scholar]
- 101.Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem. 2002;277:7076–85. doi: 10.1074/jbc.M108255200. [DOI] [PubMed] [Google Scholar]
- 102.Aaronson DS, Muller M, Neves SR, Chung WC, Jayaram G, Iyengar R, et al. An androgen-IL-6-Stat3 autocrine loop re-routes EGF signal in prostate cancer cells. Mol Cell Endocrinol. 2007;270:50–6. doi: 10.1016/j.mce.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 103.Smith PC, Keller ET. Anti-interleukin-6 monoclonal antibody induces regression of human prostate cancer xenografts in nude mice. Prostate. 2001;48:47–53. doi: 10.1002/pros.1080. [DOI] [PubMed] [Google Scholar]
- 104.Wallner L, Dai J, Escara-Wilke J, Zhang J, Yao Z, Lu Y, et al. Inhibition of interleukin-6 with CNTO328, an anti-interleukin-6 monoclonal antibody, inhibits conversion of androgen-dependent prostate cancer to an androgen-independent phenotype in orchiectomized mice. Cancer Res. 2006;66:3087–95. doi: 10.1158/0008-5472.CAN-05-3447. [DOI] [PubMed] [Google Scholar]
- 105.Dorff TB, Goldman B, Pinski JK, Mack PC, Lara PN, Jr, Van Veldhuizen PJ, Jr, et al. Clinical and correlative results of SWOG S0354: a phase II trial of CNTO328 (siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin Cancer Res. 2011;16:3028–34. doi: 10.1158/1078-0432.CCR-09-3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Veltri RW, Miller MC, Zhao G, Ng A, Marley GM, Wright GL, Jr, et al. Interleukin-8 serum levels in patients with benign prostatic hyperplasia and prostate cancer. Urology. 1999;53:139–47. doi: 10.1016/s0090-4295(98)00455-5. [DOI] [PubMed] [Google Scholar]
- 107.Inoue K, Slaton JW, Kim SJ, Perrotte P, Eve BY, Bar-Eli M, et al. Interleukin 8 expression regulates tumorigenicity and metastasis in human bladder cancer. Cancer Res. 2000;60:2290–9. [PubMed] [Google Scholar]
- 108.Seaton A, Scullin P, Maxwell PJ, Wilson C, Pettigrew J, Gallagher R, et al. Interleukin-8 signaling promotes androgen-independent proliferation of prostate cancer cells via induction of androgen receptor expression and activation. Carcinogenesis. 2008;29:1148–56. doi: 10.1093/carcin/bgn109. [DOI] [PubMed] [Google Scholar]
- 109.Araki S, Omori Y, Lyn D, Singh RK, Meinbach DM, Sandman Y, et al. Interleukin-8 is a molecular determinant of androgen independence and progression in prostate cancer. Cancer Res. 2007;67:6854–62. doi: 10.1158/0008-5472.CAN-07-1162. [DOI] [PubMed] [Google Scholar]
- 110.Singh RK, Lokeshwar BL. Depletion of intrinsic expression of Interleukin-8 in prostate cancer cells causes cell cycle arrest, spontaneous apoptosis and increases the efficacy of chemotherapeutic drugs. Mol Cancer. 2009;8:57. doi: 10.1186/1476-4598-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Traish AM, Morgentaler A. Epidermal growth factor receptor expression escapes androgen regulation in prostate cancer: a potential molecular switch for tumour growth. Br J Cancer. 2009;101:1949–56. doi: 10.1038/sj.bjc.6605376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Di Lorenzo G, Tortora G, D’Armiento FP, De Rosa G, Staibano S, Autorino R, et al. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin Cancer Res. 2002;8:3438–44. [PubMed] [Google Scholar]
- 113.Abreu-Martin MT, Chari A, Palladino AA, Craft NA, Sawyers CL. Mitogen-activated protein kinase kinase kinase 1 activates androgen receptor-dependent transcription and apoptosis in prostate cancer. Mol Cell Biol. 1999;19:5143–54. doi: 10.1128/mcb.19.7.5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–8. [PubMed] [Google Scholar]
- 115.Liu Y, Karaca M, Zhang Z, Gioeli D, Earp HS, Whang YE. Dasatinib inhibits site-specific tyrosine phosphorylation of androgen receptor by Ack1 and Src kinases. Oncogene. 2010;29:3208–16. doi: 10.1038/onc.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Peterziel H, Mink S, Schonert A, Becker M, Klocker H, Cato AC. Rapid signalling by androgen receptor in prostate cancer cells. Oncogene. 1999;18:6322–9. doi: 10.1038/sj.onc.1203032. [DOI] [PubMed] [Google Scholar]
- 117.Léotoing L, Manin M, Monté D, Baron S, Communal Y, Lours C, et al. Crosstalk between androgen receptor and epidermal growth factor receptor-signalling pathways: a molecular switch for epithelial cell differentiation. J Mol Endocrinol. 2007;39:151–62. doi: 10.1677/JME-07-0021. [DOI] [PubMed] [Google Scholar]
- 118.Sirotnak FM, She Y, Lee F, Chen J, Scher HI. Studies with CWR22 xenografts in nude mice suggest that ZD1839 may have a role in the treatment of both androgen-dependent and androgen-independent human prostate cancer. Clin Cancer Res. 2002;8:3870–6. [PubMed] [Google Scholar]
- 119.Canil CM, Moore MJ, Winquist E, Baetz T, Pollak M, Chi KN, et al. Randomized phase II study of two doses of gefitinib in hormone-refractory prostate cancer: a trial of the National Cancer Institute of Canada-Clinical Trials Group. J Clin Oncol. 2005;23:455–60. doi: 10.1200/JCO.2005.02.129. [DOI] [PubMed] [Google Scholar]
- 120.Whang YE, Armstrong AJ, Rathmell WK, Godley PA, Kim WY, Pruthi RS, et al. A phase II study of lapatinib, a dual EGFR and HER-2 tyrosine kinase inhibitor, in patients with castration-resistant prostate cancer. Urol Oncol. 2011 doi: 10.1016/j.urolonc.2010.09.018. [In Press] [DOI] [PubMed] [Google Scholar]
- 121.Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. 1998;279:563–6. doi: 10.1126/science.279.5350.563. [DOI] [PubMed] [Google Scholar]
- 122.Chen C, Lewis SK, Voigt L, Fitzpatrick A, Plymate SR, Weiss NS. Prostate carcinoma incidence in relation to prediagnostic circulating levels of insulin-like growth factor I, insulin-like growth factor binding protein 3, and insulin. Cancer. 2005;103:76–84. doi: 10.1002/cncr.20727. [DOI] [PubMed] [Google Scholar]
- 123.Harman SM, Metter EJ, Blackman MR, Landis PK, Carter HB. Serum levels of insulin-like growth factor I (IGF-I), IGF-II, IGF-binding protein-3, and prostate-specific antigen as predictors of clinical prostate cancer. J Clin Endocrinol Metab. 2000;85:4258–65. doi: 10.1210/jcem.85.11.6990. [DOI] [PubMed] [Google Scholar]
- 124.Wu JD, Haugk K, Woodke L, Nelson P, Coleman I, Plymate SR. Interaction of IGF signaling and the androgen receptor in prostate cancer progression. J Cell Biochem. 2006;99:392–401. doi: 10.1002/jcb.20929. [DOI] [PubMed] [Google Scholar]
- 125.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–65. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 126.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309–19. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 127.Kraus S, Gioeli D, Vomastek T, Gordon V, Weber MJ. Receptor for activated C kinase 1 (RACK1) and Src regulate the tyrosine phosphorylation and function of the androgen receptor. Cancer Res. 2006;66:11047–54. doi: 10.1158/0008-5472.CAN-06-0596. [DOI] [PubMed] [Google Scholar]
- 128.Asim M, Siddiqui IA, Hafeez BB, Baniahmad A, Mukhtar H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene. 2008;27:3596–604. doi: 10.1038/sj.onc.1211016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gioeli D, Mandell JW, Petroni GR, Frierson HF, Jr, Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999;59:279–84. [PubMed] [Google Scholar]
- 130.Agoulnik IU, Bingman WE, 3rd, Nakka M, Li W, Wang Q, Liu XS, et al. Target gene-specific regulation of androgen receptor activity by p42/p44 mitogen-activated protein kinase. Mol Endocrinol. 2008;22:2420–32. doi: 10.1210/me.2007-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lyons LS, Burnstein KL. Vav3, a Rho GTPase guanine nucleotide exchange factor, increases during progression to androgen independence in prostate cancer cells and potentiates androgen receptor transcriptional activity. Mol Endocrinol. 2006;20:1061–72. doi: 10.1210/me.2005-0346. [DOI] [PubMed] [Google Scholar]
- 132.Lyons LS, Rao S, Balkan W, Faysal J, Maiorino CA, Burnstein KL. Ligand-independent activation of androgen receptors by Rho GTPase signaling in prostate cancer. Mol Endocrinol. 2008;22:597–608. doi: 10.1210/me.2007-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15:4799–805. doi: 10.1158/1078-0432.CCR-08-0125. [DOI] [PubMed] [Google Scholar]
- 134.Halvorsen OJ, Haukaas SA, Akslen LA. Combined loss of PTEN and p27 expression is associated with tumor cell proliferation by Ki-67 and increased risk of recurrent disease in localized prostate cancer. Clin Cancer Res. 2003;9:1474–9. [PubMed] [Google Scholar]
- 135.Bertram J, Peacock JW, Fazli L, Mui AL, Chung SW, Cox ME, et al. Loss of PTEN is associated with progression to androgen independence. Prostate. 2006;66:895–902. doi: 10.1002/pros.20411. [DOI] [PubMed] [Google Scholar]
- 136.McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers WR. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999;59:4291–6. [PubMed] [Google Scholar]
- 137.Ayala G, Thompson T, Yang G, Frolov A, Li R, Scardino P, et al. High levels of phosphorylated form of Akt-1 in prostate cancer and non-neoplastic prostate tissues are strong predictors of biochemical recurrence. Clin Cancer Res. 2004;10:6572–8. doi: 10.1158/1078-0432.CCR-04-0477. [DOI] [PubMed] [Google Scholar]
- 138.Kreisberg JI, Malik SN, Prihoda TJ, Bedolla RG, Troyer DA, Kreisberg S, et al. Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Res. 2004;64:5232–6. doi: 10.1158/0008-5472.CAN-04-0272. [DOI] [PubMed] [Google Scholar]
- 139.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–21. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 140.King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–6. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–24. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chen CD, Sawyers CL. NF-kappa B activates prostate-specific antigen expression and is upregulated in androgen-independent prostate cancer. Mol Cell Biol. 2002;22:2862–70. doi: 10.1128/MCB.22.8.2862-2870.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ismail HA, Lessard L, Mes-Masson AM, Saad F. Expression of NF-kappaB in prostate cancer lymph node metastases. Prostate. 2004;58:308–13. doi: 10.1002/pros.10335. [DOI] [PubMed] [Google Scholar]
- 144.Lessard L, Bégin LR, Gleave ME, Mes-Masson AM, Saad F. Nuclear localisation of nuclear factor-kappaB transcription factors in prostate cancer: an immunohistochemical study. Br J Cancer. 2005;93:1019–23. doi: 10.1038/sj.bjc.6602796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jin RJ, Lho Y, Connelly L, Wang Y, Yu X, Saint Jean L, et al. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res. 2008;68:6762–9. doi: 10.1158/0008-5472.CAN-08-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nadiminty N, Lou W, Sun M, Chen J, Yue J, Kung HJ, et al. Aberrant activation of the androgen receptor by NF-kappaB2/p52 in prostate cancer cells. Cancer Res. 2010;70:3309–19. doi: 10.1158/0008-5472.CAN-09-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nazareth LV, Weigel NL. Activation of the human androgen receptor through a protein kinase A signaling pathway. J Biol Chem. 1996;271:19900–7. doi: 10.1074/jbc.271.33.19900. [DOI] [PubMed] [Google Scholar]
- 148.Sadar MD. Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. J Biol Chem. 1999;274:7777–83. doi: 10.1074/jbc.274.12.7777. [DOI] [PubMed] [Google Scholar]
- 149.Buschhorn HM, Klein RR, Chambers SM, Hardy MC, Green S, Bearss D, et al. Aurora-A over-expression in high-grade PIN lesions and prostate cancer. Prostate. 2005;64:341–6. doi: 10.1002/pros.20247. [DOI] [PubMed] [Google Scholar]
- 150.Lee EC, Frolov A, Li R, Ayala G, Greenberg NM. Targeting Aurora kinases for the treatment of prostate cancer. Cancer Res. 2006;66:4996–5002. doi: 10.1158/0008-5472.CAN-05-2796. [DOI] [PubMed] [Google Scholar]
- 151.Shu SK, Liu Q, Coppola D, Cheng JQ. Phosphorylation and activation of androgen receptor by Aurora-A. J Biol Chem. 2010;285:33045–53. doi: 10.1074/jbc.M110.121129. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 152.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–16. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 153.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375:1437–46. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–46. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]