

Abstract
Craniostenosis is a disease characterized by untimely fusion of cranial sutures resulting in a variety of craniofacial deformities and neurological sequelae due to alteration in cranial volume and restriction of brain growth. This involves vault sutures predominantly, but cranial base is not immune. Association with a variety of syndromes makes the management decision complex. These children need careful evaluation by multiple specialists to have strategic treatment options. Parental counseling is an important and integral part of the treatment. Recent advancements in the surgical techniques and concept of team approach have significantly enhanced the safety and outcome of these children. We had an opportunity of treating 57 children with craniostenosis in the last 15 years at our craniofacial service. Out of them, 40 were nonsyndromic and 17 were syndromic variety. We describe our successful results along with individualized operative technical modifications adopted based on the current understanding of the disease.
Keywords: Nonsyndromic craniostenosis, operative results, pediatric craniofacial surgery
Introduction
Craniofacial surgery is a relatively new specialty as most of the developments have occurred in the twentieth century. This surgical field encompasses several different surgical subspecialties. By virtue of their combined evolution, technology adoption and creation of a multidisciplinary teamwork have helped to overcome the complexity associated. Advances in anesthesia, surgical techniques, and technology have enhanced the good outcomes and safety. Craniofacial surgery has thus emerged from the valuable contributions of neurosurgery, plastic surgery, occuloplastic, ENT, and head and neck surgery. Teaming of such disciplines helped us to establish a credible unit with ample clinical opportunity. With the longitudinal experience, we could analyze own results and also introspect. The purpose of this article is to share our clinical experience in treating pediatric craniofacial anomalies, both syndromic and nonsyndromic, over 15 years with a review and understanding of current concepts in offering surgery.
Nonsyndromic Craniosynostosis and Deformational Plagiocephaly
Abnormal shape of the skull has evinced keen interest from time immemorial. People have attributed it to various facts like black magic, superstitions, and stellar events like eclipses as causative agents. Sommerling in 1791 reported relationship between growth and shape of the skull. In 1851, Virchow recognized that premature closure of suture line causes limitation of growth perpendicular to that suture and also expansion in the direction of the suture line.
Craniosynostosis is a pathologic condition that results from premature fusion of one or more sutures in the cranial vault, resulting in a deformity of the vault and the cranial base. Most demonstrate a sporadic pattern of occurrence. When a suture fuses prematurely, the skull restricts the growing brain beneath the suture thus allowing expansion into regions of less restriction. This “compensatory” growth of the skull occurs largely in planes parallel to the affected suture, resulting in consistent, recognizable cranial deformities. The incidence of nonsyndromic craniosynostosis has been reported as 0.4–1 per 1,000 live births.
The developments of surgical interventions and corrections have been profound in the last 30 years. Technologic advances and formation of craniofacial teams, incorporating the expertise of necessary specialties like genetics, anesthesiology, ophthalmology, otolaryngology, orthodontics, speech therapy, physical therapy, and psychology.
As the concept of cosmoses took major role, surgeons were forced to be more self-critical of their results. Strip craniectomies soon got converted to complex reconstructions. Craniostenosis is closely related to the complex craniofacial anomalies.
Understanding and development of craniofacial principles by Paul Tessier has created unlimited scope for the correction of these deformities and attracted the attention of surgeons from all over the world [Figure 1]. He asserted several basic principles: (1) correction requires a wide subperiosteal exposure of face and orbit, (2) the affected orbit can be moved safely in any direction without risking vision or occulomotor dysfunction, (3) osteotomies and repositioning of facial structures produce better results than the sole use of bone grafting, and (4) as many deformities as possible should be corrected during the same operative procedure. He was also instrumental in conceptualizing and introducing the multidisciplinary approach to craniofacial malformations.
Figure 1.

Paul Tesseier
In 1976, Hoffman and Mohr recognized the inadequacy of craniotomy for the correction of craniostenosis and introduced the concept of mobilization of supra-orbital bar with an osteotomy of the orbital process. Another significant discovery was the concept of the “floating forehead” as described by Marchac and Renier. The development of power drills, craniotomies, significantly decreased the operating time, reduced the blood loss, and eliminated cosmetically undesirable burr holes.
History and Pathogenesis
Virchow, in 1851, noted that premature fusion along the suture lines resulted in compensatory growth along a plane parallel to the fused suture and a decrease in the growth of the skull in relation to the perpendicular axis.
This hypothesis remained unchanged for nearly 100 years until Van der Klaauw, in 1946, and Moss, in 1959, proposed that the cranial base was the source of abnormal physical stress leading to dural abnormalities that yielded in premature sutural fusion, called the “functional matrix theory.” Further supportive evidence for the formation of cranial vault suture pathology in most cases of nonsyndromic craniosynostosis comes from the clinical observation by Marsh and Vannier following cranioplasty in individual sutural craniosynostosis where the surgery altered only the cranial vault structure, while previously developed cranial base abnormalities were ameliorated.[3] More recently, Opperman, Ogle, Longaker, and others have demonstrated developmental and biological abnormalities in prematurely fusing sutures.[4,5]
Both performed strip craniectomies for fused cranial vault sutures in young infants. To the present day, modifications of this approach continued to treat these children with cranial deformities. Tessier's recommendations include the use of wide exposure, intracranial approach to correct the facial deformities, liberal and exclusive use of autogenous bone grafts, and reliance on rigid bony fixation. Subsequent developments are three-dimensional radiographic visualization and precise computer-guided modeling. Improved pediatric anesthesia, critical care, and monitoring contributed to the overall safety and effectiveness while decreasing morbidity and mortality. Rigid fixation and the development of resorbable plates and screws and bone substitutes have also greatly improved the efficacy of the surgery. The development of the techniques of distraction osteogenesis applied to both the cranial vault and the facial skeleton, and have radically altered and expanded surgical options for correction of these deformities. Surgical treatment of craniosynostosis began with Lane (1892) and Lannelongue.
Cranial Anatomy and the Development of Anomalies
The cranial vault is composed of a series of bony plates, the junctions of which constitute the cranial sutures [Figure 2]. The major cranial sutures are the metopic, saggittal, coronal, and lambdoid. The minor sutures include the temporo squamosal, the fronto nasal, and the fronto sphenoidal. In some situations, brain growth is retarded by prolonged restriction of the cranial vault secondary to fusion of the overlying sutures. In majority, fusion of a solitary suture does not result in gross neurologic impairment, although more subtle learning disabilities may be more frequent.[6] When multiple sutures are involved, the possibility of compression of underlying brain becomes significant.[7] Quite often craniosynostosis may not be detectable at birth because of subtle manifestations and warrants periodic evaluation by an experienced craniofacial team where ever suspected.
Figure 2A-B.
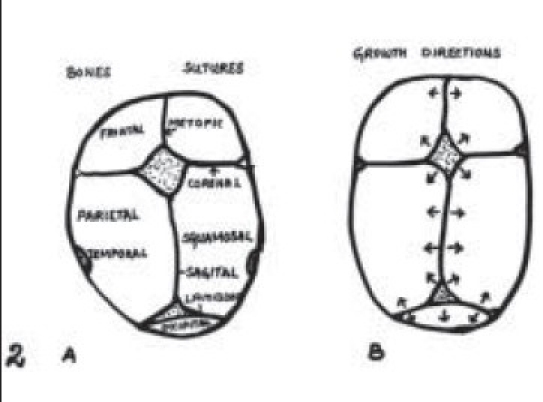
Major cranial sutures. B: Growth of the brain that is refl ected in expansion of the cranial vault is always perpendicular to cranial sutures as demonstrated by arrow
Figure 2C.
Clinical appearance
The most common types of craniosynostosis are (a) metopic synostosis producing trigonocephaly, (b) Sagittal synostosis producing scaphocephaly, (c) unilateral coronal synostosis resulting in plagiocephaly, and (d) bilateral coronal synostosis resulting in a brachycephaly. Lambdoid synostosis is distinctly an uncommon condition that also results in a form of plagiocephaly. Positional (deformational) plagiocephaly of the occiput (a non-craniosynostosis, self-correcting deformity) is frequently seen and must be distinguished from real craniosynostosis.
Materials and Methods
Forty children were treated for nonsyndromic variety of craniosynostosis at our center in the last 15 years. The age ranges from 6 months to 5 years. 16 were males and 24 were females. All of them underwent a detailed evaluation by the craniofacial team, which includes clinical, morphological, psychological, and radiological assessments. The treatment was customized to each individual variety, the plan of treatment was extensively discussed with the parents, and the outcomes were counseled appropriately.
Preoperative Considerations
Multidisciplinary team approach is essential to provide a comprehensive care for this condition. Clinical evaluation including a detailed family history is essential to make the diagnosis. Genetic testing and counseling are highly desirable in confirming the diagnosis and in counseling the family. Additional, neuropsychological evaluation can discern the adequacy of developmental milestones. The neurologic evaluation is instrumental in determining the functional abilities and to identify increased intracranial pressure (ICP) (anorexia, lethargy, or visual changes) among other complicating factors. A careful note on respiratory problems is essential in children with significant mid-facial retrusion, to assess airway compromise, and sleep apnea, Pulmonary and otolaryngologic evaluation, fiberoptic laryngoscopy, and sleep studies are done wherever necessary. Continuous positive airway pressure (CPAP) or tracheostomy may be planned in mid-facial deformities [Figure 3].
Figure 3.
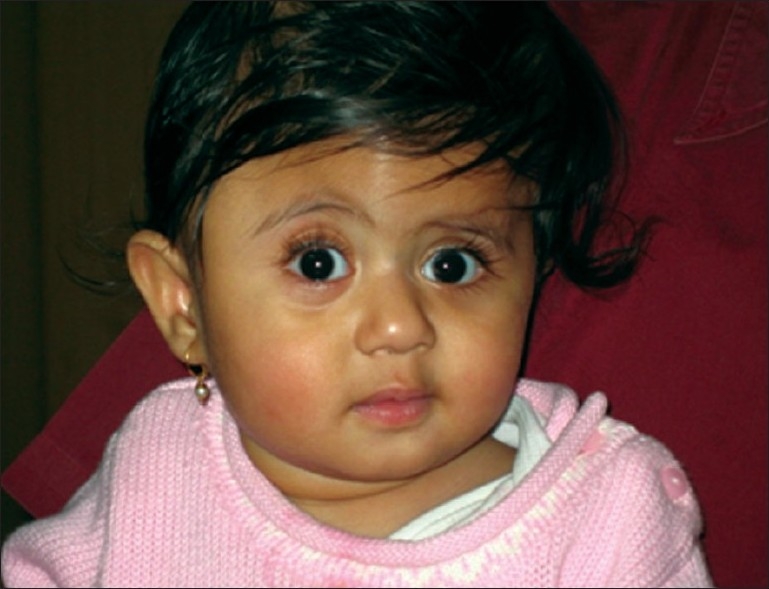
Plagiocephaly
We realized that a sensitive and supportive psychological therapist may play a significant role to the child and family. Additional evaluations by the orthodontist, otolaryngologist, and ophthalmologist are critical in syndromic variety. For those requiring extensive reconstruction, it is important to anticipate potential problems and alert anesthesiologist and pediatric intensivists accordingly. Special attention should be paid to the best use and replacement of blood products. Autologus blood products are preferred wherever feasible.
Physical examination included assessment of sutural ridging, sutural patency and skull defects, overall and regional descriptions of skull and facial configuration, and fullness and patency of both the anterior and posterior fontanels. The positions of the medial and lateral canthi, as well as presence of exophthalmos, orbital dystopia, or upper lid ptosis, need to be examined. Visual acuity, the presence of diplopia, and papilledema are tested by an ophthalmologist. Occlusal relationship and dental development are appropriately assessed by the orthodontist. Cephalometric studies and dental models are very useful. Pre- and postoperative photographs were taken to record and document progress through all phases of treatment.
Radiographic Assessment
Although a variety of radiographic examinations might be useful, plain film radiographs (anteroposterior and lateral skull) can document “sclerosis,” or closure of additional sutures, and possible widening of adjacent compensating sutures. Lateral cephalometric radiographs and orthopantographic (Panorex) radiographs are frequently taken to document the occlusal and facial relationships. Computerized tomography scanning of the cranial vault, base, and facial structures, including mandible, is important. Three-dimensional reconstruction of the cranium, orbits, and face is particularly useful in demonstrating the general volume relationships and overall shape of the underlying bony structures. Refinements in computer-aided designs are of additional benefit for patients requiring primary or secondary correction in future. Computer-generated models can be manufactured and milled from three-dimensional CT data, which can be used preoperatively to assist in planning osteotomies. Magnetic resonance imaging (MRI) is not routinely done, but in syndromic cases, it is useful to evaluate developmental abnormalities of brain [Figure 4].
Figure 4A.
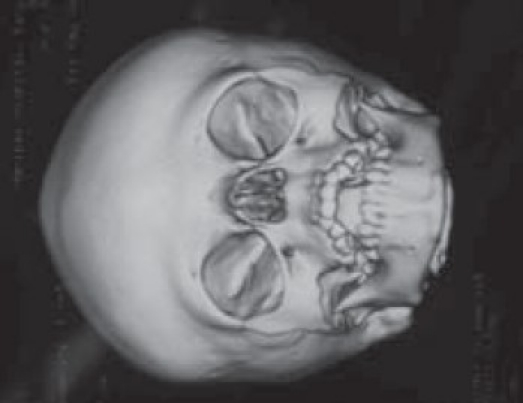
Plagiocephaly
Figure 4B.
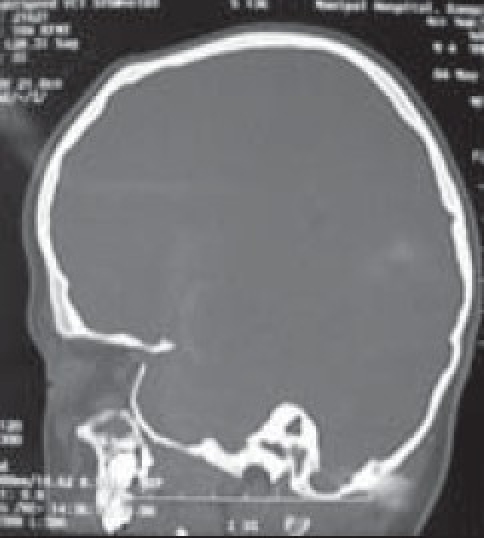
Evaluation of cranial/orbital volume
Figure 4C.
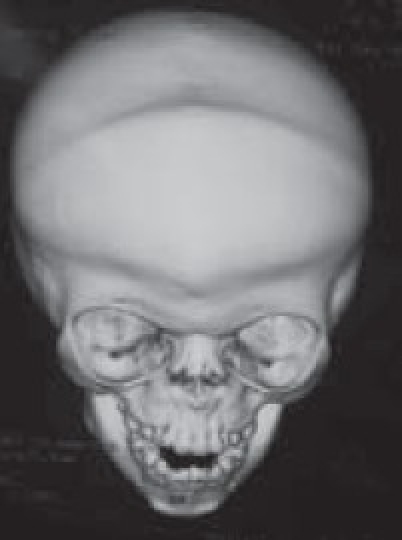
3-D OF CORONAL SUTURES (frontal)
Figure 4D.
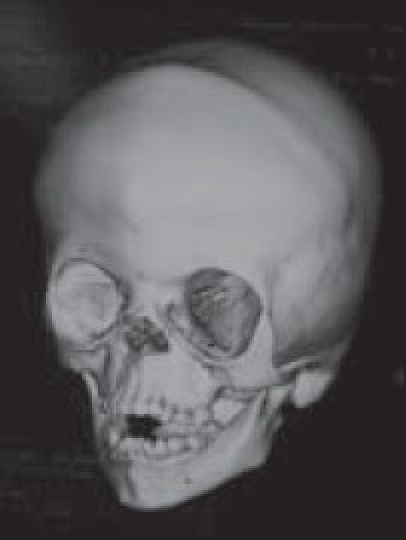
Oblique view
Figure 4E.
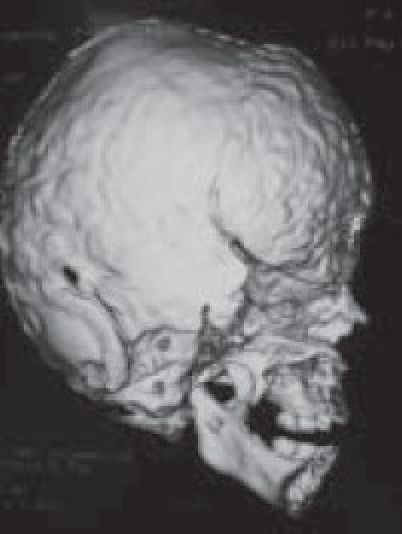
Evaluation of other sutures/defects
Figure 4F.
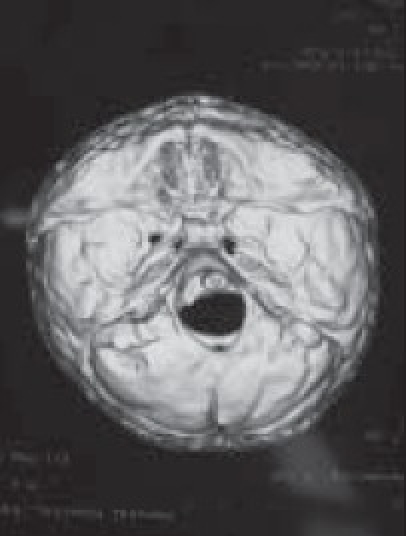
Basal view
Timing of Surgery
The optimal timing for surgical treatment of craniosynostosis is not yet clear. Our philosophy, however, is to operate as soon as the patient is able to withstand the stress of surgery, in order to capitalize on the ameliorating effects of brain growth on overall skull shape. In practice, this usually turns out to be approximately 6–9 months of age, earlier if there is evidence of increased ICP (bulging fontanel, progressive optic atrophy, or multiple-suture stenosis). The determining factor usually is the extent of surgery and anticipated blood loss. For instance if “barrel stave” osteotomy procedure is planned, it may be helpful to delay the surgery till 5–6 months of age. The NYU group proposes 9–10 months as ideal age for barrel stave osteotomy. The rationale being closer the cranium to the adult size, the less overcorrection that needs to be built into the reconstruction and better the ultimate skull shapes. There is a consensus, however, to operate before 1 year of age so as to take advantage of the ability to ossify small craniectomy defects.
Type of Surgery
The patients may be placed in the modified prone position for optimal whole-vault exposure at the time of surgery. Cervical spine films are taken to exclude craniovertebral anomalies or instability that might lead to spinal cord or brainstem dysfunction.
There is considerable variation in the surgical techniques, which includes extended strip craniectomies with or without postoperative molding helmets, regional craniectomies, as well as craniectomies associated with distraction osteogenesis and spring-mediated movements, and whole-vault cranioplasties, etc. Each type is chosen based on the clear understanding of the inherent risks and benefits associated with a careful review of various factors influencing the outcomes. The surgical technique is tailor-made for each child. The details were discussed extensively with the parents. The innovative technique of bone lengthening by Illizarov has been adapted to craniofacial surgery. Joseph McCarthy and his colleagues have applied this concept of distraction osteogenesis for the reconstruction of mandible deformities. Ortiz-Monasterio and his colleagues have refined these techniques resulting in accomplishment of well-controlled and significant craniofacial distraction in mandible and midface. Severe modified techniques for bone refinement and fixation are available. These techniques have significantly reduced the incidence of surgical relapse and complications.
Clinical Scenarios and Management
Metopic Synostosis
The metopic suture is the first cranial suture to fuse approximately at 7–8 months of age. Premature closure of this results in a well-recognized “keel”-shaped deformity known as trigonocephaly. Metopic synostosis is relatively uncommon and accounts for less than 10% of isolated, nonsyndromic craniosynostosis. Brain development is not usually impaired, although Renier has noted elevated ICP in approximately 4% of his patients. Trigonocephaly may be associated with underdevelopment of the frontal lobes as an associated primary brain anomaly.[7] Such developmental problems may be primarily due to brain maldevelopment. Hypotelorism is evident and often accompanied by upward slanting of both lateral canthi and the lateral portions of the eyebrows, as well as flattening of the supraorbital ridges.
When the metopic suture is prematurely fused, bone growth is apparently reduced all along the margins of the frontal bones adjacent to the stenotic suture. There is a compensatory increase in bone deposition at the abutting Sagittal suture, resulting in enlarged parietal bones. The triangular shape is a result of the flattening of the frontal bones, anterior displacement of the coronal sutures, and flaring up of parietal bones. This shape is exaggerated by the lack of lateral projection of the supraorbital rims and narrowing of the temporal regions. Degree of deformity can vary widely. An individualized and properly timed correction is ideal. The basic operative management includes overcorrection of the bilateral dimensions wherever required [Figure 5].
Figure 5.
Metopic stenosis
Operative Technique
Early correction (younger than 1 year) is performed with the child in the supine position. A bifrontal craniotomy is performed, and the frontal and parietal bones are removed as a single bone graft. Remodeling of the bifrontoparietal bone graft is performed using a shaping burr and radial osteotomies. Care is taken to avoid the detachment of the orbital rims at the frontozygomatic suture. Our current technique advances the orbital rims by the bowstring method. This appears to reduce the potential to compromise the supraorbital rim blood supply and allows angulations of the orbital rim pivoting on the fronto zygomatic suture. We believe that this step is important for supporting the orbital rim and frontal bone, preventing a superior flattening of the supraorbital/frontal region, which may yield a less-satisfactory result. We maintain the temporalis muscle attachment to the underlying bone and squamous bone overlay, attempting to prevent the “temporal hollowing.” This is usually seen following techniques that detach the muscle fully, and then reattach with suture. The anterior temporal bone and muscle are outfractured as a composite unit. The frontal bone graft is secured to the supraorbital rims either with wire or suture fixation. As a general rule, bone grafting is required when the defect is greater than the size of a quarter. Otherwise the small defects ossify spontaneously in children less than 1 year of age.
In children between 1 and 3 years, the bony remodeling techniques are altered to allow reshaping of the more mature cranial vault. The bone in this age group is more brittle, making bending less effective. The major difference in technique in this age group involves selective weakening of the bone by the placement of endocranial channels or “kerfs” in the bone. These children's (older than 1 year) usually require bone grafting, as these defects tend to be larger.
In children older than 3 years, surgical correction requires further modifications. Cranial vault and brain growth are approximately 85% complete by 3 years. At this point, there is greater emphasis on direct reshaping and fixation of formed bone segments in their “normal” adult positions. These basic principles of bone remodeling and cranial fixation apply to the treatment of all forms of nonsyndromic craniosynostosis as well [Figure 6].
Figure 6.
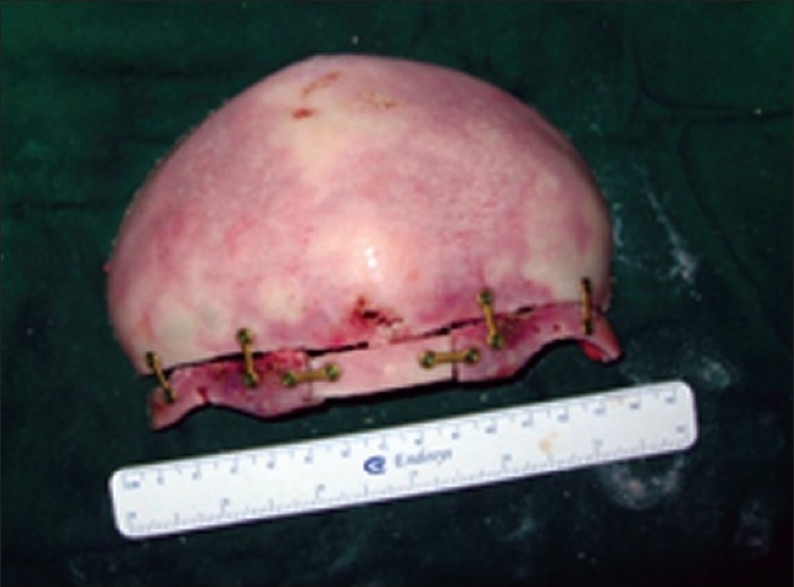
Bench remodeling fronto –orbital segment
Sagittal Synostosis
Saggittal synostosis is the most common form of craniosynostosis associated with a deformity known as scaphocephaly. The skull shape is the result of premature fusion of the Saggittal suture, and characterized by a skull with increased anteroposterior (AP) length and decreased width, yielding a “boatlike” shape. Saggittal synostosis is typically sporadic, with only a 2% genetic or familial predisposition. The male-to-female incidence is 4:1. Following Saggittal suture fusion, the coronal and lambdoid sutures, which are adjacent to the restricted parietal bone plate, are compensated by enhanced bone deposition at the edges (in the frontal and occipital bones, respectively). The metopic suture compensates with symmetric bone expansion along its sutural borders. The compensatory growth process produces the characteristic frontal and occipital prominence seen in Sagittal synostosis. Because the squamosal sutures are distant to the fused suture, they do not participate significantly in the compensatory growth process and, as such, there is no prominent projection of the temporal (squamous) regions bilaterally. The entire Sagittal suture might not be involved so that deformity may be predominantly anterior, posterior, or both [Figure 7].
Figure 7.
Sagittal stenosis
Operative Technique
Treatment of Sagittal synostosis varies according to the age. Children younger than 1 year with malleable cranial vault bones can be easily reshaped. Reshaping is more difficult in older age group because of increased “brittleness” of the bones. The primary operative goals are to release the stenosis and reshape the cranial vault by increasing the parietal and temporal width and decreasing its AP dimension.
If an access to both anterior and posterior deformities is necessary, the child is placed in the modified prone position. Supra periosteal dissection is performed extending from the glabella anteriorly to the posterior lip of the foramen magnum posteriorly. After appropriate burr holes placement, craniotomies are performed to separate the bifrontal and biparietal-occipital fragments. The narrowing of the low temporal and parietal regions is addressed by placement and outfracturing of parallel vertical “barrel stave” osteotomies. The cone-shaped occiput is remodeled after radial osteotomies and bending of the bone to provide a more gradual convex curvature. The bifrontal fragment is similarly reshaped after placement of radial osteotomies. Shortening of the AP length of the skull is accomplished by resecting a portion of the frontal and parietal bones at the midline. The remaining parietal bone fragments are remodeled with a goal of increasing the lateral convexity, particularly in the parietal regions [Figure 8].
Figure 8.
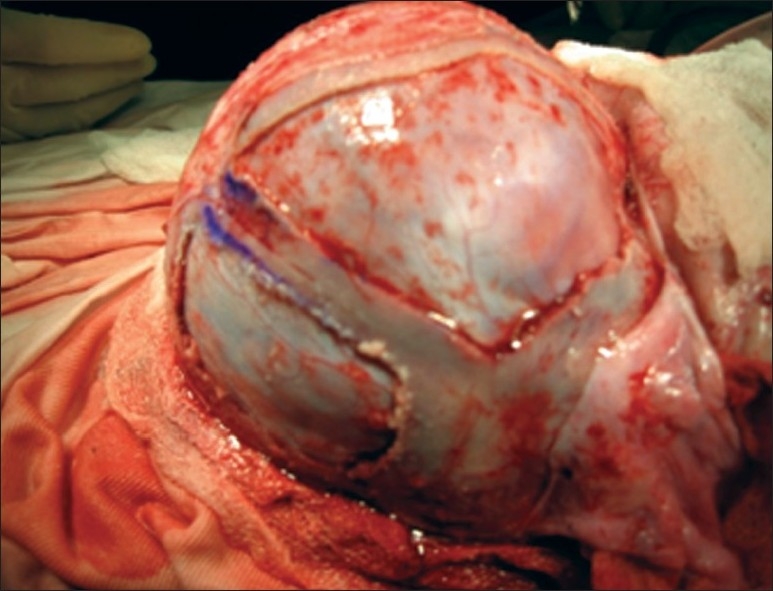
Barrel Steeve osteotomies
Once the bone fragments are remodeled satisfactorily, they are secured with wire or suture fixation in the midline. The frontal segment is secured anteriorly to the superior orbital rims, and posteriorly to the parietal bone segment overlying the Sagittal sinus. The occipital segment is attached to the basal occiput posteriorly and to the parietal bone anteriorly. The remodeled parietal bone segments are attached to the underlying dura but are not fixed to the adjacent bone so as to encourage further widening of the skull postoperatively.
Modifications, Kerfs, or channels, placed on the internal surface of the bone-oriented perpendicular to the long axis of the bone segment, allow for selective weakening of the bone and easier reshaping and molding. In older children, the techniques of bone remodeling require several modifications [Figure 9].
Figure 9A.
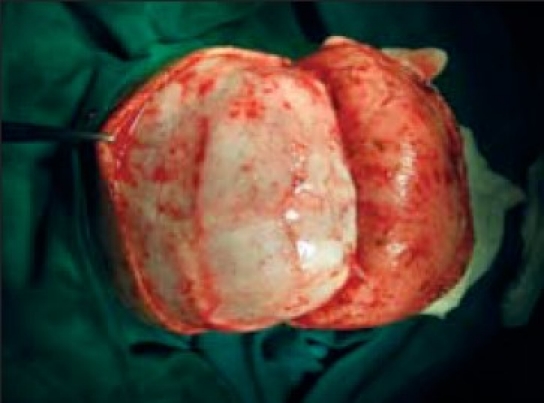
Exposure-Sagittal stenosis
Figure 9B.
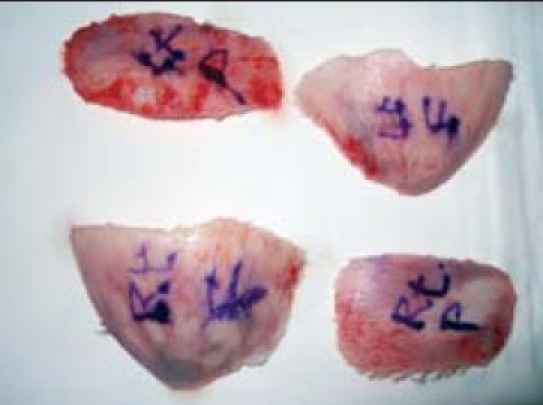
Bench remodelling
Figure 9C.
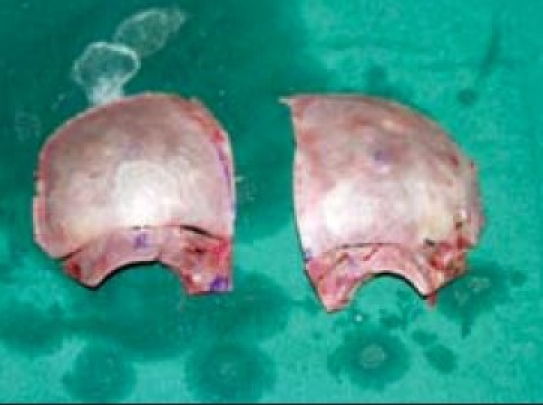
Bench osteo synthesis
Figure 9D.
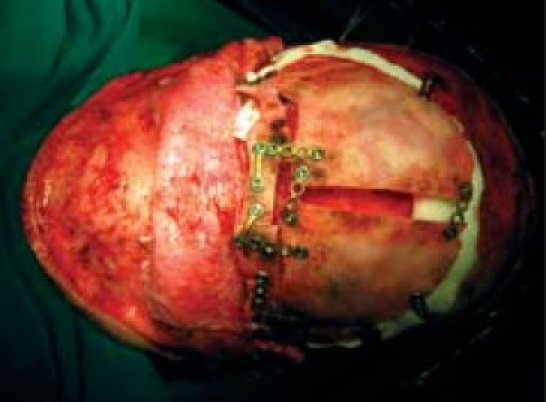
Final osteo synthesis and fixation
Unilateral Coronal Synostosis
Premature fusion of one of the coronal sutures results in plagiocephaly (a Greek term denoting oblique skull). This is an uncommon disorder, occurring in 1 in 10,000 live births. In our series, this group constituted majority of patients.
Operative Technique in Coronal Synostosis
Reduction in AP skull length is achieved frontally by removing a segment of midline bone. Triangular wedges of frontal bone are removed just cephalad to the supraorbital margins to allow for posterior inclination of the forehead. The frontal bone laterally is remodeled to create a neocoronal suture. As the wires are cinched down frontally, bulging occurs in the parietal region, for which the parietal bone is reshaped to add increased contour laterally.
In unilateral coronal synostosis, fusion at the coronal suture results in a single frontoparietal bone plate ipsilateral to the fused suture, with an apparent reduced growth potential. This impairs the ventral expansion of the anterior cranial fossa, resulting in a shortened anterior cranial fossa ipsilateral to the fused suture. Growth superiorly results in elongation of the forehead, whereas inferiorly directed growth produces deformity of the middle cranial fossa with ventral bowing of the greater wing of the sphenoid. The deformity of the sphenoid results in effacement of the temporal fossa, which, combined with shortening of the lateral wall of the orbit, produces proptosis of the globe. The “harlequin” orbit seen on AP radiographs is pathognomonic for unilateral coronal synostosis and is secondary to lack of descent of the greater wing of the sphenoid during development. Compensatory overgrowth of bone occurs asymmetrically at all perimeter sutures that border the now fused single frontoparietal bone plate that incorporates the coronal suture. This accounts for the bulge in the ipsilateral squamous portion of the temporal bone, contralateral frontal and parietal bones, and, to a much lesser degree, contralaterally in the occipital bone. The fused coronal suture may demonstrate prominent ridging, and the ipsilateral frontal and parietal bones are flattened.
Clinical facial features include widening of the ipsilateral palpebral fissure, superior and posterior displacement of the ipsilateral orbital rim and eyebrow, and deviation of the nasal root toward the flattened side of the frontal bone. The chin point deviates to the contralateral side, and the malar eminence is frequently anterior on the same side as the flattened side forehead (and fused coronal suture) when compared to the contralateral malar eminence.
Operative Technique
The child is placed supine and a bifrontal craniotomy is performed. The greater wing of the sphenoid, which is thickened and displaced superiorly, is rongeured to the level of the lateral supraorbital fissure. A supraorbital rim osteotomy is performed bilaterally, and the orbital rims are contoured to symmetry and then advanced. Wherever the prominence of the brain and dura is increased, dural plication can be performed gently with the bipolar cautery or with suture. In unilateral coronal synostosis, the dura is plicated in the frontal bone region contralateral to the fused coronal suture to achieve frontal region symmetry. An advancement of the supraorbital rim is performed unilaterally. The affected side must be overcorrected to avoid reversal to the preoperative asymmetry. The frontal bone is remodeled using radially oriented osteotomies, and selective fractures are performed to achieve the desired form. The frontal bone plate is attached to the orbital rims superiorly and laterally, but not posteriorly so as to allow further growth of skull at the “neo”-coronal suture.
Bilateral Coronal Synostosis
Bilateral coronal synostosis is characterized by a tower-shaped, but short, head or turribrachycephaly. The skull is shortened anteroposteriorly, widened mediolaterally, and elongated vertically. The anterior cranial base is shortened, and the orbital rims are retrusive. The superior frontal and squamous temporal bones are protuberant, and the occiput is usually flattened. There may be ridging of the coronal suture and flattening of the caudal portion of the frontal bones and supraorbital ridges. Radiographically, bilateral harlequin abnormalities may be present.
Operative Technique
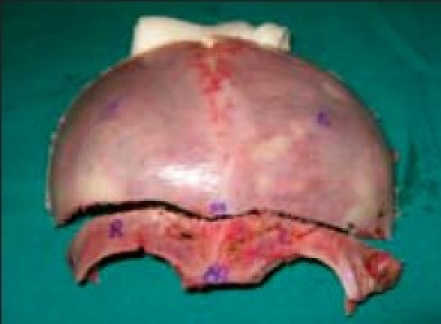
The child is placed in either supine or in a modified prone position. If the primary focus of involvement is frontal bar/orbital rim with minimal involvement of the occipital region, then the supine position is preferred. If there is absolute elevation of the height of the skull associated with shortening in an AP direction, modified prone position is preferred to reduce the height of the skull and elongate the AP dimension. A biparietal-occipital bone graft is elevated and reshaped by a combination of radial osteotomies and controlled fractures of the bone segments. Barrel stave osteotomies are placed in the occipital region, which are “outfractured” to increase the bony “capacity” of the intracranial compartment. The thickened and elevated superior portion of the greater wing of the sphenoid is removed by rongeur to the level of the supraorbital fissure. Osteotomies can be done in both orbits to increase orbital rim projection bilaterally. The remaining parietal bone struts are severed and shifted posteriorly approximately 1–2 cm. The height of the skull is reduced, while ICP is monitored. Throughout the procedure, the ICP can be monitored in order to maintain an adequate cerebral perfusion pressure. The reshaped bone segments are replaced and a postoperative skull molding cap is used to guide skull shape for 2–3 months [Figures 11 and 12].
Figure 11.
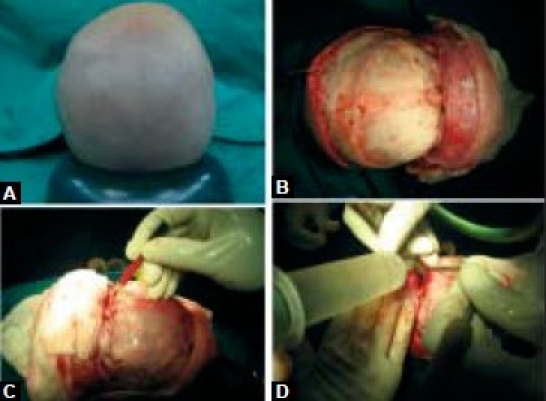
(A) positioning for surgery, (B) bicoronal fl ap exposure, (C) marking, bifrontal craniotomy, (D) fronto orbital craniotomy
Figure 12 A.
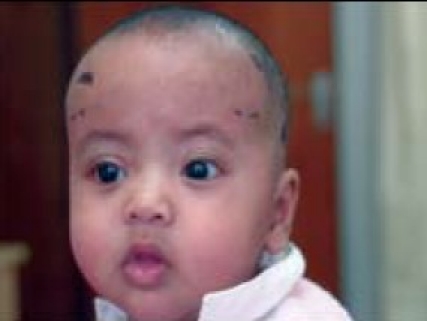
Early post-op
Figure 11 E.
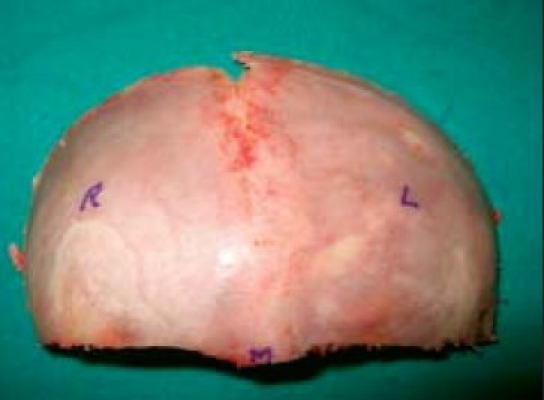
Removal frontal bone fl ap and marking FIG
Figure 11 F.
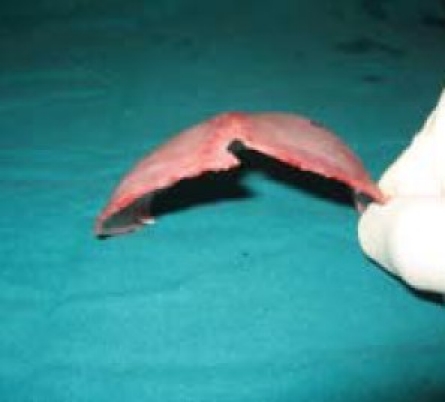
Deformity
Figure 11 G.
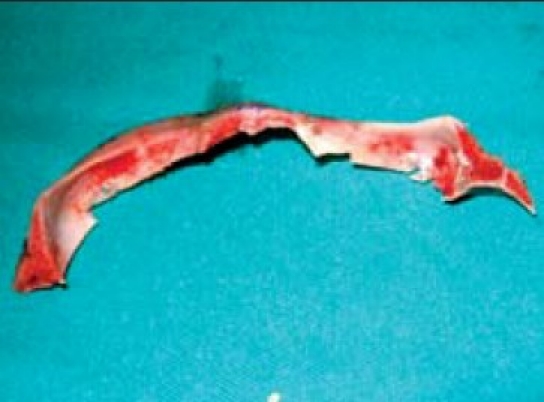
Removal of orbital ridge
Figure 11 H.
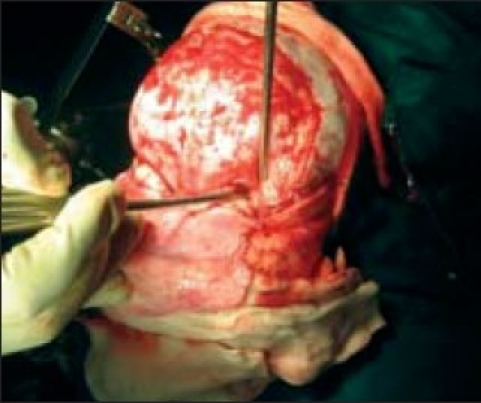
Hypo plastic orbital ridge
Figure 11 I.
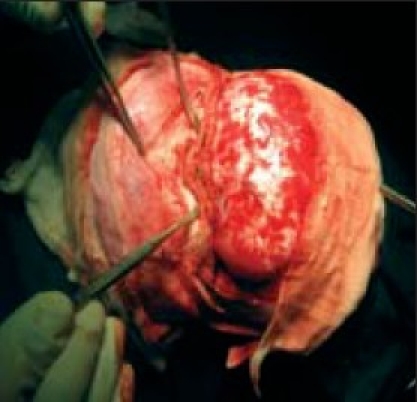
Expanding middle cranial fossa
Figure 11 J.
Bench remodeling
Figure 11K.
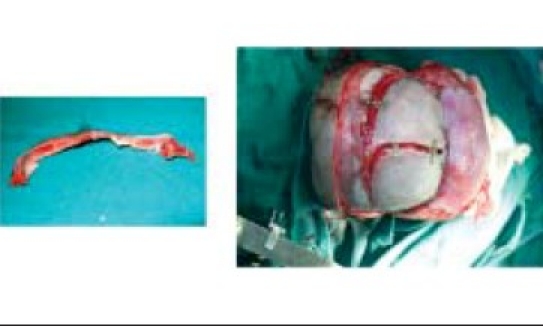
(A,B) Bone flap repositioning
Figure 11L.
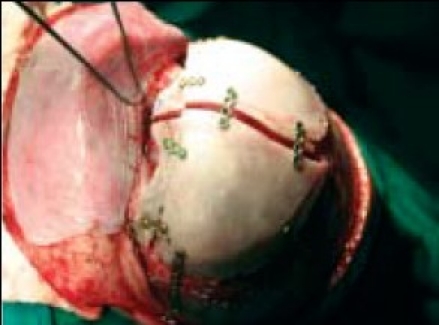
Final fixation
Figure 12B.

Long term follow-up
Lambdoid Synostosis
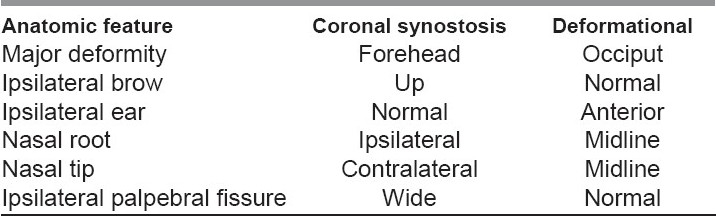
True lambdoid synostosis is extremely uncommon, and represents the least-common form of craniosynostosis. It is characterized by bony fusion of the lambdoid suture, flattening of the ipsilateral occiput, posterior/inferior displacement of the ear ipsilateral to the fused suture, and distortion of the posterior cranial base. It is to be distinguished from deformational plagiocephaly, which is common and characterized by a “parallelogram” skull deformity. In both deformational plagiocephaly and unilateral lambdoid synostosis, the occiput is asymmetrically flattened. In patients with lambdoid synostosis, the ear is positioned posteriorly and inferiorly on the side of suture fusion and occipital flattening. In patients with deformational skull deformity (plagiocephaly), the ear is positioned anteriorly (compared to the opposite side ear) on the side of occipital flattening [Figure 13].
Figure 13.
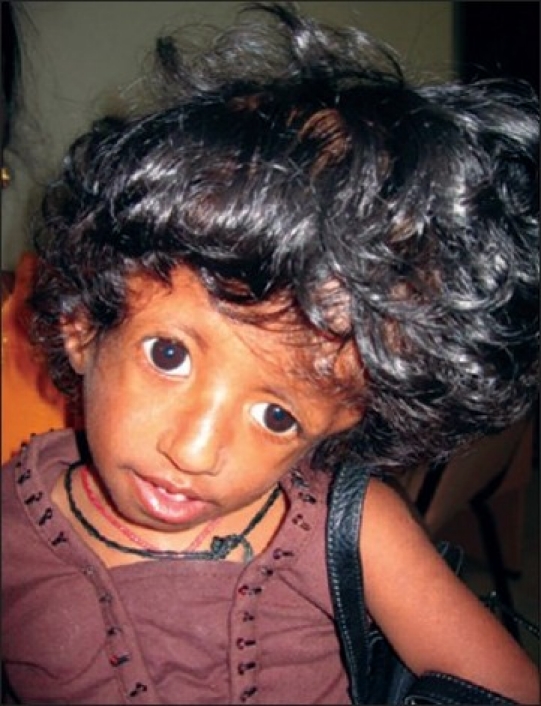
Complex Lambdoid stenosis
Radiographically, lambdoid synostosis is characterized by bony bridging of the lambdoid suture and deviation of the foramen magnum toward the side of the fused suture. In deformational plagiocephaly, the cranial base midline is unaltered and there is no bony bridging of the lambdoid suture. Premature infants tend to have positional deformities more often simply because they are less mobile because of gestational age. The distinction between deformational plagiocephaly and lambdoid synostosis is important because deformational plagiocephaly ordinarily will improve without surgery, whereas lambdoid synostosis skull deformity will not improve without surgery. The occipital flattening deformity (i.e., deformational plagiocephaly), when unilateral, is usually seen on the right side, which is most likely related to consistent decubitus positioning in infancy. The decision to recommend surgical repair for unilateral lambdoid synostosis depends on the severity of the deformity. The treatment varies depending on unilateral or bilateral, but the operative exposure and craniotomy principles are similar [Table 1].
Table 1.
Anatomic features that differentiate coronal synostosis from deformational plagiocephaly[17]
Operative Technique
The child is placed in the prone position and the occipital bone is fully exposed to the level of the foramen magnum. A bilateral parieto-occipital bone segment is elevated. Barrel stave osteotomies are performed bilaterally in the flattened basal occipital bone to increase the convex projection of the occipital bone locally. In patients with moderate unilateral lambdoid synostosis deformity, the barrel staves are placed primarily ipsilateral to the fused suture in the occipital bone. In more severe cases, bilateral barrel staves are performed, “in fracturing” the normally convex, and “outfracturing” the flattened portion of occipital bone. The biparietal-occipital bone graft is cut radially and remodeled to achieve a normally rounded, symmetric posterior skull. Cranial bone struts, secured by resorbable plates maintain the normal occipital convexity and symmetry postoperatively. The dura is plicated in areas of excess projection before bone remodeling. The bone graft is replaced and secured to the dura without attaching it to surrounding bone. If the parieto-occipital abnormality is accompanied by significant frontal abnormality (a rare clinical presentation), a bifrontal craniotomy is also performed. The frontal bone is remodeled by radial osteotomies and controlled fractures are described earlier for the parietal segments. The frontal bone is secured to the surrounding bone with nonabsorbable suture material.
Our Experience
We have operated on 40 children with nonsyndromic craniostenosis [Figure 10, Table 2].
Figure 10.
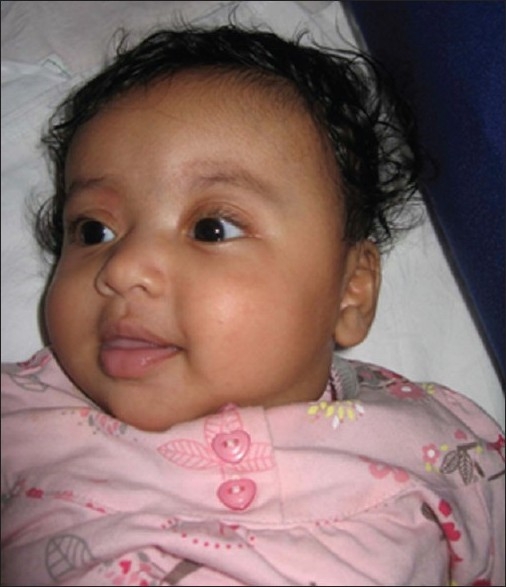
Plagiocephaly
Table 2.
Non Syndromic Craniosynostosis: Our experience

Functional Aspects
Increased Intracranial Pressure
Despite extensive studies, elevated ICP in craniosynostosis remains controversial. Lannelongue (1890) suggested that craniosynostosis results in microcephaly with secondary mental retardation. He also noted that excision of the fused sutures could reverse or prevent later intellectual impairment. Shillito and Matson advocated early craniectomy, in infancy, as they postulated that the constricting effect of untreated craniosynostosis would cause elevated ICP, and subsequent brain damage. Several authors advocated combining suture release with more formal cranio-orbital reshaping procedures to allow decompression of the brain with simultaneous reshaping of the calvarium.
It is well known that changes in skull shape are influenced by changes in the shape of the underlying brain. Premature fusion of the cranial sutures and continued brain growth seems logically reduce the volumes and lead to elevated ICP and possibly mental retardation. There are beliefs contradicting this theory as well.
Renier measured the ICP in 121 children using sensor[7] and found that elevated ICP occurred in 42% of multiple-suture involvement and in 13% with single-suture involvement. They have also noted a decrease in ICP after the corrective surgery. Technique of accurate intracranial volume measurements using computed tomographic (CT) scan to judge the adequacy of intracranial volume for the growing brain is possible.[8,9] Gault et al. demonstrated that raised ICP was most frequent in those children with more than one suture fused prematurely predominant in syndromic variety (complex, oxycephaly, Crouzon, brachycephaly, and Apert syndromes).[8]
In contrast to multiple-suture involvement, children with single-suture synostosis are more likely to have normal ICP. Intracranial volume measurement does not always give a reliable indication of the ICP (i.e., small cranial volume does not necessarily mean increased ICP); however, when ICP is elevated, skull volume is almost always significantly reduced. This observation is exclusive of conditions with primary brain maldevelopment, such as Apert syndrome.
Hydrocephalus
Hydrocephalus occurs infrequently in both syndromic and nonsyndromic craniosynostosis, but is much more common in syndromic variety. The development in imaging techniques has provided highly accurate and noninvasive method of assessing ventricular size. Assessment of ventricular size alone may not give a true picture of hydrocephalus. Ventriculomegaly, for instance, as seen in Apert syndrome, is usually unrelated to increased ICP; rather, an indicator of disordered (dystrophic) development of the brain. Measurement of serial ICPs, progressive ventricular enlargement on serial CT scans, and periventricular lucency as a result of transependymal flow of cerebrospinal fluid (CSF) are more reliable signs of sympyomatic hydrocephalus.
Mental Retardation
The true incidence of mental retardation in craniosynostosis is not clear. Historically, many with craniosynostosis syndromes were considered mentally retarded solely because of their facial appearance. The risk of mental retardation in craniosynostosis, however, is generally higher than that in normal population. Several factors have been incriminated most notable of which are increased ICP leading to cerebral atrophy, hydrocephalus, infection, intracranial developmental abnormalities, prematurity, and a family history of mental retardation (genetic).
Kapp-Simon et al. evaluated the effect of surgery on the mental development of infants with nonsyndromic craniosynostosis.[10] They concluded that surgery did not favorably affect mental development, but improved the deformity. (An additional conclusion of the study was that children with simple craniosynostosis are unlikely to experience increased ICP.) These findings were disputed by Marchac and Renier,[7] who observed relationship between ICP and volume in craniosynostosis, and increased ICP can be seen in single-suture craniosynostosis also. They proposed early surgery to improve the mental development. Additional data suggest that the recognition of mental abnormalities is dependent on the sensitivity of testing tools used, and more subtle forms of mental dysfunction (e.g., perceptual abnormalities) require different and detailed assessments (i.e., psychological tests) rather than relying on crude IQ scores alone.
Camfield and Camfield studied the relationship of underlying brain malformations and mental retardation and suggested that the mental retardation (IQ < 70) in children with single-suture craniosynostosis is likely due to the primary brain malformation rather than brain distortion caused by skull maldevelopment.[11]
Visual Abnormalities
Optic atrophy and papilledema are not uncommon findings in untreated multi-suture craniosynostosis. Optic atrophy has been attributed to the compression of the nerve by bony overgrowth of the optic canal or orbital roof, secondary stretching of the nerve, compression by the carotid vessels, or due to post-papilledemic atrophy. Of which increased ICP is believed to be the most probable cause. Syndromic craniosynostosis, especially untreated Crouzon syndrome, may be associated with optic nerve dysfunction. In contrast, patients with isolated single-suture craniosynostosis are much less affected by visual symptoms.
Complications
Complications are relatively infrequent and may be divided into those that are immediate and delayed. Immediate ones complications include blood loss, air embolism, dural tear with CSF leak, infection, and respiratory complications. Blood loss occurs nearly continuously intraoperatively and accounts for most complications. Consequently, vigilant attention to accurate blood replacement is of paramount importance to avoid coagulopathy. Blood loss can also be rapid and hemodynamically significant with a tear of the venous sinuses or major cortical veins. Abnormal transosseous veins in some syndromes, such as the Carpenter syndrome, in the region of the torcula may be torn during the bone dissection and require immediate control. In the presence of sutural fusion, the dural connection to the underlying sinus is often attenuated and the risk of a tear of the sinus is generally reduced. Air embolism has been documented in children undergoing cranioplasty and may occur in any operative position, including supine. Hence it is appropriate to do precordial Doppler monitors as well as end-tidal CO2 monitors for early detection and central venous lines for blood volume assessment and air aspiration. Blood loss may continue for 12–24 h following cranioplasty and monitoring in ICU is cruicial. Dural tears may result in CSF leak with or without infection. Immediate repair is indicated wherever possible. Postoperative CSF leak can be controlled with a lumbar drain, but if leak persists it may require surgical repair. Loss of continuity of the osteogenic dura may result in a thinning of the overlying bone, resulting in a cranial defect. These infections, though uncommon, can be potentially life-threatening. Infection generally is associated with a CSF leak and persistent communication of the intracranial cavity to exterior. Whitaker et al. reported an approximate 7% infection rate for patients undergoing craniofacial surgery.[13]
Acute airway problems may be related to inadvertent transection of the nasotracheal tube during frontofacial advancement. This may require immediate changing to an oral endotracheal tube or possibly tracheostomy. In a retrospective analysis of 542 patients undergoing 579 craniofacial procedures at the Hospital for Sick Children, Crysdale et al. reported 18% patients requiring tracheostomy for the maintenance of airway.[14] Those patients who were intubated for longer than 24 h experienced considerably more complications. Currently, the use of small-caliber fiberoptic flexible bronchoscopy greatly facilitates nasotracheal intubation in those patients previously considered incapable of endotracheal intubation.
Delayed complications generally associated as a sequelle of abnormal bone healing and impaired bone growth. Children older than 1 year have decreased ability to fill cranial defects compared to younger ones following cranioplasty in 592 patients after 1 year. Prevot et al. reported that 5% of patients had inadequate ossification.[15] Generally, defects greater than 2 cm (in children older than 1 year) should be filled with split calvarial bone grafts in order to prevent postoperative cranial defects. Significant bone loss requiring bone grafting may occur in the setting of infection and subsequent resorption.
From its inception, the use of miniplate and microplate rigid fixation has greatly improved the outcomes of craniofacial surgery. However, Persing et al. have reported significant transcranial plate migration in growing children.[16] Although no harmful effects have been reported, the potential risk and difficulty posed for reoperations have been noted. We do not have similar experience and are happy with the titanium miniplate and miniscrew rigid osteosynthesis.
With complete and adequate cranioplasty, relapse and recrudescence to the original defect is uncommon, provided there is normal brain growth and development. However, in the region of the orbits and midface, if operated on at a very young age, bone contour may relapse. The degree of relapse depends on the severity of the phenotype as well as the continued effect of cranial base restriction. The constricting force of the soft-tissue envelope and resorption of bone grafts may contribute to further relapse. The advent of distraction osteogenesis is of particular interest in the prevention of relapse as soft-tissue expansion is combined with new bone development. Orthodontic occlusal management is also important in ensuring stability in the final result in the older children [Figure 14, Table 3].
Figure 14.
CSF leak treated conservatively
Table 3.
Complications (our series)

Overall the morbidity and mortality from the treatment of craniofacial syndromes is quite low. Mortality has been reported to range between 1.5% and 2%. In 1979, Whitaker et al. reported an experience of six craniofacial centers with 13 deaths representing, 1.6% mortality.[13] It is likely that ongoing advances in monitoring and anesthetic techniques, as well as refinements in surgical techniques, will continue to yield similar or better results.
In spite of significant problems that these children and their families face, the future continues to be bright, with the technical advances providing a multiplicity of diagnostic and treatment alternatives that will impact our ability to provide better infrequent care to these children.
Conclusions
Nonsyndromic craniosynostoses is relatively common and can cause gross brain damage as a consequence of restriction of skull growth. More subtle abnormalities in learning are now being detected often. Surgical correction to reshape skulls to normal contours is indicated wherever the deformity is significant. The surgical techniques need to be individualized based on the merits. The complication rate is usually low with an experienced team and a state-of-the-art facility.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Persing JA, Babler WJ, Jane JA, Duckworth PF. Experimental unilateral coronal synostosis in rabbits. Plast Reconstr Surg. 1986;77:369–76. doi: 10.1097/00006534-198603000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Mooney MP, Losken HW, Tschakaloff A, Siegel MI, Losken A, Lalikos JF. Congenital bilateral coronal suture synostosis in a rabbit and craniofacial growth comparisons with experimental models. Cleft Palate Craniofac J. 1993;30:121–8. doi: 10.1597/1545-1569_1993_030_0121_cbcssi_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 3.Marsh JL, Vannier MW. Cranial base changes following surgical treatment of craniosynostosis. Cleft Palate J. 1986;23:9. [PubMed] [Google Scholar]
- 4.Opperman LA, Sweeney TM, Redmon J, Persing JA, Ogle RC. Tissue interactions with dura mater inhibit osseous obliteration of developing cranial sutures. Dev Dyn. 1993;198:312–22. doi: 10.1002/aja.1001980408. [DOI] [PubMed] [Google Scholar]
- 5.Duncan BW, Adzick NS, Moelleken BR, Chua J, Bradley SM, Longaker MT, et al. An in-utero model of craniosynostosis. J Craniofac Surg. 1992;3:70–8. doi: 10.1097/00001665-199209000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Magee SN, Westerveld M, Pruzinsky T, Persing JA. Long-term neuropsychological effects of sagittal craniosynostosis on child development. J Craniofac Surg. 2002;13:99–104. doi: 10.1097/00001665-200201000-00023. [DOI] [PubMed] [Google Scholar]
- 7.Renier D. Intracranial pressure in craniosynostosis: Pre-and postoperative recordings-correlation with functional results. In: Persing JA, Edgerton MT, Jane JA, editors. Scientific foundations and surgical treatment of craniosynostosis. Baltimore: Williams and Wilkins; 1989. p. 263. [Google Scholar]
- 8.Gault DT, Renier D, Marchac D, Jones BM. Intracranial pressure and intracranial volume in children with craniosynostosis. Plast Reconstr Surg. 1992;90:377–81. doi: 10.1097/00006534-199209000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Posnick JC, Armstrong D, Bite U. Metopic and sagittal synostosis: Intracranial volume measurements prior to and after cranio-orbital reshaping in childhood. Plast Reconstr Surg. 1992;89:299–309. [PubMed] [Google Scholar]
- 10.Kapp-Simon KA, Figueroa A, Jocher CA, Schafer M. Longitudinal assessment of mental development in infants with nonsyndromic craniosynostosis with and without cranial release and reconstruction. Plast Reconstr Surg. 1993;92:831–9. [PubMed] [Google Scholar]
- 11.Camfield PR, Camfield CS. Neurologic aspects of craniosynostosis. In: Cohen MM, editor. Craniosynostosis: Diagnosis, evaluation, and management. New York: Raven; 1986. p. 215. [Google Scholar]
- 12.Turk AE, McCarthy JG, Thorne CH, Wisoff JH. The “back to sleep campaign” and deformational plagiocephaly: Is there cause for concern? J Craniofac Surg. 1996;7:12–8. doi: 10.1097/00001665-199601000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Whitaker LA, Munro IR, Salyer KE. Combined report on problems and complications in 793 craniofacial operations. Plast Reconstr Surg. 1979;64:198. doi: 10.1097/00006534-197908000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Crysdale WS, Kohli-Dang N, Mullind GC, Pullerits J, Munro IR, Burke RC. Airway management in craniofacial surgery: Experience in 542 patients. J Otolaryngol. 1987;16:207–15. [PubMed] [Google Scholar]
- 15.Prevot M, Renier D, Marchac D. Lack of ossification after cranioplasty for craniosynostosis: A review of relevant factors in 592 consecutive patients. J Craniomaxillofac Surg. 1976;4:131. [PubMed] [Google Scholar]
- 16.Persing JA, Posnick J, Magee S, Spinelli HM, Wolfe SA, Munro I, et al. Cranial plate and screw fixation in infancy: An assessment of risk. J Craniofac Surg. 1996;7:267–70. doi: 10.1097/00001665-199607000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Hansen M, Mulliken JE. Frontal plagiocephaly: Diagnosis and treatment. Clin Plast Surg. 1994;21:547. [PubMed] [Google Scholar]