

Abstract
Ewing's sarcoma is a malignant, small, round cell neoplasm that normally affects the long bones of the limbs or the pelvis. It is a relatively frequent malignant bone tumor in children. The occurrence of the primary tumor in the facial region is approximately 2%, with most of the cases affecting the mandible. Primary zygoma involvement is rare and as per the available literature only three cases have been reported.
A case of Ewing's sarcoma that originated in the zygoma of a 15-year-old girl in reported. Bearing in mind its neuroectodermal origin, immunohistochemical studies are essential to distinguish Ewing's sarcoma from other small round cell tumors.
Keywords: Ewing's sarcoma, primitive neuroectodermal tumor, zygoma
INTRODUCTION
Ewing's sarcoma is a highly malignant, small round cell bone tumor that was first described by James Ewing in 1921.[1] An extraskeletal form of this tumor has been described by Angervall and Enzinger in 1975 and termed as Ewing's sarcoma of soft tissue.[2]
The Ewing's sarcoma is a part of the Ewing's sarcoma family of tumors (ESFT), which includes Ewing's sarcoma, peripheral primitive neuroectodermal tumor, neuroepithelioma, and Askin tumor (tumor of the chest wall).[3,4] ESFT are thought to be derived from cells of the neural crest and are found to contain the same reciprocal translocation between chromosomes 11 and 22, t(11;22).[3]
Ewing's sarcoma represents 4–7% of all primary bone tumors and is the second most common malignant bone tumor in children, surpassed only by osteosarcoma.[5,6] The lesion usually involves the diaphyses of the long bones, but ribs and pelvis may be affected as well.[1,5] Only 2–3% of the lesions are seen in the head and neck region.[1,6]
Here, a case of Ewing's sarcoma primarily affecting the zygoma in a 15-year-old girl is discussed. This will add to the previously reported three cases of Ewing's sarcoma in the zygoma.
CASE REPORT
A 15-year-old girl reported to the Department of Oral Pathology and Microbiology, in January 2007, with the complaint of a painless, slow growing swelling on the right side of her face, which she had first noticed one month ago. The general condition of the patient was good on initial clinical examination.
The physical examination showed an immobile, nontender mass, approximately 4 × 3.5cm, in the right zygomatic region extending up to the lateral canthus of the right eye. The ocular mobility was preserved and no other ophthalmic manifestations were present. The borders of the swelling were poorly defined. The consistency of the swelling was relatively soft below the lateral canthus of the eye and hard elsewhere. On intraoral examination, no findings of clinical interest were seen. There was no regional lymphatic involvement.
On a computed tomography scan an expansile, lytic lesion, measuring 4 × 3 × 2.5cm was seen, which involved the right zygomatic bone and lateral wall of the right orbit. The lesion had a heterogeneous matrix with soft tissue density. It had resulted in cortical thinning with discontinuity of the cortex at multiple sites [Figure 1]. There was displacement of the lateral rectus muscle medially and compression of the right eyeball. The rest of the orbital structures were normal.
Figure 1.
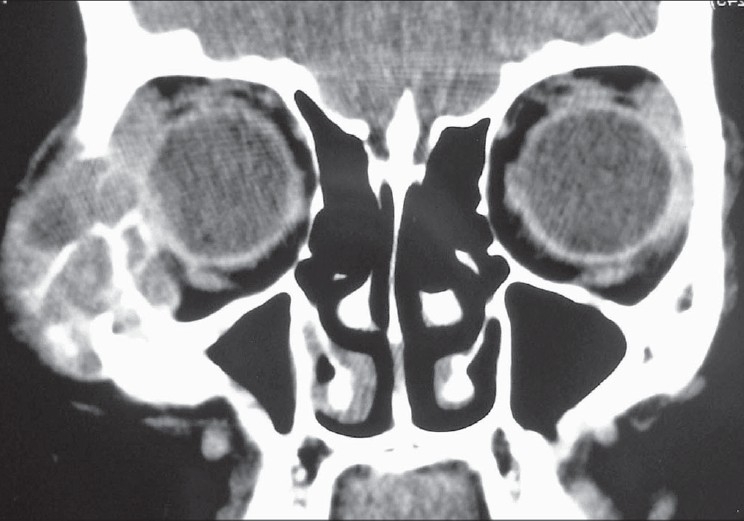
An expansile, lytic mass involving right zygoma and lateral wall of right orbit (Contrast enhancing computed tomography)
A complete haemogram, chest radiograph, and ultrasonography of the abdomen revealed no abnormal findings. Fine needle aspiration cytology (FNAC) of the lesion showed few clusters of pleomorphic round cells with mostly round nuclei, irregular nuclear chromatin, and nucleolus at places. Cells showed scanty amount of cytoplasm with nuclear pleomorphism [Figure 2]. The overall impression was suggestive of round cell malignancy.
Figure 2.
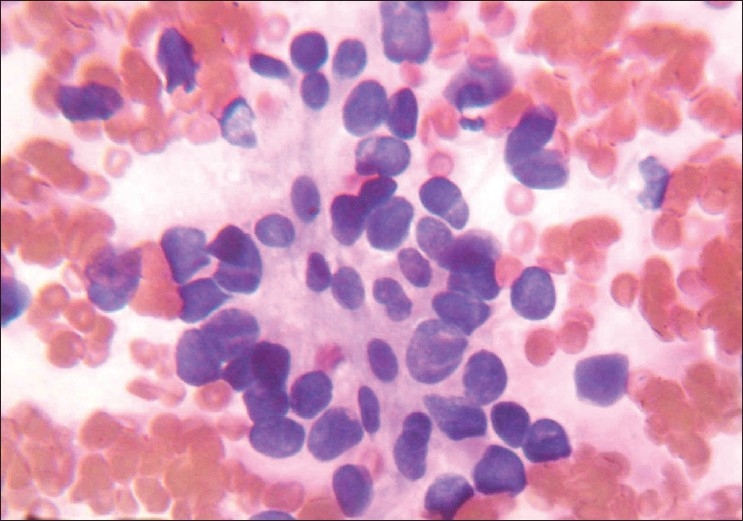
Cytological smear showing pleomorphic round cells with pleomorphic nuclei and scanty cytoplasm (PAP stain, 1000×)
Considering the size of the lesion and to avoid repeated surgical trauma, the surgeons decided to excise the lesion with wide surgical margins without performing incisional biopsy. The gross specimen was of size 5 × 4 × 3 cm, irregular in shape and reddish-white in color with areas of hemorrhage. Internally it showed irregular locules, which contained soft, yellowish-white material.
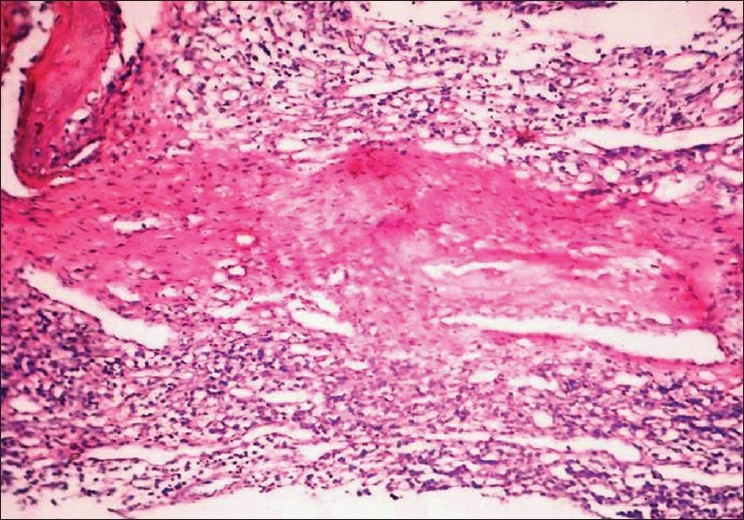
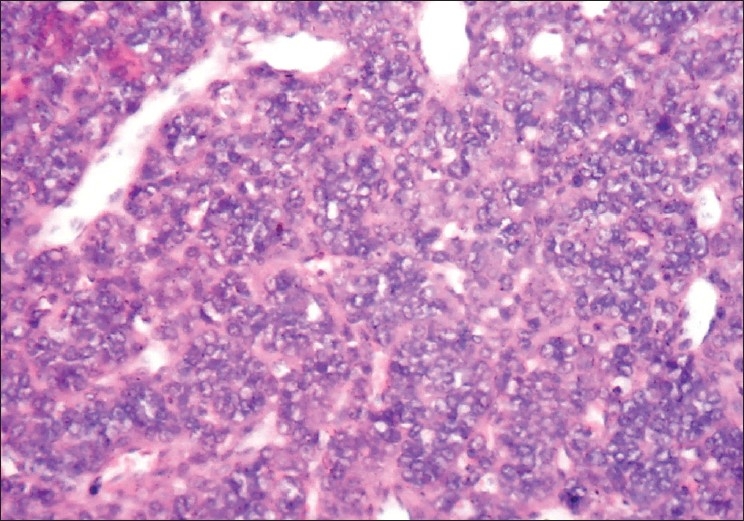
The microscopic examination of the H and E stained tissue sections showed sheets of pleomorphic round cells with scanty cytoplasm and a poorly defined cellular boundary. The nuclei were large, vesicular, and almost occupying the cell [Figure 3]. Thin connective tissue septa with capillaries were seen in between sheets of round cells [Figure 4]. The section of the peripheral tumor tissue showed connective tissue with few inflammatory cells, some bony spicules, infiltrated by small, round neoplastic cells [Figure 5]. This histopathological picture was suggestive of round cell malignancy, probably Ewing's sarcoma. For confirmation of the diagnosis, immunohistochemistry was done. The neoplastic cells were immunopositive for vimentin and MIC-2 [Figure 6, Figure 7]. On the basis of clinical, radiological, and histopathological features, and immunohistochemistry, the diagnosis of “Ewing's sarcoma of the zygoma” was rendered.
Figure 3.
Round blue tumor cells infi ltrating the peripheral tissue with few infl ammatory cells. A bony spicule is seen on the top left side (H and E, 40×)
Figure 4.
Small sheets of round blue tumor cells with thin connective tissue septa and capillaries in between (H and E, 100×)
Figure 5.
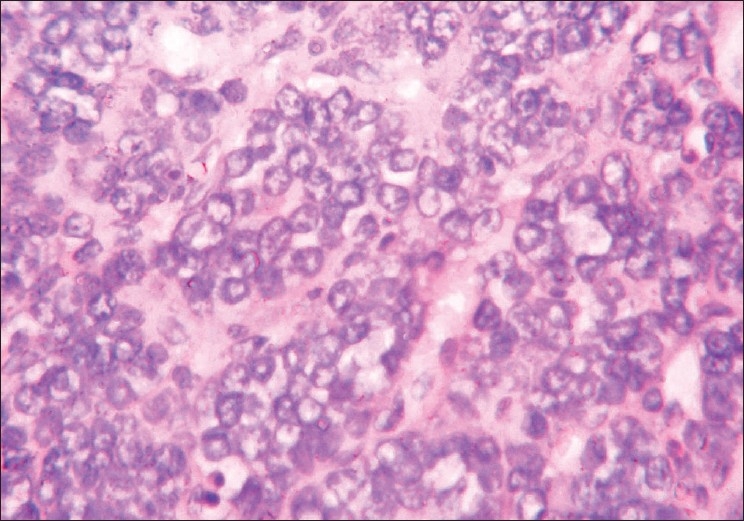
High power field demonstrates pleomorphic round cells with large vesicular nuclei and scanty cytoplasm (H and E, 400×)
Figure 6.
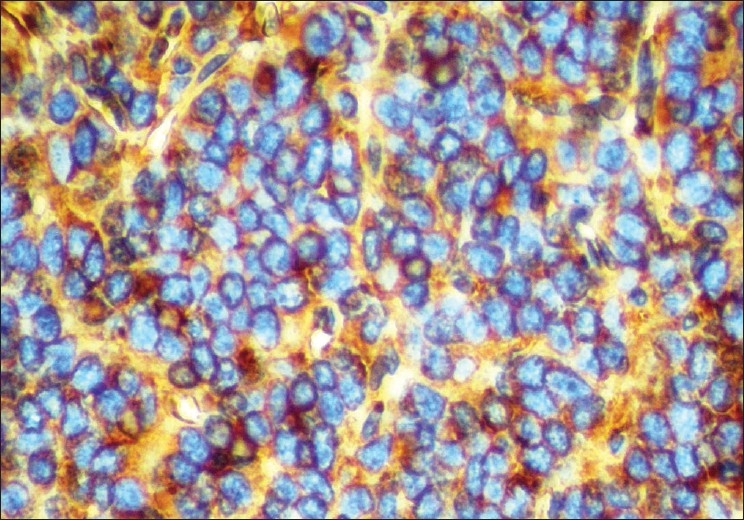
Round cells showing immunopositivity for vimentin (IHC, 400×)
Figure 7.
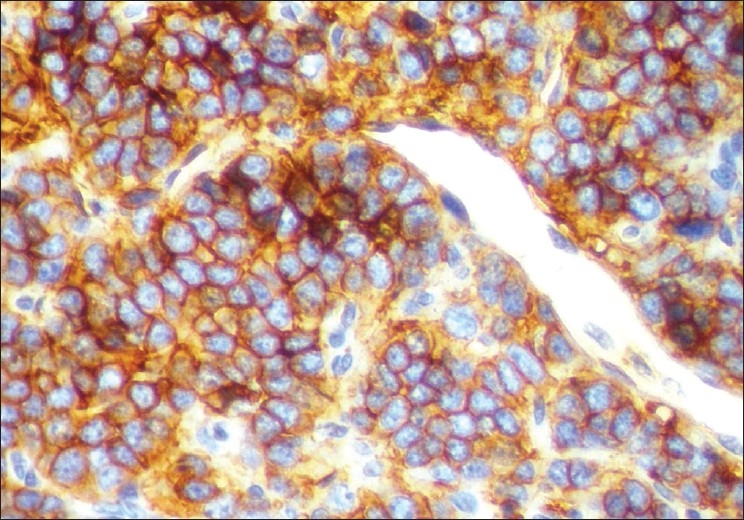
Immunohistochemical expression of MIC-2 in cytoplasm of tumor cells (IHC, MIC-2 400×)
The patient was followed up for the period of six months after surgery and no recurrence was noted. After that, unfortunately, the patient was not able to report for a follow-up.
DISCUSSION
Ewing's sarcoma is an uncommon malignant neoplasm, which occurs as a primary destructive lesion of the bone. The exact origin of the cells of Ewing's sarcoma has been questioned ever since the original description of the tumor by Ewing. He believed that the cells were derived from marrow endothelium, while some authors insisted that the tumor arose from undifferentiated cells of the reticuloendothelial system.[2] In recent times, Ewing's sarcoma has been thought to be neuroectodermal in origin and derived from cells of the neural crest.[1,3]
An osseous and extraosseous subtype of Ewing's sarcoma exists. The osseous type accounts for most cases of Ewing's sarcoma and it has a predilection for the long bones (i.e., femur, tibia, humerus) and pelvic girdle.[6] Only 2–3% of the cases of osseous Ewing's sarcoma arise in the head and neck region, where the calvaria and the mandible are most frequently affected.[1,6] Primary maxillary involvement is rare, having appeared in the literature in about 30 cases until now.[1] Primary involvement isolated to zygoma is even rarer with only three cases reported.[1,5] The extraosseous type accounts for 4–7% of all cases of Ewing's sarcoma and it most commonly affects the thigh, pelvis, and paravertebral soft tissues.[6]
Most Ewing's sarcomas occur predominantly between the age of five and 25 years; the median age of occurrence is 13 years.[7] Overall, a slight male predominance exists, but in the head and neck region the sex distribution is equal.[6] In the present case, the demographic data matches that described in the literature, with the lesion seen in a young girl, 15 years of age.
Radiological studies characteristically demonstrate an osteolytic lesion, and may show bone expansion and erosion of cortical bone; however, none of these findings is specific to Ewing's sarcoma.[6] The characteristic periosteal “onion-skin” appearance, seen in the long bones, rarely appears in the bones of the skull.[1] Osteophyte formation may also be visible on the roentgenogram constituting the “sun-ray” image, as seen in osteosarcoma.[7] The CT scan is considered superior to plain radiography for demonstrating the disease extent.[1,6]
Considering the age of the patient and clinical presentation, other malignant and proliferative processes should also be given consideration. Among these the diagnosis of eosinophilic granuloma was more probable followed by hemangioma.[5] The diagnosis of these lesions is established without significant difficulty. Other much rarer tumors of zygoma, such as, chondrosarcoma, lymphoma, and giant cell tumor have been considered as well. The precise diagnosis of these tumors is possible only after histological examination of the tumor tissue.[5]
Ewing's sarcoma belongs to a group of neoplasm called “small round cell tumors”, which are typically seen in childhood and adolescence.[1] Histologically, Ewing's sarcoma is a highly cellular lesion composed of narrow sheets of small, round, and densely packed cells with scanty stroma. Cells show uniformly appearing, small, round-to-oval, hyperchromatic nuclei, and scanty cytoplasm; thus, it is designated as a “small round blue cell tumor”.[1,6] The cytoplasmic border of cells is poorly defined. The cells are positive for glycogen and are diastase resistant.[7] Many tiny vascular channels and hemorrhagic areas may be seen. On electron microscopy, neurosecretory granules and neurite-like cytoplasmic processes can be recognized.[4] These are the features of neural differentiation which further emphasize the neural crest origin of the tumor cells.
Results of immunohistochemical staining in Ewing's sarcoma are positive with vimentin, MIC-2, and neuron-specific enolase; they are negative with muscle-specific actin, myogenin, and desmin.[6] Immunohistochemical staining, especially with MIC-2 (CD-99) confirms the diagnosis.[1] The MIC2 gene is expressed in more than 90% of Ewing's sarcomas, as are the MIC2 gene glycoprotein product p30–32 and the CD99 antigen.[6] In the present case, the neoplastic cells express immunopositivity for vimentin and MIC-2.
Ewing's sarcoma must be histologically differentiated from other tumors that also have a small, round, blue cell appearance, in particular, lymphoma, rhabdomyosarcoma and neuroblastoma.[1,6] It is not easy to distinguish Ewing's sarcoma from these lesions by focusing only on histological studies. However, immunohistochemistry improves the diagnostic certainty as none of these tumors express positive reaction with MIC-2 antibody.[6] Other tumors that have to be differentiated from Ewing's sarcoma are reticulum cell sarcoma, small cell osteosarcoma, and mesenchymal chondrosarcoma.[7]
The lesion that is very difficult to distinguish from Ewing's sarcoma is primitive neuroectodermal tumor (PNET). Both lesions are thought to be closely related members of the same family of tumors. It is likely that they actually represent the same tumor type, but with varying levels of differentiation. Histologically, there are differences between these two tumors. PNET tends to represent a somewhat more differentiated end of the tumor spectrum. The cells show eosinophilic cytoplasm, coarse chromatin, and prominent nucleoli. Homer-Wright rosettes containing a central core of neurofibrillary material are common, as are Flexner-Wintersteiner rosettes containing a central lumen. Approximately 40% of the cases are PAS-positive. Ewing's sarcoma represents the poorly differentiated end of the spectrum. The cells show scanty pale cytoplasm, fine dusty chromatin, and inconspicuous nucleoli. These cells also contain glycogen, are generally PAS positive and have fewer and less distinctive Homer-Wright rosettes than PNET.[4,8]
The t(11;22)(q24;q12) translocation and MIC2 gene are also expressed in PNET; these findings suggest that Ewing's sarcoma is histogenetically related to PNET.[4] The distinction between Ewing's sarcoma and PNET is more difficult because of the lack of immunohistochemical markers that can be used to distinguish the two.[6]
Cytogenetic studies in Ewing's sarcoma reveal a typicalEws-Fli-1fusion transcript in more than 90% of the cases.[3,4,6] Ewing's sarcoma carries chromosomal translocations - t (11; 22) (q24; q12), which result in the fusion of the Fli-1 gene on 11q24 and the Ews gene on 22q12.[3,4] The fusion gene is designated as EWS/FL-1(t (11; 22) (q24q12)) and the monoclonal antibodies to the fusion gene protein product are termed CD-99.[7] This is the most specific feature for the diagnosis of Ewing's sarcoma.
The tumors in the ESFT are treated in a similar way, that is, on the basis of their clinical presentation (e.g., metastatic or localized) rather than on their histological subtype.[3] Multimodality therapy for Ewing's sarcoma is associated with markedly improved survival rate. Surgery followed by adjuvant radiotherapy and chemotherapy dramatically improves the survival rate as compared to a single or even dual modality therapy.[6] The use of radiotherapy is debatable, as it promotes the development of secondary malignancies and disorders of growth on the facial skeleton in young patients.[1,6] On the other hand, local surgical excision is strongly recommended in all completely resectable lesions followed by postoperative chemotherapy.[1,5] Radiotherapy should be reserved for initially unresectable lesions or residual disease.[1] In the present case, the lesion was easily accessible, and hence, resected completely with safety margins. Subsequent chemotherapy was not given, as patient was unable to report back for recall.
The prognosis appears to be dependent on the location of the primary tumor and the presence of distant metastasis. Survival rate in patients with Ewing's sarcoma of the head and neck are significantly better than those of patients with tumor in other locations.[6] The five-year survival rate for patients with extraosseous Ewing's sarcoma is approximately 50–70%.[1] Metastasis is present in fewer than 20% of the cases and primarily involves the lungs, but can also be seen in other bones and lymph nodes.[6,7] Local recurrence has also been reported.
CONCLUSION
A case of Ewing's sarcoma in the zygoma of a young girl has been reported. Primary Ewing's sarcoma of the maxilla and zygoma constitutes a diagnostic challenge, due to its rarity, scarcity of relevant symptoms, atypical characteristics, and absence of pathognomic radiographic signs. An immunohistochemical study is the key to specific diagnosis. Though rare, the lesion should be considered in the differential diagnosis of round cell tumors of the face, in patients under 20 years of age, showing bone destruction in the maxilla and zygoma. A multimodal treatment approach and strict follow-up is essential for the successful management of Ewing's sarcoma.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Infante-Cossio P, Gutierrez-Perez JL, Garcia-Perla A, Noguer-Mediavilla M, Gavilan-Carrasco F. Primary Ewing's sarcoma of the maxilla and zygoma: Report of a case. J Oral Maxillofac Surg. 2005;63:1539–42. doi: 10.1016/j.joms.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 2.Shafer WG, Hine MK, Levy BM, Tomich CE. 4th ed. WB Saunders: Philadelphia; 1983. A textbook of oral pathology. [Google Scholar]
- 3.Toretsky JA. Ewing sarcoma and primitive neuroectodermal tumours. [last updated on 2008 Jun 17]. Available from: http://www.emedicine.com/ped/topic2589.htm .
- 4.Jayakumar S, Power D. Ewing's sarcoma / PNET:A histopathological review. Internet J Orthop Surg. 2006;3:1–9. [Google Scholar]
- 5.Postovsky S, Daitzchman M, Elhasid R, Arush MW. Ewing's sarcoma of the facial zygomatic area bones in a young child: A case presentation. Am J Otolaryngol. 2000;21:213–5. doi: 10.1016/s0196-0709(00)85027-9. [DOI] [PubMed] [Google Scholar]
- 6.McMains KC, Gourin CG. Pathology - sarcomas of the head and neck. [last updated on 2007 Oct 16]. Available from: http://www.emedicine.com/ent/topic675.htm .
- 7.Neville BW, Damm DD, Allen CA, Bouquot JE. 2nd ed. Philadelphia: WB Saunders; Oral and maxillofacial pathology. [Google Scholar]
- 8.Votta TJ, Fantuzzo JJ, Boyd BC. Peripheral primitive neuroectodermal tumour associated with the anterior mandible: A case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:592–7. doi: 10.1016/j.tripleo.2005.03.015. [DOI] [PubMed] [Google Scholar]