

Abstract
Protein kinase C (PKC) is a family of kinases that are implicated in a plethora of diseases, including cancer and cardiovascular disease. PKC isoforms can have different, and sometimes opposing, effects in these disease states. Toll-like receptors (TLRs) are a family of pattern recognition receptors that bind pathogens and stimulate the secretion of cytokines. It has long been known that PKC inhibitors reduce LPS-stimulated cytokine secretion by macrophages, linking PKC activation to TLR signaling. Recent studies have shown that PKC-α, -δ, -ε, and -ζ are directly involved in multiple steps in TLR pathways. They associate with the TLR or proximal components of the receptor complex. These isoforms are also involved in the downstream activation of MAPK, RhoA, TAK1, and NF-κB. Thus, PKC activation is intimately involved in TLR signaling and the innate immune response.
1. Introduction
Protein kinase C (PKC) is a family of protein serine/threonine kinases centrally involved in intracellular signal transduction. The PKC isoforms are divided into 3 subfamilies based on their activation requirements: the conventional isoforms, PKC-α, -βI, -βII, and -γ, require calcium, diacylglycerol, and phosphatidylserine; the novel isoforms, PKC-δ, -ε, -η, and –θ, require diacylglycerol and phosphatidylserine but are calcium independent; the atypical isoforms, PKC-ζ and λ/ι, require only phosphatidylserine [1]. Different isoforms of PKC are involved in such pivotal functions as cell growth, differentiation, apoptosis, motility, and secretion. Accordingly, these enzymes have been implicated in many disease states including cancer and cardiovascular disease [2–6]. The role of PKC in cancer is complicated by the tissue-specific, and often opposing, effects of the different isoforms on cell cycle and apoptosis. Similarly, the role of PKC in heart disease is complex because components of the disease (myocyte hypertrophy, cardiac function, fibrosis, and inflammation) are influenced in different ways by the different isoforms.
Toll-like receptors (TLRs) are a family of pattern recognition receptors that are critical for the effective innate immune response to infection [7–9]. Signaling from different TLRs varies but is initiated by the recruitment of TIR-containing adaptor proteins (e.g., TIRAP recruits MyD88, TRAM recruits TRIF). MyD88 recruits IRAK4, IRAK1, IRAK2, and TRAF6. Phosphorylation and degradation of IRAK1 releases this complex into the cytoplasm where it binds and activates TAK1 downstream. Through an as yet unknown, and possibly indirect, mechanism, TAK1 activates the IKKβ → IkB-α → NF-κB pathway for induction of proinflammatory genes. TAK1 also activates the MAPK cascades that influence gene expression. TLR binding of TRIF recruits TRAF6, βRIP1, and TAK1 for activation of MAPK, IRF3, NF-κB, and transcription of interferon-β. The TRIF pathway also stimulates the secretion of proinflammatory cytokines although to a lesser degree than the MyD88 pathway.
Initial evidence for the involvement of PKC in TLR signaling came from observations that altering PKC activity in cells of the innate immune system affected cytokine secretion. Subsequently, LPS and other TLR ligands were shown to activate most of the PKC isoforms expressed in monocytes, macrophages, dendritic cells, and neutrophils [10–14]. A large number of studies have shown that pharmacological inhibition of PKC, or its depletion by long-term treatment with phorbol esters, decreases LPS-stimulated cytokine secretion [10, 15–17]. Accordingly, acute activation of PKC with phorbol esters increases cytokine secretion [11, 15, 17, 18].
Results from studies performed over the last decade have identified four PKC isoforms that impact different steps in TLR signaling. This paper will summarize recent advances detailing the role of PKC-α, -δ, -ε, and -ζ in activation of the initial TLR signaling complex, activation of RhoA, and transcription factors (Figure 1).
Figure 1.
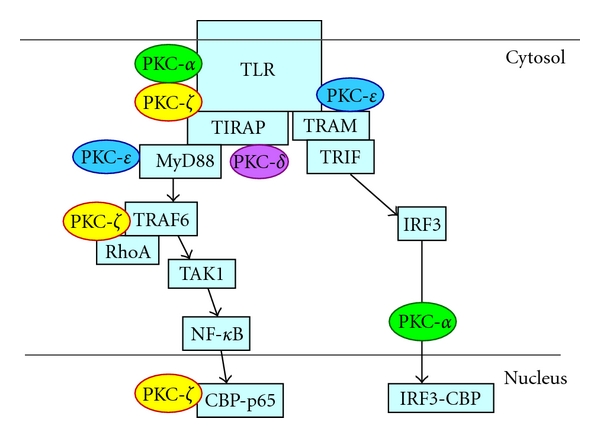
PKC isoforms act at many levels in TLR signaling. PKC-α associates with the TLR2 signaling complex in a MyD88-dependent manner. PKC-α is also required for TLR3-mediated IRF-3 binding to CBP and IFN-β gene induction. Interaction of PKC-δ with TIRAP is required for the downstream activation of NF-κB. PKC-ε associates with complexes of MyD88 and TLR2 or TLR4 and is necessary for downstream signaling. PKC-ε is also required for the phosphorylation of TRAM. PKC-ζ is activated upon ligation of TLR2 or TLR4. During signaling by these receptors, active PKC-ζ binds to TLR2, associates with TRAF6 and RhoA, and is required for full transcriptional activation of p65.
2. PKC Isoforms Involved in TLR Signaling and Host Defense
2.1. PKC-α
Conventional PKC isoforms have been implicated in cytokine secretion by several studies showing that Gö6976, which inhibits PKC-α and β, blocks TLR-stimulated cytokine secretion by macrophages [14, 19–22]. A series of studies found that 264.7 RAW macrophages expressing a dominant negative (DN) PKC-α have reduced LPS-stimulated TNF-α, IL-1β, iNOS, and NF-IL6 (CAAT/enhancer-binding protein β) induction [23, 24]. These cells also had defects in phagocytosis, phagosome maturation, and killing of intracellular pathogens, suggesting that PKC-α is also involved in other, non-TLR-mediated aspects of innate immunity [25–28]. A further link between PKC-α and TLR-mediated responses is the finding that PPARγ modulates phorbol ester-induced NF-κB activation and TNF-α secretion by preventing the activation of PKC-α [18].
Findings by Langlet et al. [29] revealed that, in human DC, PKC-α inhibition blocked IL-12p40 secretion induced by TLR2/6, TLR 2/1, TLR5, and IL-1R, but not TLR3. The role of PKC-α was dissected using Gö6976, expression of DN PKC-α, and DC from PKC-α −/− mice. Using these approaches, it was determined that activation of PKC-α is required for TLR2/1-mediated activation of MAPK, NF-κB, and AP-1 as well as secretion of TNF-α, IL-6, and IL-10 by DC. That immunoprecipitation of PKC-α from TLR2/1-activated DC captured TLR2 provides additional evidence that PKC-α is linked to TLR2 signaling. As TLR2 was not found in PKC-α immunoprecipitates of cells from MyD88−/− mice suggests that MyD88 links TLR2 with PKC-α.
Johnson et al. [30] found that poly (I : C), a TLR3 ligand, activated PKC-α. Downregulation of PKC-α with siRNA, or expression of DN PKC-α, blocked TLR3-stimulated IFN-β production in dendritic cells (DCs). Interfering with PKC-α activity did not change the activation of IRF3 in terms of phosphorylation, dimerization, nuclear translocation, or DNA binding but did inhibit IRF-3 transcriptional activity induced by TRIF and TBK1 overexpression. This latter effect is due to decreased IRF-3 binding to the coactivator, CREB binding protein (CBP) which requires PKC-α activation.
2.2. PKC-δ
Several studies have implicated PKC-δ in TLR-mediated cytokine secretion [12, 31–34]. Inhibition of PKC-δ with Rottlerin, or its downregulation, consistently decreases activation of NF-κB, secretion of inflammatory cytokines, and production of nitric oxide by cells of the innate immune system. Results from Kubo-Murai et al. [35] suggest that PKC-δ is involved in TLR signaling through its interaction with TIRAP. Specifically, PKC-δ in macrophage lysates bound to immobilized TIRAP via the TIR domain of TIRAP. Additionally, PKC-δ depletion results in the loss of kinase activity in immobilized TIRAP implicating PKC-δ as the relevant kinase for propagating signaling from the TIRAP complex. That downregulation of PKC-δ severely depresses the activation of p38 MAPK and NF-κB by ligands for TLR4, and TLR2 underscores its importance in signaling via these receptors. To date, this is the only report demonstrating a direct role for PKC-δ in TLR signaling.
Cecal ligation and puncture (CLP) is an established animal model for sepsis and has been used to study TLR signaling in vivo. Recently, it was shown that, when administered intratracheally, a PKC-δ-inhibiting peptide reduced the lung injury associated with CLP-induced sepsis in rats [36]. This inhibitor blocked sepsis-induced phosphorylation of PKC-δ. Animals given the inhibitor prior to CLP had reduced levels of the chemokines CINC-1 and MIP-2 in lung lavage and blood samples. At the same time, there was reduced infiltration of inflammatory cells into the lungs and less pulmonary edema. The authors linked the protective effects of the inhibitor to reduced NF-κB activation and chemokine production by macrophages, endothelial cells, and epithelial cells [37–39]. This is consistent with a previous study showing that PKC-δ −/− mice had reduced cytokine production, neutrophil infiltration, and lung injury due to asbestos [40].
Sphingosine kinase 1 (SphK1) has been implicated in inflammation through the formation of sphingosine 1 phosphate [41–44]. SphK1 is induced and activated by several immune stimuli including LPS. In LPS-treated macrophages, PKC-δ lies downstream of SphK1 activation and is required for the activation of NF-κB [37]. Furthermore, the inhibition of SphK1 in vivo reduced LPS- and sepsis-stimulated cytokine secretion and mortality. These studies support a TLR4→SphK1→PKC-δ→NF-κB→cytokine pathway. It remains to be determined if the same pathway links SphK1 to TNFR1 and IL-1R [43, 44].
Beyond the scope of this paper are the effects of this PKC isoform in several TLR-independent aspects of inflammation including signaling via TNFR1, neutrophil activation, and endothelial cell function [43–48].
2.3. PKC-ε
A role for PKC-ε in host defense was apparent when Castrillo et al. [49] observed that PKC-ε −/− mice were difficult to breed due to infections of the uterus. Additionally, these animals had a 2- to 3-fold reduction in survival time after infection with Gram-negative or Gram-positive bacteria. Several aspects of the immune system in these animals were normal although macrophages were defective in the production of LPS-stimulated TNF-α, IL-1β, PGE2, and nitric oxide. There were also deficits in LPS-stimulated MAPK and NF-κB activation. Other studies, using PKC-ε-specific inhibitors, depletion with antisense oligonucleotides, and macrophages from PKC-ε −/− mice, verified that PKC-ε is critical for LPS-stimulated TNF-α and IL-12 secretion by DC and macrophages [10, 50, 51]. In vivo administration of a PKC-ε inhibitor reduced the inflammation associated with a murine model of cardiac transplantation [52]. Thus, PKC-ε has a likely role in inflammation and host defense [53, 54].
More recent studies have sought to clarify the role of PKC-ε in TLR signaling. PKC-ε is phosphorylated by all TLRs that signal through MyD88, that is, TLR1 through 9 except TLR3 in macrophages [55]. Upon TLR4 ligation with LPS, PKC-ε is phosphorylated on Ser-346 and 368 and binds to 14-3-3β. Association with 14-3-3β requires the presence of MyD88. Phosphorylation of these serines is critical for PKC-ε signaling as cells expressing PKC-ε S346A/S368A fail to activate an NK-κB reporter in response to ligands for TLR4 and TLR2. These findings suggest that the complex of TLR, MyD88, 14-3-3β, and PKC-ε is required for gene induction. Of note, since PKC-ε can be phosphorylated by PKC-α, it is possible that the effects of PKC-α on cytokine secretion are shared by PKC-ε [56, 57].
In addition to its role in MyD88-dependent signaling, PKC-ε is also involved in TLR4 activation via the TRAM pathway. The TRAM pathway primarily stimulates the production of IFN-β and RANTES. Phosphorylation of TRAM in LPS-stimulated macrophages allows it to dissociate from the membrane and bridge TLR4 with TRIF. McGettrick et al. [11] found that recombinant PKC-ε, but not PKC-ζ, phosphorylates TRAM on Ser-16, and TRAM−/− macrophages reconstituted with TRAM S16A do not signal. That this phosphorylation of TRAM did not occur in PKC-ε −/− cells and that macrophages from PKC-ε −/− mice have reduced production of IFN-β places PKC-ε in the TLR4→ PKC-ε → Ser-16 TRAM→ IFN-β pathway.
2.4. PKC-ζ
This atypical PKC isoform is a component of the signaling pathways for IL-1R and TNFR [58–60]. More recently, PKC-ζ has been shown to be involved in the activation of TLR, IRAK, RhoA, and NF-κB [61–64].
PKC-ζ is downstream of TLR2 in human macrophages stimulated with Mycobacterium tuberculosis (MTB) [64]. Specific inhibitors of PKC-ζ or its downregulation blocked ERK activation and TNF-α secretion stimulated by MTB. PKC-ζ was also found to associate with immunoprecipitated TLR2 but not TLR4 after stimulation with MTB or peptidoglycan. Finally, PKC-ζ did not associate with TLR2 in THP-1 cells expressing DN PKC-ζ. These results suggest a TLR2 → PKC-ζ→ ERK pathway for production of TNF-α in response to MTB.
IRAK phosphorylation is an early event in TLR signaling. Phosphorylated IRAK is subsequently degraded which acts as a negative feedback control on the signaling pathway. Using a panel of protein kinase inhibitors, Hu et al. [61] demonstrated that the phosphorylation of IRAK by TLR4 is mediated by PKC in THP-1 cells. PKC-ζ was found in IRAK immunoprecipitates from LPS-stimulated cells. Furthermore, macrophages from wild-type mice responded to LPS with activation of PKC-ζ and phosphorylation of IRAK, but macrophages from C3H/HeJ mice that have a nonfunctional TLR4 did not. Although not yet directly tested, these results suggest that IRAK is a PKC-ζ substrate.
Several studies have highlighted a role for RhoA in TLR signaling [65–69]. Two papers have directly implicated PKC-ζ in TLR2 and TLR4 signaling through the activation of RhoA and subsequent activation of NF-κB [62, 63]. Teusch et al. [63] found that full transcriptional activation of p65 by TLR2 ligands in monocytic cells requires the activity of both RhoA and PKC-ζ. Inhibition of PKC-ζ, or expression of its dominant negative mutant, reduces the phosphorylation of p65 on serine 311 and its transcriptional activity. PKC-ζ transiently associates with RhoA with a time course similar to that of RhoA activation. The authors concluded that PKC-ζ mediates at least part of the effects of RhoA on gene transcription.
Further refinement of the position of PKC-ζ in the TLR→ RhoA → NF-κB pathway has been elucidated by the studies of Huang et al. [62]. They reported that treatment of macrophages with LPS activates PKC-ζ and that interfering with PKC-ζ activation blocks LPS-stimulated activation of NF-κB and cytokine production. As in the TLR2 studies above, PKC-ζ was present in anti-RhoA or anti-TRAF6 immunoprecipitates of cell lysates from LPS-stimulated macrophages. Inhibition of RhoA or TRAF6 blocked PKC-ζ activation, placing PKC-ζ downstream of RhoA and TRAF6. Similar experiments place TAK1 between PKC-ζ and NF-κB. That is, inhibition of PKC-ζ blocked TAK1 phosphorylation, and constitutively active PKC-ζ fails to activate NF-κB in TAK1-depleted cells. Together, these studies are consistent with the following model: TLR2/4→ RhoA/TRAF6→ PKC-ζ→ TAK1 → s311 p65 → cytokine induction.
Cigarette smoke is a major cause of lung inflammation which is exacerbated by its common contamination with LPS. Yao et al. [70] recently demonstrated that PKC-ζ −/− mice exposed to smoke/LPS had less lung inflammation than wild-type mice. Bronchoalveolar cells from mice exposed to smoke/LPS had activated PKC-ζ that translocated to the nucleus in association with p65 and CBP. This was associated with the phosphorylation (Ser311) of p65 and induction of cytokines. All of these events were reduced in PKC-ζ −/− mice.
Surfactant protein A (SP-A) limits several aspects of inflammation in the lung, partly due to stabilization of IkB-α [71]. Moulakakis et al. [72] found that PKC-ζ is required for this effect. SP-A activates PKC-ζ, limits its translocation to the nucleus, and stabilizes IkB-α. In alveolar macrophages from PKC-ζ −/− mice, these protective effects were lost. That is, in PKC-ζ −/− macrophages, SP-A failed to inhibit LPS-stimulated IkB-α activation, and NF-κB DNA binding was nearly normal. It was concluded that the effect of SP-A → PKC-ζ axis may function as a brake on the inflammatory response of alveolar macrophages.
3. Perspective
The establishment of PKC in TLR signaling as well as in other inflammatory processes may provide a means to treat inflammatory diseases. Inhibition of a kinase required for so many important biological functions may be expected to result in substantial side effects. However, several inhibitors of PKC have proven to be well tolerated in clinical trials [73–75].
The studies detailed above provide convincing evidence that PKC is directly involved in TLR signaling. In most cases, further studies are needed to determine the nature of the interaction between PKC isoforms and other proteins in the signaling cascades and then to identify the specific PKC substrates. Another important area of investigation is the integration of the TLR→PKC axis with the involvement of these same isoforms (PKC-α, -δ, and -ζ) and possible interaction of the isoforms in non-TLR-dependent aspects of innate immunity. PKC-α may contribute to phagocytosis and pathogen killing [25–28], PKC-δ is involved in signaling via TNFR1, neutrophil activation, and endothelial cell function [43–48], and PKC-ζ participates in signaling through TNFR and IL-1R [58–60]. Thus, PKC isoforms likely sit at different nodes in the signaling web. Understanding their contribution to individual pathways as well as their position in the web is necessary to avoid misinterpreting results of experiments such as one in which a PKC inhibitor appears to reduce the TNF-α response to a TLR ligand, but, in fact, the inhibitor blocked the secondary augmentation of TNF-α secretion mediated through the TNFR. Finally, understanding the role of TLR-mediated activation of specific PKC isoforms will immediately provide insights into the mechanisms of the disease.
One of the more important advances during the 20th century was the alleviation of a great deal of the suffering from infectious diseases. We may hope for a similar advance with regard to inflammatory diseases in the 21st century.
Acknowledgments
The authors acknowledge the secretarial assistance of Wendy Vienneau and funding by NIH GM090325, HL095971 and HL089730 (MRL).
References
- 1.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochemical Journal. 2003;370(2):361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali AS, Ali S, El-Rayes BF, Philip PA, Sarkar FH. Exploitation of protein kinase C: a useful target for cancer therapy. Cancer Treatment Reviews. 2009;35(1):1–8. doi: 10.1016/j.ctrv.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Palaniyandi SS, Sun L, Ferreira JC, Mochly-Rosen D. Protein kinase C in heart failure: a therapeutic target? Cardiovascular Research. 2009;82(2):229–239. doi: 10.1093/cvr/cvp001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonelli A, Mischiati C, Guerrini R, Voltan R, Salvadori S, Zauli G. Perspectives of protein kinase C (PKC) inhibitors as anti-cancer agents. Mini-Reviews in Medicinal Chemistry. 2009;9(4):498–509. doi: 10.2174/138955709787847967. [DOI] [PubMed] [Google Scholar]
- 5.Bynagari-Settipalli YS, Chari R, Kilpatrick L, Kunapuli SP. Protein kinase C—possible therapeutic target to treat cardiovascular diseases. Cardiovascular & Hematological Disorders—Drug Targets. 2010;10(4):292–308. doi: 10.2174/187152910793743869. [DOI] [PubMed] [Google Scholar]
- 6.Churchill E, Budas G, Vallentin A, Koyanagi T, Mochly-Rosen D. PKC isozymes in chronic cardiac disease: possible therapeutic targets? Annual Review of Pharmacology and Toxicology. 2008;48:569–599. doi: 10.1146/annurev.pharmtox.48.121806.154902. [DOI] [PubMed] [Google Scholar]
- 7.Gay NJ, Gangloff M. Structure and function of Toll receptors and their ligands. Annual Review of Biochemistry. 2007;76:141–145. doi: 10.1146/annurev.biochem.76.060305.151318. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature Immunology. 2010;11(5):373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 9.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends in Molecular Medicine. 2007;13(11):460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Fronhofer V, Lennartz MR, Loegering DJ. Role of PKC isoforms in the Fc(gamma)R-mediated inhibition of LPS-stimulated IL-12 secretion by macrophages. Journal of Leukocyte Biology. 2006;79(2):408–415. doi: 10.1189/jlb.0805438. [DOI] [PubMed] [Google Scholar]
- 11.McGettrick AF, Brint EK, Palsson-McDermott EM, et al. Trif-related adapter molecule is phosphorylated by PKC{epsilon} during Toll-like receptor 4 signaling. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(24):9196–9201. doi: 10.1073/pnas.0600462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kontny E, Kurowska M, Szczepanska K, Maslinski W. Rottlerin, a PKC isozyme-selective inhibitor, affects signaling events and cytokine production in human monocytes. Journal of Leukocyte Biology. 2000;67(2):249–258. doi: 10.1002/jlb.67.2.249. [DOI] [PubMed] [Google Scholar]
- 13.Zhou X, Yang W, Li J. Ca2+- and protein kinase C-dependent signaling pathway for nuclear factor-kappaB activation, inducible nitric-oxide synthase expression, and tumor necrosis factor-alpha production in lipopolysaccharide-stimulated rat peritoneal macrophages. The Journal of Biological Chemistry. 2006;281(42):31337–31347. doi: 10.1074/jbc.M602739200. [DOI] [PubMed] [Google Scholar]
- 14.Asehnoune K, Strassheim D, Mitra S, Yeol KJ, Abraham E. Involvement of PKCalpha/beta in TLR4 and TLR2 dependent activation of NF-kappaB. Cellular Signalling. 2005;17(3):385–394. doi: 10.1016/j.cellsig.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 15.West MA, Lemieur T, Clair L, Bellingham J, Rodriguez JL. Protein kinase C regulates macrophage tumor necrosis factor secretion: direct protein kinase C activation restores tumor necrosis factor production in endotoxin tolerance. Surgery. 1997;122(2):204–212. doi: 10.1016/s0039-6060(97)90010-6. [DOI] [PubMed] [Google Scholar]
- 16.Labeta MO, Durieux JJ, Spagnoli G, Fernandez N, Wijdenes J, Herrmann R. CD14 and tolerance to lipopolysaccharide: biochemical and functional analysis. Immunology. 1993;80(3):415–423. [PMC free article] [PubMed] [Google Scholar]
- 17.Cuschieri J, Billigren J, Maier RV. Endotoxin tolerance attenuates LPS-induced TLR4 mobilization to lipid rafts: a condition reversed by PKC activation. Journal of Leukocyte Biology. 2006;80(6):1289–1297. doi: 10.1189/jlb.0106053. [DOI] [PubMed] [Google Scholar]
- 18.von Knethen A, Soller M, Tzieply N, et al. PPARgamma1 attenuates cytosol to membrane translocation of PKCalpha to desensitize monocytes/macrophages. The Journal of Cell Biology. 2007;176(5):681–694. doi: 10.1083/jcb.200605038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foey AD, Brennan FM. Conventional protein kinase C and atypical protein kinase Czeta differentially regulate macrophage production of tumour necrosis factor-alpha and interleukin-10. Immunology. 2004;112(1):44–53. doi: 10.1111/j.1365-2567.2004.01852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Catley MC, Cambridge LM, Nasuhara Y, et al. Inhibitors of protein kinase C (PKC) prevent activated transcription: role of events downstream of NF-kappaB DNA binding. The Journal of Biological Chemistry. 2004;279(18):18457–18466. doi: 10.1074/jbc.M400765200. [DOI] [PubMed] [Google Scholar]
- 21.Foreback JL, Sarma V, Yeager NR, Younkin EM, Remick DG, Ward PA. Blood mononuclear cell production of TNF-alpha and IL-8: engagement of different signal transduction pathways including the p42 MAP kinase pathway. Journal of Leukocyte Biology. 1998;64(1):124–133. doi: 10.1002/jlb.64.1.124. [DOI] [PubMed] [Google Scholar]
- 22.Loegering DJ, Lennartz MR. Signaling pathways for Fc gamma receptor-stimulated tumor necrosis factor-alpha secretion and respiratory burst in RAW 264.7 macrophages. Inflammation. 2004;28(1):23–31. doi: 10.1023/b:ifla.0000014708.87440.45. [DOI] [PubMed] [Google Scholar]
- 23.St Denis A, Chano F, Tremblay P, St Pierre Y, Descoteaux A. Protein kinase C-alpha modulates lipopolysaccharide-induced functions in a murine macrophage cell line. The Journal of Biological Chemistry. 1998;273(49):32787–32792. doi: 10.1074/jbc.273.49.32787. [DOI] [PubMed] [Google Scholar]
- 24.Chano F, Descoteaux A. Modulation of lipopolysaccharide-induced NF-IL6 activation by protein kinase C-alpha in a mouse macrophage cell line. The European Journal of Immunology. 2002;32(10):2897–2904. doi: 10.1002/1521-4141(2002010)32:10<2897::AID-IMMU2897>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 25.St Denis A, Caouras V, Gervais F, Descoteaux A. Role of protein kinase C-alpha in the control of infection by intracellular pathogens in macrophages. Journal of Immunology. 1999;163(10):5505–5511. [PubMed] [Google Scholar]
- 26.Breton A, Descoteaux A. Protein kinase C-alpha participates in FcgammaR-mediated phagocytosis in macrophages. Biochemical and Biophysical Research Communications. 2000;276(2):472–476. doi: 10.1006/bbrc.2000.3511. [DOI] [PubMed] [Google Scholar]
- 27.Ng Yan Hing JD, Desjardins M, Descoteaux A. Proteomic analysis reveals a role for protein kinase C-alpha in phagosome maturation. Biochemical and Biophysical Research Communications. 2004;319(3):810–816. doi: 10.1016/j.bbrc.2004.05.054. [DOI] [PubMed] [Google Scholar]
- 28.Holm A, Tejle K, Gunnarsson T, Magnusson KE, Descoteaux A, Rasmusson B. Role of protein kinase C alpha for uptake of unopsonized prey and phagosomal maturation in macrophages. Biochemical and Biophysical Research Communications. 2003;302(4):653–658. doi: 10.1016/s0006-291x(03)00231-6. [DOI] [PubMed] [Google Scholar]
- 29.Langlet C, Springael C, Johnson J, et al. PKC-alpha controls MYD88-dependent TLR/IL-1R signaling and cytokine production in mouse and human dendritic cells. The European Journal of Immunology. 2010;40(2):505–515. doi: 10.1002/eji.200939391. [DOI] [PubMed] [Google Scholar]
- 30.Johnson J, Albarani V, Nguyen M, Goldman M, Willems F, Aksoy E. Protein kinase Calpha is involved in interferon regulatory factor 3 activation and type I interferon-beta synthesis. The Journal of Biological Chemistry. 2007;282(20):15022–15032. doi: 10.1074/jbc.M700421200. [DOI] [PubMed] [Google Scholar]
- 31.Bhatt KH, Pandey RK, Dahiya Y, Sodhi A. Protein kinase Cdelta and protein tyrosine kinase regulate peptidoglycan-induced nuclear factor-kappaB activation and inducible nitric oxide synthase expression in mouse peritoneal macrophages in vitro. Molecular Immunology. 2010;47(4):861–870. doi: 10.1016/j.molimm.2009.10.029. [DOI] [PubMed] [Google Scholar]
- 32.Kim DC, Kim SH, Jeong MW, Baek NI, Kim KT. Effect of rottlerin, a PKC-delta inhibitor, on TLR-4-dependent activation of murine microglia. Biochemical and Biophysical Research Communications. 2005;337(1):110–115. doi: 10.1016/j.bbrc.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 33.Ikewaki N, Fujii N, Onaka T, Ikewaki S, Inoko H. Immunological actions of Sophy beta-glucan (beta-1,3-1,6 glucan), currently available commercially as a health food supplement. Microbiology and Immunology. 2007;51(9):861–873. doi: 10.1111/j.1348-0421.2007.tb03982.x. [DOI] [PubMed] [Google Scholar]
- 34.Platten M, Eitel K, Wischhusen J, Dichgans J, Weller M. Involvement of protein kinase Cdelta and extracellular signal-regulated kinase-2 in the suppression of microglial inducible nitric oxide synthase expression by N-[3,4-dimethoxycinnamoyl]-anthranilic acid (tranilast) Biochemical Pharmacology. 2003;66(7):1263–1270. doi: 10.1016/s0006-2952(03)00449-0. [DOI] [PubMed] [Google Scholar]
- 35.Kubo-Murai M, Hazeki K, Sukenobu N, et al. Protein kinase Cdelta binds TIRAP/Mal to participate in TLR signaling. Molecular Immunology. 2007;44(9):2257–2264. doi: 10.1016/j.molimm.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 36.Kilpatrick LE, Standage SW, Li H, et al. Protection against sepsis-induced lung injury by selective inhibition of protein kinase C-{delta} ({delta}-PKC) Journal of Leukocyte Biology. 2011;89(1):3–10. doi: 10.1189/jlb.0510281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puneet P, Yap CT, Wong L, et al. SphK1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science. 2010;328(5983):1290–1294. doi: 10.1126/science.1188635. [DOI] [PubMed] [Google Scholar]
- 38.Kilpatrick LE, Lee JY, Haines KM, Campbell DE, Sullivan KE, Korchak HM. A role for PKC-delta and PI 3-kinase in TNF-alpha-mediated antiapoptotic signaling in the human neutrophil. The American Journal of Physiology—Cell Physiology. 2002;283(1):C48–C57. doi: 10.1152/ajpcell.00385.2001. [DOI] [PubMed] [Google Scholar]
- 39.Cummings R, Zhao Y, Jacoby D, et al. Protein kinase Cdelta mediates lysophosphatidic acid-induced NF-kappaB activation and interleukin-8 secretion in human bronchial epithelial cells. The Journal of Biological Chemistry. 2004;279(39):41085–41094. doi: 10.1074/jbc.M404045200. [DOI] [PubMed] [Google Scholar]
- 40.Shukla A, Lounsbury KM, Barrett TF, et al. Asbestos-induced peribronchiolar cell proliferation and cytokine production are attenuated in lungs of protein kinase C-delta knockout mice. The American Journal of Pathology. 2007;170(1):140–151. doi: 10.2353/ajpath.2007.060381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nayak D, Huo Y, Kwang WX, et al. Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience. 2010;166(1):132–144. doi: 10.1016/j.neuroscience.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 42.Di A, Kawamura T, Gao XP, et al. A novel function of sphingosine kinase 1 suppression of JNK activity in preventing inflammation and injury. The Journal of Biological Chemistry. 2010;285(21):15848–15857. doi: 10.1074/jbc.M109.075549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nature Reviews Molecular Cell Biology. 2008;9(2):139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 44.Snider AJ, Orr Gandy KA, Obeid LM. Sphingosine kinase: role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie. 2010;92(6):707–715. doi: 10.1016/j.biochi.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu ZG, Liu H, Yamaguchi T, Miki Y, Yoshida K. Protein kinase Cδ activates RelA/p65 and nuclear factor-κB signaling in response to tumor necrosis factor-alpha. Cancer Research. 2009;69(14):5927–5935. doi: 10.1158/0008-5472.CAN-08-4786. [DOI] [PubMed] [Google Scholar]
- 46.Chou WH, Messing RO. Protein kinase C isozymes in stroke. Trends in Cardiovascular Medicine. 2005;15(2):47–51. doi: 10.1016/j.tcm.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 47.Kim JH, Kim JH, Jun HO, Yu YS, Kim KW. Inhibition of protein kinase C delta attenuates blood-retinal barrier breakdown in diabetic retinopathy. The American Journal of Pathology. 2010;176(3):1517–1524. doi: 10.2353/ajpath.2010.090398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buder-Hoffmann SA, Shukla A, Barrett TF, MacPherson MB, Lounsbury KM, Mossman BT. A protein kinase Cdelta-dependent protein kinase D pathway modulates ERK1/2 and JNK1/2 phosphorylation and Bim-associated apoptosis by asbestos. The American Journal of Pathology. 2009;174(2):449–459. doi: 10.2353/ajpath.2009.080180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castrillo A, Pennington DJ, Otto F, Parker PJ, Owen MJ, Boscá L. Protein kinase Cepsilon is required for macrophage activation and defense against bacterial infection. Journal of Experimental Medicine. 2001;194(9):1231–1242. doi: 10.1084/jem.194.9.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aksoy E, Amraoui Z, Goriely S, Goldman M, Willems F. Critical role of protein kinase C epsilon for lipopolysaccharide-induced IL-12 synthesis in monocyte-derived dendritic cells. The European Journal of Immunology. 2002;32(11):3040–3049. doi: 10.1002/1521-4141(200211)32:11<3040::AID-IMMU3040>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 51.Comalada M, Xaus J, Valledor AF, Lopez-Lopez C, Pennington DJ, Celada A. PKC epsilon is involved in JNK activation that mediates LPS-induced TNF-alpha, which induces apoptosis in macrophages. The American Journal of Physiology—Cell Physiology. 2003;285(5):C1235–C1245. doi: 10.1152/ajpcell.00228.2003. [DOI] [PubMed] [Google Scholar]
- 52.Koyanagi T, Noguchi K, Ootani A, Inagaki K, Robbins RC, Mochly-Rosen D. Pharmacological inhibition of epsilon PKC suppresses chronic inflammation in murine cardiac transplantation model. Journal of Molecular and Cellular Cardiology. 2007;43(4):517–522. doi: 10.1016/j.yjmcc.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 53.Aksoy E, Goldman M, Willems F. Protein kinase C epsilon: a new target to control inflammation and immune-mediated disorders. The International Journal of Biochemistry and Cell Biology. 2004;36(2):183–188. doi: 10.1016/s1357-2725(03)00210-3. [DOI] [PubMed] [Google Scholar]
- 54.Tan SL, Parker PJ. Emerging and diverse roles of protein kinase C in immune cell signalling. Biochemical Journal. 2003;376(3):545–552. doi: 10.1042/BJ20031406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Faisal A, Saurin A, Gregory B, Foxwell B, Parker PJ. The scaffold MyD88 acts to couple protein kinase Cepsilon to Toll-like receptors. The Journal of Biological Chemistry. 2008;283(27):18591–18600. doi: 10.1074/jbc.M710330200. [DOI] [PubMed] [Google Scholar]
- 56.Saurin AT, Durgan J, Cameron AJ, Faisal A, Marber MS, Parker PJ. The regulated assembly of a PKCepsilon complex controls the completion of cytokinesis. Nature Cell Biology. 2008;10(8):891–901. doi: 10.1038/ncb1749. [DOI] [PubMed] [Google Scholar]
- 57.Durgan J, Cameron AJ, Saurin AT, et al. The identification and characterization of novel PKCepsilon phosphorylation sites provide evidence for functional cross-talk within the PKC superfamily. Biochemical Journal. 2008;411(2):319–331. doi: 10.1042/bj20071348. [DOI] [PubMed] [Google Scholar]
- 58.Hirai T, Chida K. Protein kinase Czeta (PKCzeta): activation mechanisms and cellular functions. The Journal of Biochemistry. 2003;133(1):1–7. doi: 10.1093/jb/mvg017. [DOI] [PubMed] [Google Scholar]
- 59.Duran A, Diaz-Meco MT, Moscat J. Essential role of RelA Ser311 phosphorylation by zetaPKC in NF-kappaB transcriptional activation. The EMBO Journal. 2003;22(15):3910–3918. doi: 10.1093/emboj/cdg370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leitges M, Sanz L, Martin P, et al. Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Molecular Cell. 2001;8(4):771–780. doi: 10.1016/s1097-2765(01)00361-6. [DOI] [PubMed] [Google Scholar]
- 61.Hu J, Jacinto R, McCall C, Li L. Regulation of IL-1 receptor-associated kinases by lipopolysaccharide. Journal of Immunology. 2002;168(8):3910–3914. doi: 10.4049/jimmunol.168.8.3910. [DOI] [PubMed] [Google Scholar]
- 62.Huang X, Chen LY, Doerner AM, et al. An atypical protein kinase C (PKC zeta) plays a critical role in lipopolysaccharide-activated NF-kappa B in human peripheral blood monocytes and macrophages. Journal of Immunology. 2009;182(9):5810–5815. doi: 10.4049/jimmunol.0804073. [DOI] [PubMed] [Google Scholar]
- 63.Teusch N, Lombardo E, Eddleston J, Knaus UG. The low molecular weight GTPase RhoA and atypical protein kinase Czeta are required for TLR2-mediated gene transcription. Journal of Immunology. 2004;173(1):507–514. doi: 10.4049/jimmunol.173.1.507. [DOI] [PubMed] [Google Scholar]
- 64.Yang CS, Lee JS, Song CH, et al. Protein kinase C zeta plays an essential role for Mycobacterium tuberculosis-induced extracellular signal-regulated kinase 1/2 activation in monocytes/macrophages via Toll-like receptor 2. Cellular Microbiology. 2007;9(2):382–396. doi: 10.1111/j.1462-5822.2006.00797.x. [DOI] [PubMed] [Google Scholar]
- 65.Chen Q, Gupta S, Pernis AB. Regulation of TLR4-mediated signaling by IBP/Def6, a novel activator of Rho GTPases. Journal of Leukocyte Biology. 2009;85(3):539–543. doi: 10.1189/jlb.0308219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen LY, Zuraw BL, Liu FT, Huang S, Pan ZK. IL-1 receptor-associated kinase and low molecular weight GTPase RhoA signal molecules are required for bacterial lipopolysaccharide-induced cytokine gene transcription. Journal of Immunology. 2002;169(7):3934–3939. doi: 10.4049/jimmunol.169.7.3934. [DOI] [PubMed] [Google Scholar]
- 67.Shibolet O, Giallourakis C, Rosenberg I, Mueller T, Xavier RJ, Podolsky DK. AKAP13, a RhoA GTPase-specific guanine exchange factor, is a novel regulator of TLR2 signaling. The Journal of Biological Chemistry. 2007;282(48):35308–35317. doi: 10.1074/jbc.M704426200. [DOI] [PubMed] [Google Scholar]
- 68.Manukyan M, Nalbant P, Luxen S, Hahn KM, Knaus UG. RhoA GTPase activation by TLR2 and TLR3 ligands: connecting via Src to NF-kappa B. Journal of Immunology. 2009;182(6):3522–3529. doi: 10.4049/jimmunol.0802280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin H, Xiao Y, Chen G, et al. HMG-CoA reductase inhibitor simvastatin suppresses Toll-like receptor 2 ligand-induced activation of nuclear factor kappa B by preventing RhoA activation in monocytes from rheumatoid arthritis patients. Rheumatology International. 2010:1–8. doi: 10.1007/s00296-010-1510-6. [DOI] [PubMed] [Google Scholar]
- 70.Yao H, Hwang JW, Moscat J, et al. Protein kinase Czeta mediates cigarette smoke/aldehyde- and lipopolysaccharide-induced lung inflammation and histone modifications. The Journal of Biological Chemistry. 2010;285(8):5405–5416. doi: 10.1074/jbc.M109.041418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sorensen GL, Husby S, Holmskov U. Surfactant protein A and surfactant protein D variation in pulmonary disease. Immunobiology. 2007;212(4-5):381–416. doi: 10.1016/j.imbio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 72.Moulakakis C, Adam S, Seitzer U, Schromm AB, Leitges M, Stamme C. Surfactant protein A activation of atypical protein kinase C zeta in IkappaB-alpha-dependent anti-inflammatory immune regulation. Journal of Immunology. 2007;179(7):4480–4491. doi: 10.4049/jimmunol.179.7.4480. [DOI] [PubMed] [Google Scholar]
- 73.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circulation Research. 2010;106(8):1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sommerer C, Zeier M. AEB071—a promising immunosuppressive agent. Clinical Transplantation. 2009;23(supplement 21):15–18. doi: 10.1111/j.1399-0012.2009.01104.x. [DOI] [PubMed] [Google Scholar]
- 75.Roffey J, Rosse C, Linch M, Hibbert A, McDonald NQ, Parker PJ. Protein kinase C intervention: the state of play. Current Opinion in Cell Biology. 2009;21(2):268–279. doi: 10.1016/j.ceb.2009.01.019. [DOI] [PubMed] [Google Scholar]