

Abstract

A mild and metal-free method for the chlorodeboronation of organotrifluoroborates using trichloroisocyanuric acid (TCICA) was developed. Aryl-, heteroaryl-, alkenyl-, alkynyl- and alkyltrifluoroborates were converted into the corresponding chlorinated products in good yields. This method proved to be tolerant of a broad range of functional groups.
Introduction
Aryl chlorides are found in many pharmaceuticals and natural products and have been employed as important synthetic intermediates in carbon–carbon bond-forming reactions, such as Suzuki–Miyaura cross-couplings.1 Among the methods utilized for the synthesis of chlorinated arenes, the Sandmeyer2 reaction and direct electrophilic aromatic substitution3 are the most utilized. The direct halogenation of aromatics with electrophilic halogenating agents via electrophilic aromatic substitution is the classic way to introduce chlorine into aromatic and heteroaromatic substrates.4 However, this method has obvious limitations in terms of both chemoselectivity and regioselectivity. In particular, the site selectivity of these chlorinations relies on directing functional groups in the substrate, and certain regioisomers are often unattainable. The halogenation of boron compounds, particularly those synthesized by complementary methods such as C–H activation5 and ortho metalation,6 has emerged as an alternative to circumvent this problem.7 Specifically, the chlorodeboronation of boronic acids and boronate esters has been described.8 In the absence of a metal-based catalyst or promoter, the scope of these reactions appears limited, and moderate yields have been described for electron deficient aryl substrates.8d The use of transition metal complexes, such as copper salts, as catalysts for the chlorination of aromatic boronic acids and boronate esters improved the yield for electron-poor aromatic systems.8a Although many boronic acids and boronate esters are commercially available, their susceptibility to protodeboronation as well as their ability to react with commonly employed organic reagents, such as bases, nucleophiles and oxidants, make them prone to undesirable side reactions. Over the past years organotrifluoroborates have emerged as an alternative to other organoboron species. 9 The stability of this boron functional group allows molecular complexity to be built into a molecule while leaving the carbon-boron bond intact.10 Thus the use of organotrifluoroborates would allow extensive elaboration of a simple substructure, with subsequent late stage chlorination. To the best of our knowledge, no chlorodeboronation of organotrifluoroborates has been reported; consequently, we were prompted to investigate a mild and convenient method for the synthesis of aryl chlorides. Herein we report a metal-free chlorodeboronation of organotrifluoroborates using trichloroisocyanuric acid (TCICA).
Results and Discussion
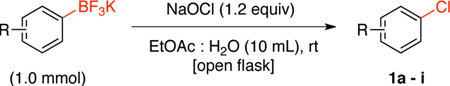
Electrophilic chlorinating agents have been reported as efficient reagents for the chlorination of boronic acids and boronate esters.8 Our investigations were initiated by exploring the chlorodeboronation of potassium naphthalen-1-yltrifluoroborate with sodium hypochlorite (NaOCl, 6.15%, Clorox®), because this is a widely available and inexpensive chlorinating agent. A screening of common solvents revealed that EtOAc:H2O (1:1) was a good solvent system. Thus the reaction of 1a and 1.2 equiv of NaOCl provided the desired product in 92% yield (Table 1) after 30 min (monitored by 11B NMR). The scope of the reaction for various aryltrifluoroborates was investigated (Table 1).
Table 1.
Chlorination of Potassium Aryltrifluoroborate Using Sodium Hypochlorite
 | ||||
|---|---|---|---|---|
| entry | product | reaction time |
% isolated yield |
|
| 1 | 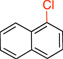 |
1a | 40 min | 92 |
| 2 | 1b | 4 h | 53 | |
| 3 | 1c | 1 h | 75 | |
| 4 | 1d | 40 min | 71 | |
| 5 | 1e | 40 min | 91 | |
| 6 | 1f | 1 h | 73 | |
| 7 | 1g | 40 min | 81 | |
| 8 | 1h | 1 h | 64 | |
| 9 | 1i | 1 h | 53 | |
The reaction with electron-rich aryltrifluoroborates proceeded in good yields, and most transformations were complete in 40 minutes or 1 hour (entries 1 – 7). Halogen-containing aryltrifluoroborates also underwent chlorodeboronation to afford the desired aryl chlorides in modest yields (entries 8 and 9). Unfortunately, electron deficient aryltrifluoroborates were not reactive under these conditions, and the starting material was completely recovered. In an attempt to obtain complete reaction conversion, an excess of NaOCl was utilized (5 equiv); however, the use of a large amount of this reagent afforded a mixture of the desired chlorinated product (2a) along with protodeboronation, dichlorination and boronic acid side-products (eq 1). All efforts to optimize the conditions for aryltrifluoroborates containing electron withdrawing groups (e.g., ester, ketone or nitro) with NaOCl were unsuccessful.
| (1) |
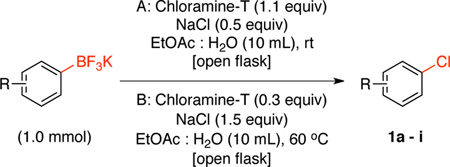
Next we investigated the use of Chloramine-T as the chlorinating reagent, because it has been used as an oxidant for the bromination5c and iodination5d of aryltrifluoroborates. After extensive optimization we determined that the reaction of potassium naphthalen-1-yltrifluoroborate with 1.1 equiv of Chloramine-T and 0.5 equiv of NaCl in EtOAc:H2O at rt (condition A), afforded the desired chlorinated product (1a) in 94% yield (Table 2). Importantly, the reactions with 0.3 equiv of Chloramine-T and 1.5 equiv of NaCl in EtOAc:H2O at 60 °C (condition B), also afforded the desired chlorinated product (1a) in 83% yield and 100% conversion.
Table 2.
Chlorination of Potassium Aryltrifluoroborate Using Chloramine-T
 | ||||
|---|---|---|---|---|
| entry | product | reaction time |
% isolated yield |
|
| 1 | 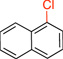 |
1a | 40 min | A: 94 B: 83 |
| 2 | 1b | 4 h | A: 72 B: 70 |
|
| 3 | 1c | 1 h | A: 71 B: 68 |
|
| 4 | 1d | 40 min | A: 78 B: 65 |
|
| 5 | 1e | 40 min | A: 87 B: 74 |
|
| 6 | 1f | 1 h | A: 82 B: 71 |
|
| 7 | 1g | 40 min | A: 85 B: 71 |
|
| 8 | 1h | 1 h | A: 54 B: 15 |
|
| 9 | 1i | 1 h | A: 57 B: 21 |
|
As illustrated in Table 2, the reaction of electron rich aryltrifluoroborates with Chloramine-T afforded the desired chlorinated products in good yield (entries 1 – 7). However, the chlorodeboronation of halogen-containing aryltrifluoroborates proceeded in low yields (entries 8 and 9). Once more, the chlorination of aryltrifluoroborates bearing electron withdrawing groups (e.g., ester, ketone or nitro) did not afford the desired product in good yield.
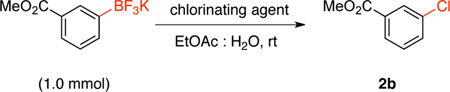
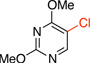
To improve the yield of this reaction for electron withdrawing groups, other chlorinating agents were tested (Table 3). The chlorodeboronation of potassium (3-methoxycarbonyl)phenyltrifluoroborate with N-chlorosuccinimide (NCS) afforded the desired chlorinated product (3a) in only 21% yield. Chloramine-T improved the yield to only 30% after 24 h in a mixture with protodeboronation product, whereas 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) and sodium hypochlorite (NaOCl) were inefficient in this transformation. However, when 1.0 equivalent of trichloroisocyanuric acid (TCICA) was utilized, we were pleased to find that methyl 3-chlorobenzoate (3a) was obtained in 92% yield in only 1 hour at room temperature. Because TCICA is a widely available and inexpensive material ($11.00/mol catalog price), all subsequent reactions were carried out using this electrophilic chlorinating agent.
Table 3.
Chlorination of Potassium (3-methoxycarbonyl)phenyltrifluoroborate Using Various Chlorinating Agents
 | ||||
|---|---|---|---|---|
| entry | oxidant | (mmol) | reaction time |
yield (%) |
| 1 | NCS | 1.2 | 24 h | 21 |
| 2 | Chloramine-T | 1.2 | 24 h | 30 |
| 3 | DCDMH | 1.2 | 24 h | only S.M. recovered |
| 4 | NaOCl | 1.2 | 2 h | 15 |
| 5 | TCICA | 1.0 | 1 h | 92 |
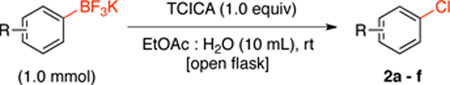
With optimized conditions in hand, the scope of the reaction for aryltrifluoroborates containing electron electron withdrawing groups was investigated (Table 4).
Table 4.
Chlorodeboronation of Electron-Poor Potassium Aryltrifluoroborate with TCICA
 | ||||
|---|---|---|---|---|
| entry | product | reaction time |
% isolated yield |
|
| 1 | 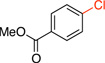 |
2a | 2 h | 87 |
| 2 | 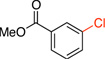 |
2b | 1 h | 92 |
| 3 | 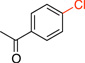 |
2c | 30 min | 82 |
| 4 | 2d | 40 min | 80 | |
| 5a | 2e | 4 h | 85 | |
| 6a | 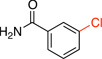 |
2f | 6 h | 89 |
reaction run at 80 °C.
The reaction with a variety of available electron-poor aryltrifluoroborates proceeded in good yields with 1 equivalent of TCICA. 1-Chloro-3-nitrobenzene (2e, entry 5) and 3-chlorobenzamide (2f, entry 6) were obtained in high yields, although heating to 80 °C was required. Furthermore, 2f was obtained in 89% yield with no observed chlorination at the nitrogen of the amide.11 Importantly, the reaction with potassium (4-methoxycarbonyl)phenyltrifluoroborate afforded 2b (entry 2) in 87% yield. This regiochemistry was previously unattainable by the chlorination of simple arenes.4
Next, TCICA was applied as the chlorinating agent in the reaction with electron-rich aryltrifluoroborates (Table 5).
Table 5.
Chlorodeboronation of Electron-Rich and Halogen-Containing Potassium Aryltrifluoroborate with TCICA
 | ||||
|---|---|---|---|---|
| entry | product | reaction time |
% isolated yield |
|
| 1 | 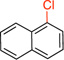 |
1a | 40 min | A: 95 B: 82 |
| 2 | 1b | 40 min | A: 81a B: 85 |
|
| 3 | 1c | 1 h | A: 94 B: 87 |
|
| 4 | 1d | 40 min | A: 98 B: 91 |
|
| 5 | 1e | 40 min | A: 92 B: 89 |
|
| 6 | 1f | 1 h | A: 91 B: 86 |
|
| 7 | 1g | 40 min | A: 92 B: 80 |
|
| 8 | 1h | 1 h | A: 81 B: 67 |
|
| 9 | 1i | 1 h | A: 86 B: 54 |
|
| 10 | 1j | 1 h | A: 84 B: 76 |
|
5 mmol scale, 15 mL of solvent mixture
The reaction with electron-rich aryltrifluoroborates proceeded in good yields using only 0.33 equivalents of TCICA at rt (condition A) or 0.16 equivalents of TCICA and 1.5 equiv of NaCl at 60 °C (condition B), and all were complete in 1 h or less (entries 1 – 7). It is important to mention that the reaction with electron donating groups in the para position (e.g., compounds 1b – 1e and 1g) can be run at higher concentrations (e.g., 0.33 M). However, for compounds such as 1a and 1h – 1j, higher concentrations lead to a mixture of regioisomers. Nonetheless, the reaction with potassium biphenyl-4-yltrifluoroborate was carried out on a 5 mmol scale (1.3 g) with 0.33 equivalents of TCICA and 15 mL of solvent (0.33 M) providing product 1b in 81% yield (entry 2). Halogen-containing aryltrifluoroborates also underwent chlorodeboronation to afford the desired aryl chloride in moderate to good yields (entries 8 – 9) depending on the condition utilized. Unfortunately, this method was unsuccessful for the chlorodeboronation of meta-substituted electron-rich aryl systems. Inexplicably, only starting material or protodeboronation products were obtained for the reactions of TCICA with potassium 3-methoxyphenyltrifluoroborate, potassium 3-(benzyloxy)phenyltrifluoroborate and potassium 3,5-diisopropylphenyltrifluoroborate.
The mechanism of these transformations is enigmatic, particularly in view of the fact that less than one equivalent of electrophilic chlorine can be employed along with NaCl in an oxygenated atmosphere. We considered the possibility that the reaction transpired via a version of the rare SON1 mechanism (Scheme 1),12 where the various chlorinating agents initially served as oxidants of the trifluoroborate. To investigate the possibility of radical intermediates, potassium [2-(allyloxy)phenyl]trifluoroborate was subjected to both reaction conditions (Table 5, entry 10). However, no cyclization product was observed, and the reaction afforded only the ortho-substituted chlorinated product in 84% or 76% yield, respectively. Therefore, it seems unlikely that the reaction proceeds by a radical mechanism,13 and perhaps an electrophilic aromatic substitution with ipso attack is more likely (Scheme 2).14
Scheme 1.

Scheme 2.

Moving forward, the scope of the reaction for heteroaryl systems was also examined. To the best of our knowledge, the chlorodeboronation of heteroarylboron compounds in the literature is limited to one example using stoichiometric copper(II) chloride as the chlorinating agent.8b Hence, diverse heteroaryltrifluoroborates were examined under two different reaction conditions with TCICA (Table 6).
Table 6.
Chlorodeboronation of Potassium Heteroaryltrifluoroborate With TCICA
 | ||||||
|---|---|---|---|---|---|---|
| entry | product | method | reaction time |
% isolated yield |
||
| 1 | 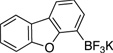 |
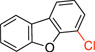 |
3a | A | 40 min | 95 |
| 2 | 3b | A | 6 h | 80 | ||
| 3 | 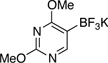 |
 |
3c | A | 2 h | 91 |
| 4 | 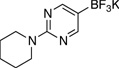 |
 |
3d | A | 2 h | 90 |
| 5 | 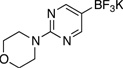 |
 |
3e | A | 2 h | 89 |
| 6 | 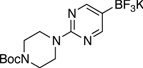 |
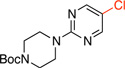 |
3f | A | 2 h | 95 |
| 7 | 3g | B | 1 h | 88 | ||
| 8 | 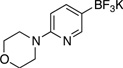 |
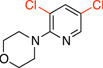 |
3h | B | 1 h | 91 |
| 9 | 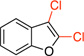 |
3i | B | 30 min | 86 | |
The majority of the heteroaryl chlorides obtained were not commercially available or had limited commercial availability. Organotrifluoroborate derivatives containing the dibenzofuranyl, quinolinyl, benzofuranyl, pyrimidinyl and pyridinyl subunits were successfully converted into the corresponding chlorinated product in good yields. However, the use of only 0.33 equivalents of TCICA (method A) in the reaction with the pyridine and benzofuran derivatives (entries 7 – 9) afforded mixtures of mono and dichlorinated compounds (1 : 1). The use of less than 0.33 equivalents of TCICA with or without NaCl did not improve the selectivity of the reaction. When method B (1 equivalent of TCICA) was applied to these substrates, only dichlorination products were observed in good yields. Under the developed conditions, heteroaryls such as thiophenes, furans and indoles afforded complex product mixtures of monochlorinated regioisomers, as well as dichlorinated and protodeboronated compounds.
The formation of dichlorinated compounds from monotrifluoroborates heteroaryls represented an unexpected reactivity. To elucidate the source of these products, we examined the possibility of a chlorodeboronation and subsequent chlorination of the monochloride intermediate 3j (eq 2). Thus, we applied our general method B (1 equivalent of TCICA) in the reaction with 2-chlorobenzofuran (3j, eq 2). Surprisingly, none of the dichlorinated product was observed under these conditions, and only starting material 3j was recovered. Although the developed protocol is an efficient method for the synthesis of these dichlorinated heterocycles, the mechanistic pathway for their formation is again puzzling.
 |
(2) |
Encouraged by the results obtained with aryl- and heteroaryltrifluoroborates, we examined the feasibility of applying the process to alkyl-, alkenyl- and alkynyltrifluoroborates (Table 7). The use of only 0.33 equivalents of TCICA was sufficient to afford the desired chlorinated products in good yields. Although for many substrates the process worked very well, the protocol is somewhat capricious, and thus attempts to promote the chlorodeboronation of secondary alkyltrifluoroborates as well as Z-alkenyltrifluoroborates were unsuccessful.
Table 7.
Chlorodeboronation of Potassium Alkyl-, Alkenyl- and Alkynyltrifluoroborate with TCICA
 | ||||
|---|---|---|---|---|
| entry | product | reaction time |
% isolated yield |
|
| 1 | 4a | 30 min | 85 | |
| 2 | 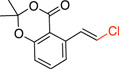 |
4b | 30 min | 92 |
| 3 | 4c | 40 min | 82 | |
| 4 | 4d | 2 h | 81 | |
Finally, based on the work of Kabalka and co-workers7c where the bromination of aryltrifluoroborates was described by using Chloramine-T and sodium bromide, we demonstrated that the use of 0.33 equivalents of TCICA in the presence of 1 equivalent of sodium bromide afforded the desired brominated product (5a) in 94% yield in only 30 minutes (eq 3).
 |
(3) |
In conclusion, we have developed the first metal-free method for the chlorodeboronation of organotrifluoroborates utilizing commercially available TCICA. Under our mild conditions aryl-, heteroaryl-, alkyl-, alkenyl- and alkynyltrifluoroborates bearing a variety of functional groups afforded the corresponding chlorinated product in good yields. The mechanism of these reactions is unclear, leading to surprising and perplexing results in some cases. We are attempting to elucidate the nature of these and other reactions that transpire under oxidative conditions15 in our continuing studies of the organotrifluoroborates.
Experimental Section
General Procedure for Chlorination of Organotrifluoroborates with NaOCl
To a 50 mL round bottom flask containing a solution of potassium organotrifluoroborate (1 mmol) in EtOAc : H2O (1:1, 10 mL, 0.1 M) was added NaOCl [1.5 mL of Clorox® Ultra (6.15% NaOCl), 1.2 mmol, 1.2 equiv] in one portion. The reaction was stirred open to air at rt until 11B NMR indicated completion of the reaction. The reaction was quenched with 10% aq. Na2SO3 (10 mL). The aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic layers were washed with 1 N NaOH (3 × 10 mL) to remove any unreacted starting material. The Et2O layer was dried (Na2SO4), filtered, concentrated, and dried in vacuo. In general the product obtained was pure. Trace impurities were removed by column chromatography using Et2O/pentanes to afford the desired pure product. Compounds 1a – 1i.
General Procedure for Chlorination of Organotrifluoroborates with Stoichiometric Chloramine-T
To a 50 mL round bottom flask containing a solution of potassium organotrifluoroborate (1 mmol) in EtOAc : H2O (1:1, 10 mL, 0.1 M) was added NaCl (1M in H2O, 0.5 mL, 0.5 mmol, 0.5 equiv) and Chloramine-T. 3 H2O (310 mg, 1.1 mmol, 1.1 equiv) in one portion. The reaction was stirred open to air at rt until 11B NMR indicated completion of the reaction. The reaction was quenched with 10% aq. Na2SO3 (10 mL). The aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic layers were washed with 1 N NaOH (3 × 10 mL) to remove any unreacted starting material. The Et2O layer was dried (Na2SO4), filtered, concentrated, and dried in vacuo. In general the product obtained was pure. Trace impurities were removed by column chromatography using Et2O/pentanes to afford the desired pure product. Compounds 1a – 1i.
General Procedure for Chlorination of Organotrifluoroborates with Substoichiometric Chloramine-T
To a 50 mL round bottom flask containing a solution of potassium organotrifluoroborate (1 mmol) in EtOAc : H2O (1:1, 10 mL, 0.1 M) was added NaCl (1M in H2O, 1.5 mL, 1.5 mmol, 1.5 equiv) and Chloramine-T. 3 H2O (85 mg, 0.3 mmol, 0.3 equiv) in one portion. The reaction was stirred open to air at rt until 11B NMR indicated completion of the reaction. The reaction was quenched with 10% aq. Na2SO3 (10 mL). The aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic layers were washed with 1 N NaOH (3 × 10 mL) to remove any unreacted starting material. The Et2O layer was dried (Na2SO4), filtered, concentrated, and dried in vacuo. In general the product obtained was pure. Trace impurities were removed by column chromatography using Et2O/pentanes to afford the desired pure product. Compounds 1a – 1i.
General Procedure for Chlorination of Organotrifluoroborates with Trichloroisocyanuric acid
General Procedure A
To a 50 mL round bottom flask containing a solution of potassium organotrifluoroborate (1 mmol) in EtOAc : H2O (1:1, 10 mL, 0.1 M) was added trichloroisocyanuric acid (76.7 mg, 0.33 mmol, 0.33 equiv) in one portion. The reaction was stirred open to air at rt until 11B NMR indicated completion of the reaction. The reaction was quenched with 10% aq. Na2SO3 (10 mL). The aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic layers were washed with 1 N NaOH (3 × 10 mL) to remove any unreacted starting material. The Et2O layer was dried (Na2SO4), filtered, concentrated, and dried in vacuo. In general the product obtained was pure. Trace impurities were removed by column chromatography using Et2O/pentanes to afford the desired pure product. Compounds 1a – 1j, 3a – 3f and 4a – 4d.
General Procedure B
To a 50 mL round bottom flask containing a solution of potassium organotrifluoroborate (1 mmol) in EtOAc : H2O (1:1, 10 mL, 0.1 M) was added NaCl (1M in H2O, 1.5 mL, 1.5 mmol, 1.5 equiv) and trichloroisocyanuric acid (37 mg, 0.16 mmol, 0.16 equiv) in one portion. The reaction was stirred open to air at rt until 11B NMR indicated completion of the reaction. The reaction was quenched with 10% aq. Na2SO3 (10 mL). The aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic layers were washed with 1 N NaOH (3 × 10 mL) to remove any unreacted starting material. The Et2O layer was dried (Na2SO4), filtered, concentrated, and dried in vacuo. In general the product obtained was pure. Trace impurities were removed by column chromatography using Et2O/pentanes to afford the desired pure product. Compounds 1a – 1j.
General Procedure C
To a 50 mL round bottom flask containing a solution of potassium organotrifluoroborate (1 mmol) in EtOAc : H2O (1:1, 10 mL, 0.1 M) was added trichloroisocyanuric acid (232.4 mg, 1 mmol, 1 equiv) in one portion. The reaction was stirred open to air at rt until 11B NMR indicated completion of the reaction. The reaction was quenched with 10% aq. Na2SO3 (10 mL). The aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic layers were washed with 1 N NaOH (3 × 10 mL) to remove any unreacted starting material. The Et2O layer was dried (Na2SO4), filtered, concentrated, and dried in vacuo. In general the product obtained was pure. Trace impurities were removed by column chromatography using Et2O/pentanes to afford the desired pure product. Compounds 2a – 2f and 3g – 3i.
1-Chloronaphthalene (1a).16
General procedure A was employed using potassium naphthalen-1-yltrifluoroborate (0.23 g, 1 mmol), and the reaction was complete in 40 min. The desired pure product was obtained in 95% yield (0.15 g, 0.94 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 8.24 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.69 (d, J = 8.0 Hz, 1H), 7.56 – 7.52 (m, 2H), 7.48 (m, 1H), 7.31 (t, J = 8.0 Hz, 1H). 13C NMR (125.8 MHz, CDCl3) δ 134.8, 132.1, 131.0, 128.4, 127.4, 127.2, 126.9, 126.4, 125.9, 124.6.
4-Chlorobiphenyl (1b). 17
General procedure A was employed using potassium biphenyl-4-yltrifluoroborate (1.3 g, 5 mmol), and 15 mL of the solvent mixture, and the reaction was complete in 40 min. The desired pure product was obtained in 81% yield (0.76 g, 4.05 mmol) as a white solid, mp 75 – 77 °C (lit. 76 – 78 °C)11. 1H NMR (500 MHz, CDCl3) δ 7.54 – 7.52 (m, 2H), 7.49 (d, J = 8.5 Hz, 2H), 7.43 – 7.40 (m, 2H), 7.38 (d, J = 8.5 Hz, 2H), 7.34 (m, 1H). 13C NMR (125.8 MHz, CDCl3) δ 140.2, 139.8, 133.6, 129.1, 129.0, 128.6, 127.8, 127.2.
1-tert-Butyl-4-chlorobenzene (1c).18
General procedure A was employed using potassium 4-tert-butylphenyltrifluoroborate (0.24 g, 1 mmol), and the reaction was complete in 1 h. The desired pure product was obtained in 94% yield (0.16 g, 0.94 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.33 (d, J = 9 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H), 1.32 (s, 9H). 13C NMR (125.8 MHz, CDCl3) δ 149.7, 131.3, 128.2, 126.9, 34.6, 31.4.
1-Chloro-4-methoxybenzene (1d). 19
General procedure A was employed using potassium 4-methoxyphenyltrifluoroborate (0.21 g, 1 mmol), and the reaction was complete in 40 min. The desired pure product was obtained in 98% yield (0.14 g, 0.98 mmol) as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.22 (d, J = 9.0 Hz, 2H), 6.81 (d, J = 9.0 Hz, 2H), 3.77 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 158.3, 129.4, 125.7, 115.3, 55.6.
1-(Benzyloxy)-4-chlorobenzene (1e).20
General procedure A was employed using potassium 4-(benzyloxy)phenyltrifluoroborate (0.25 g, 1 mmol), and the reaction was complete in 40 min. The desired pure product was obtained in 92% yield (0.20 g, 0.92 mmol) as a light yellow solid, mp 65 – 67 °C (lit. 65 – 67 °C).21 1H NMR (500 MHz, CDCl3) δ 7.37 – 7.33 (m, 4H), 7.31 (m, 1H), 7.19 (d, J = 9 Hz, 2H) 6.85 (d, J = 9.0 Hz, 2H), 4.98 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 157.3, 136.6, 129.3, 128.6, 128.1, 127.4, 125.8, 116.1, 70.2.
1-(Benzyloxy)-2-chlorobenzene (1f).20
General procedure A was employed using potassium 2-(benzyloxy)phenyltrifluoroborate (0.25 g, 1 mmol), and the reaction was complete in 1 h. The desired pure product was obtained in 94% yield (0.21 g, 0.94 mmol) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.37 – 7.33 (m, 4H), 7.31 (m, 1H), 7.19 (d, J = 9 Hz, 2H) 6.85 (d, J = 9.0 Hz, 2H), 4.98 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 154.3, 136.6, 130.4, 128.7, 128.0, 127.8, 127.1, 123.3, 121.7, 114.1, 70.8.
5-(Benzyloxy)-2-chlorobenzaldehyde (1g)
General procedure A was employed using potassium (4-(benzyloxy)-2-formylphenyl)trifluoroborate (0.32 g, 1 mmol), and the reaction was complete in 40 min. The desired pure product was obtained in 91% yield (0.22 g, 0.91 mmol) as a light yellow solid, mp 55 – 57 °C. 1H NMR (500 MHz, CDCl3) δ 10.43 (s, 1H), 7.49 (d, J = 3 Hz, 1H), 7.43 – 7.38 (m, 4H), 7.39 – 7.34 (m, 2H), 7.16 (m, 1H) 5.09 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 189.7, 157.7, 135.9, 132.9, 131.5, 129.9, 128.7, 128.3, 127.5, 123.4, 113.0, 70.5. FT – IR (neat) 1696, 1230, 1004, 748 cm−1. HRMS (ESI) m/z calcd. for C14H11O2NaCl (M+Na)+ 269.0345, found 269.0347.
1-Bromo-4-chlorobenzene (1h).7b
General procedure A was employed using potassium (4-bromophenyl)trifluoroborate (0.26 g, 1 mmol), and the reaction was complete in 1 h. The desired pure product was obtained in 81% yield (0.15 g, 0.81 mmol) as a colorless solid, mp 64 – 66 °C (lit. 65 – 66 °C).22 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 8.5 Hz, 2H), 7.20 (d, J = 8.5 Hz, 2H). 13C NMR (125.8 MHz, CDCl3) δ 133.2, 132.7, 130.2, 120.2.
1,4-Dichlorobenzene (1i). 23
General procedure A was employed using potassium (4-chlorophenyl)trifluoroborate (0.22 g, 1 mmol), and the reaction was complete in 1 h. The desired pure product was obtained in 86% yield (0.13 g, 0.86 mmol) as a colorless solid, mp 47 – 50 °C (lit. 46 – 49 °C).24 1H NMR (500 MHz, CDCl3) δ 7.27 (s, 4H). 13C NMR (125.8 MHz, CDCl3) δ 132.5, 129.8.
1-(Allyloxy)-2-chlorobenzene (1j). 25
General procedure A was employed using potassium (2-(allyloxy)phenyl)trifluoroborate (0.24 g, 1 mmol), and the reaction was complete in 1 h. The desired pure product was obtained in 84% yield (0.14 g, 0.84 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.37 (m, 1H), 7.19 (m, 1H), 6.93 – 6.88 (m, 2H), 6.08 (m, 1H), 5.47 (m, 1H), 5.31 (m, 1H), 4.63 – 4.61 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 154.1, 132.7, 130.3, 127.6, 123.1, 121.5, 117.8, 113.8, 69.7.
Methyl 4-Chlorobenzoate (2a). 26
General procedure C was employed using potassium (4-methoxycarbonyl)phenyltrifluoroborate (0.24 g, 1 mmol), and the reaction was complete in 2 h. The desired pure product was obtained in 87% yield (0.15 g, 0.87 mmol) as a light yellow solid mp 40 – 43 °C (lit.10 40 – 42 °C). 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H), 3.92 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 166.2, 139.4, 131.0, 128.7, 128.6, 52.3.
Methyl 3-Chlorobenzoate (2b). 27
General procedure C was employed using potassium (3-methoxycarbonyl)phenyltrifluoroborate (0.24 g, 1 mmol), and the reaction was complete in 1 h. The desired pure product was obtained in 92% yield (0.16 g, 0.92 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 8.01 (m, 1H), 7.92 (m, 1H), 7.52 (m, 1H), 7.38 (m, 1H), 3.92 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 165.8, 134.5, 132.9, 131.8, 129.6, 129.6, 127.6, 52.3.
1-(4-Chlorophenyl)ethanone (2c). 28
General procedure C was employed using potassium (4-acetylphenyl)trifluoroborate (0.23 g, 1 mmol), and the reaction was complete in 30 min. The desired pure product was obtained in 82% yield (0.13 g, 0.82 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 8.5 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H), 2.56 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 196.7, 139.5, 135.4, 129.6, 128.8, 26.5.
3-Chloro-4-fluorobenzaldehyde (2d)
General procedure A was employed using potassium 2-fluoro-5-formylphenyltrifluoroborate (0.23 g, 1 mmol), and the reaction was complete in 40 min. The desired pure product was obtained in 80% yield (0.13 g, 0.80 mmol) as a colorless solid, mp 83 – 85 °C. 1H NMR (500 MHz, CDCl3) δ 9.94 (s, 1H), 7.97 (m, 1H), 7.81 (m, 1H), 7.32 (t, J = 8.5 Hz, 1H). 13C NMR (125.8 MHz, CDCl3) δ 189.3, 161.8 (d, J = 7.3 Hz), 133.5, 132.1, 130.0 (d, J = 8.9 Hz), 122.7 (d, J = 18.9 Hz), 117.4 (d, J = 22.1 Hz). 19F NMR (470.8 MHz, CDCl3) δ −104.7. FT – IR (neat) 1698, 1264, 1058, 708 cm−1. HRMS (ESI) m/z calcd. for C7H4OFCl (M)+ 157.9935, found 157.9941.
1-Chloro-3-nitrobenzene (2e).8a
General procedure C was employed using potassium 3-nitrophenyltrifluoroborate (0.23 g, 1 mmol), the reaction was run at 80 °C and it was complete in 4 h. The desired pure product was obtained in 85% yield (0.13 g, 0.85 mmol) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 8.22 (t, J = 2.1 Hz, 1H), 8.14 (m, 1H), 7.69 (m, 1H), 7.52 (t, J = 8.1 Hz, 1H). 13C NMR (125.8 MHz, CDCl3) δ 148.7, 135.3, 134.6, 130.3, 123.8, 121.6.
3-Chlorobenzamide (2f).8a
General procedure C was employed using potassium (3-carbamoylphenyl)trifluoroborate (0.23 g, 1 mmol), the reaction was run at 80 °C and it was complete in 6 h. The desired pure product was obtained in 89% yield (0.14 g, 0.89 mmol) as a white solid, mp 125 – 127 °C (lit.17 125 – 128 °C). 1H NMR (500 MHz, CDCl3) δ 7.81 (t, J = 2.0 Hz, 1H), 7.68 (m, 1H), 7.51 (m, 1H), 7.39 (t, J = 8.0 Hz, 1H), 6.02 (brs, 2H). 13C NMR (125.8 MHz, CDCl3) δ 167.9, 135.1, 134.9, 132.0, 129.9, 127.7, 125.4.
4-Chlorodibenzo[b,d]furan (3a)
General procedure A was employed using potassium dibenzo[b,d]furan-4-yltrifluoroborate (0.27 g, 1 mmol), and the reaction was complete in 2 h. The desired pure product was obtained in 95% yield (0.19 g, 0.95 mmol) as a white solid, mp 64 – 66 °C. 1H NMR (500 MHz, CDCl3) δ 7.92 (d, J = 7.7 Hz, 1H), 7.82 (d, J = 7.7 Hz, 1H), 7.64 (d, J = 8.3 Hz, 1H), 7.49 (t, J = 7.8 Hz, 1H), 7.45 (d, J = 7.9 Hz, 1H), 7.36 (t, J = 7.5 Hz, 1H), 7.25 (d, J = 8.3 Hz, 1H). 13C NMR (125.8 MHz, CDCl3) δ 156.1, 151.9, 127.8, 127.1, 125.9, 124.0, 123.6, 123.2, 120.9, 119.0, 117.1, 112.1. FT – IR (neat) 1420, 1197, 1040, 870, 744, 683 cm−1. HRMS (ESI) m/z calcd. for C12H8OCl (M+H)+ 203.0264, found 203.0263.
2,3-Dichloroquinoline (3b)
General procedure A was employed using potassium (2-chloroquinolin-3-yl)trifluoroborate (0.27 g, 1 mmol), and the reaction was complete in 6 h. The desired pure product was obtained in 80% yield (0.16 g, 0.80 mmol) as a white solid, mp 97 – 99 °C. 1H NMR (500 MHz, CDCl3) δ 8.22 (s, 1H), 8.00 (m, 1H), 7.75 – 7.71 (m, 2H), 7.59 (m, 1H). 13C NMR (125.8 MHz, CDCl3) δ 148.2, 145.8, 137.2, 130.6, 128.5, 127.9, 127.6, 127.1, 126.6. FT – IR (neat) 1150, 975, 757, 655 cm−1. HRMS (ESI) m/z calcd. for C9H5NCl2 (M)+ 196.9799, found 196.9799.
5-Chloro-2,4-dimethoxypyrimidine (3c)
General procedure A was employed using potassium (2,4-dimethoxypyrimidin-5-yl)trifluoroborate (0.25 g, 1 mmol), and the reaction was complete in 2 h. The desired pure product was obtained in 91% yield (0.16 g, 0.91 mmol) as a white solid, mp 70 – 72 °C (lit.29 72 – 73 °C). 1H NMR (500 MHz, CDCl3) δ 8.20 (s, 1H), 4.07 (s, 3H), 3.99 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 166.0, 163.4, 156.4, 110.3, 55.2, 54.7. FT – IR (neat) 1560, 1400, 1275, 1004, 780, 690 cm−1. HRMS (ESI) m/z calcd. for C6H8N2O2Cl (M+H)+ 175.0274, found 175.0281.
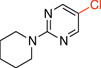
5-Chloro-2-(piperidin-1-yl)pyrimidine (3d)
General procedure A was employed using potassium 2-(piperidin-1-yl)pyrimidin-5-yltrifluoroborate (0.27 g, 1 mmol), and the reaction was complete in 2 h. The desired pure product was obtained in 90% yield (0.18 g, 0.90 mmol) as a white solid, mp 45 – 47 °C. 1H NMR (500 MHz, CDCl3) δ 8.19 (s, 2H), 3.75 (t, J = 5.5 Hz, 4H), 1.67 – 1.66 (m, 2H), 1.61 – 1.58 (m, 4H). 13C NMR (125.8 MHz, CDCl3) δ 159.9, 155.7, 117.3, 45.1, 25.6, 24.7. FT – IR (neat) 1584, 1508, 1441 1256, 782 cm−1. HRMS (ESI) m/z calcd. for C9H13N3Cl (M+H)+ 198.0798, found 198.0792.
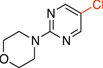
4-(5-Chloropyrimidin-2-yl)morpholine (3e)
General procedure A was employed using potassium 2-morpholinopyrimidin-5-yltrifluoroborate (0.27 g, 1 mmol), and the reaction was complete in 2 h. The desired pure product was obtained in 89% yield (0.18 g, 0.89 mmol) as a white solid, mp 70 – 72 °C. 1H NMR (500 MHz, CDCl3) δ 8.24 (s, 2H), 3.76 – 3.75 (m, 8H). 13C NMR (125.8 MHz, CDCl3) δ 160.0, 155.9, 118.6, 66.7, 44.4. FT – IR (neat) 2922, 1585, 1494, 1253, 1112, 953, 787, 667 cm−1. HRMS (ESI) m/z calcd. for C8H11N3OCl (M+H)+ 200.0591, found 200.0589.
tert-Butyl 4-(5-Chloropyrimidin-2-yl)piperazine-1-carboxylate (3f)
General procedure A was employed using potassium (2-(4-(tert-butoxycarbonyl)piperazin-1-yl)pyrimidin-5-yl)trifluoroborate (0.37 g, 1 mmol), and the reaction was complete in 2 h. The desired pure product was obtained in 95% yield (0.28 g, 0.95 mmol) as a white solid, mp 102 – 105 °C. 1H NMR (500 MHz, CDCl3) δ 8.24 (s, 2H), 3.77 (t, J = 5.0 Hz, 4H), 3.49 (t, J = 5.0 Hz, 4H), 1.49 (s, 9H). 13C NMR (125.8 MHz, CDCl3) δ 159.9, 156.0, 118.7, 80.2, 44.0, 28.6. FT – IR (neat) 1677, 1585, 1514, 1249, 1131, 993, 784 cm−1. HRMS (ESI) m/z calcd. for C13H19N4O2NaCl (M+Na)+ 321.1094, found 321.1099.
3,5-Dichloro-N,N-dimethylpyridin-2-amine (3g)
General procedure C was employed using potassium (6-(dimethylamino)pyridin-3-yl)trifluoroborate (0.23 g, 1 mmol), and the reaction was complete in 1 h. The desired pure product was obtained in 88% yield (0.17 g, 0.88 mmol) as a white solid, mp 24 – 26 °C. 1H NMR (500 MHz, CDCl3) δ 8.07 (d, J = 2 Hz, 1H), 7.54 (d, J = 2 Hz, 1H), 2.98 (s, 6H). 13C NMR (125.8 MHz, CDCl3) δ 157.5, 143.9, 138.3, 122.7, 120.9, 41.5. FT – IR (neat) 1491, 1414, 1176, 1049, 837, 753 cm−1. HRMS (ESI) m/z calcd. for C7H10N2Cl (M+H)+ 157.0533, found 157.0533.
4-(3,5-Dichloropyridin-2-yl)morpholine (3h)
General procedure C was employed using potassium 6-morpholinopyridin-3-yltrifluoroborate (0.27 g, 1 mmol), and the reaction was complete in 1 h. The desired pure product was obtained in 91% yield (0.21 g, 0.91 mmol) as a white solid, mp 83 – 85 °C. 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 2.5 Hz, 1H), 7.60 (d, J = 2.5 Hz, 1H), 3.85 (t, J = 5.0 Hz, 4H), 3.33 (t, J = 5.0 Hz, 4H). 13C NMR (125.8 MHz, CDCl3) δ 156.6, 144.3, 138.3, 124.5, 122.6, 66.8, 49.5. FT – IR (neat) 1435, 1243, 1111, 944, 823, 709 cm−1. HRMS (ESI) m/z calcd. for C9H11N2OCl2 (M+H)+ 233.0248, found 233.0257.
2,3-Dichlorobenzofuran (3i)
General procedure C was employed using potassium benzofuran-2-yltrifluoroborate (0.22 g, 1 mmol), and the reaction was complete in 30 min. The desired pure product was obtained in 86% yield (0.16 g, 0.86 mmol) as a white solid, mp 25 – 27 °C. 1H NMR (500 MHz, CDCl3) δ 7.51 (m, 1H), 7.42 (m, 1H), 7.35 – 7.29 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 152.3, 137.7, 126.5, 125.4, 123.9, 118.4, 111.3, 108.4. FT – IR (neat) 1449, 1155, 1034, 742 cm−1. HRMS (ESI) m/z calcd. for C8H5OCl2 (M+H)+ 186.9717, found 186.9721.
(E)-(2-Chlorovinyl)benzene (4a). 30
General procedure A was employed using potassium (E)-styryltrifluoroborate (0.21 g, 1 mmol), and the reaction was complete in 40 min. The desired pure product was obtained in 85% yield (0.12 g, 0.85 mmol) as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.30 – 7.24 (m, 5H), 6.81 (d, J = 13.5 Hz, 1H), 6.61 (d, J = 13.5 Hz, 1H).13C NMR (125.8 MHz, CDCl3) δ 134.8, 133.2, 128.7, 128.1, 126.1, 118.6.
(E)-5-(2-Chlorovinyl)-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one (4b)
General procedure A was employed using potassium (E)-(2-(2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-5-yl)vinyl)trifluoroborate (0.18 g, 0.5 mmol), and the reaction was complete in 30 min. The desired pure product was obtained in 92% yield (0.11 g, 0.92 mmol) as a light yellow solid, mp 86–88 °C. 1H NMR (500 MHz, CDCl3) δ 7.90 (d, J = 13.5, 1H), 7.46 (t, J = 7.5 Hz, 1H), 7.12 (d, J = 7.5 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.63 (d, J = 13.5 Hz, 1H), 1.71 (s, 6H). 13C NMR (125.8 MHz, CDCl3) δ 134.8, 133.2, 128.7, 128.1, 126.1, 118.6. FT – IR (neat) 1722, 1273, 1042, 924, 780, 691 cm−1. HRMS (ESI) m/z calcd. for C12H11O3NaCl (M+Na)+ 261.0294, found 261.0297.
(5-Chloropent-4-yn-1-yl)benzene (4c)
General procedure A was employed using potassium 5-phenylpent-1-yn-1-yltrifluoroborate (0.25 g, 1 mmol), and the reaction was complete in 40 min. The desired pure product was obtained in 82% yield (0.15 g, 0.82 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.40 – 7.37 (m, 2H), 7.31 – 7.27 (m, 3H), 2.80 (t, J = 8 Hz, 2H), 2.30 – 2.26 (m, 2H), 1.95 – 1.90 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 141.6, 128.7, 128.6, 126.2, 69.5, 57.8, 34.9, 30.2, 18.4. FT – IR (neat) 1496, 1082, 744, 698 cm−1. HRMS (ESI) m/z calcd. for C11H11Cl (M)+ 178.0549, found 178.0553.
6-Chlorohexyl benzoate (4d). 31
General procedure A was employed using potassium (6-(benzoyloxy)hexyl)trifluoroborate (0.31 g, 1 mmol), and the reaction was complete in 2 h. The desired pure product was obtained in 81% yield (0.19 g, 0.82 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 8.05 - 803 (m, 2H), 7.54 (m, 1H), 7.45 – 7.42 (m, 2H), 4.32 (t, J = 6.5 Hz, 2H), 3.65 (t, J = 6.5 Hz, 2H), 1.81 – 1.76 (m, 2H), 1.63 – 1.59 (m, 2H), 1.49 – 1.43 (m, 4H). 13C NMR (125.8 MHz, CDCl3) δ 166.9, 133.0, 130.6, 129.7, 128.5, 65.1, 62.9, 32.7, 28.9, 26.0, 25.6.
4-Bromobiphenyl (5a).32
To a 50 mL round bottom flask containing a mixture of trichloroisocyanuric acid (76.7 mg, 0.33 mmol, 1 equiv) and NaBr (103 mg, 1 mmol, 1 equiv) in EtOAc : H2O (1:1, 10 mL, 0.1 M) was added potassium biphenyl-4-yltrifluoroborate (260 mg, 1 mmol) in one portion. The reaction was stirred open to air at rt until 11B NMR indicated completion of the reaction (30 min). The reaction was quenched with 10% aq. Na2SO3 (10 mL). The aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with 1 N NaOH (3 × 10 mL) to remove any unreacted starting material. The EtOAc layer was dried (Na2SO4), filtered, concentrated, and dried in vacuo. The desired pure product was obtained in 94% yield (0.84 g, 4.45 mmol) as a white solid, mp 88 – 90 °C (lit. 89 °C)25. 1H NMR (500 MHz, CDCl3) δ 7.57 – 7.54 (m, 4H), 7.46 – 7.42 (m, 4H), 7.36 (m, 1H). 13C NMR (125.8 MHz, CDCl3) δ 140.1, 140.0, 131.8, 128.9, 128.7, 127.6, 126.9, 121.5.
Supplementary Material
Acknowledgment
This work was generously supported by the NIGMS (R01 GM086209, GM035249) and a Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) Graduate Research Fellowship to L.N.C. We also acknowledge Aldrich, BoroChem, and Frontier Scientific for their donation of the boronic acids. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining the HRMS data.
Footnotes
Supporting Information Available: Copies of 1H, 13C, and 19F spectra for all compounds prepared by the method described. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For selected examples of aryl chlorides used in Suzuki-Miyaura cross-couplings see: Molander GA, Argintaru OA, Aron I, Dreher SD. Org. Lett. 2010;12:5783. doi: 10.1021/ol102717x. Molander GA, Shin I, Jean-Gerard L. Org. Lett. 2010;12:4381. doi: 10.1021/ol101865e. Molander GA, Sandrock DL. Org. Lett. 2009;11:2369. doi: 10.1021/ol900822j. Dreher SD, Lim S-E, Sandrock DL, Molander GA. J. Org. Chem. 2009;74:3626. doi: 10.1021/jo900152n. Molander GA, Jean-Gerard L. J. Org. Chem. 2009;74:1297. doi: 10.1021/jo802453m. Dreher SD, Dormer PG, Sandrock DL, Molander GA. J. Am. Chem. Soc. 2008;130:9257. doi: 10.1021/ja8031423. Molander GA, Sandrock DL. J. Am. Chem. Soc. 2008;130:15792. doi: 10.1021/ja807076d. Molander GA, Canturk B. Org. Lett. 2008;10:2135. doi: 10.1021/ol800532p.
- 2.Sandmeyer T. Chem. Ber. 1884:1633. [Google Scholar]
- 3.Taylor R. Electrophilic Aromatic Substitution. New York: Wiley; 1990. [Google Scholar]
- 4.For selected examples see: Mo F, Yan JM, Qiu D, Li F, Zhang Y, Wang J. Angew. Chem. Int. Ed. 2010;49:2028. doi: 10.1002/anie.200906699. Qiu D, Mo F, Zheng Z, Zhang Y, Wang J. Org. Lett. 2010;12:5474. doi: 10.1021/ol102350v. Zhang Y, Shibatomi K, Yamamoto H. Synlett. 2005:2837. Bagheri M, Azizi N, Saidi MR. Can. J. Chem. 2005;83:146. Ganguly NC, De P, Dutta S. Synthesis. 2005:1103. Yadav JS, Reddy BVS, Reddy PSR, Basak AK, Narsaiah AV. Adv. Synth. Catal. 2004;346:77. Prakash GKS, Mathew T, Hoole D, Esteves PM, Wang Q, Rasul G, Olah GA. J. Am. Chem. Soc. 2004;126:15770. doi: 10.1021/ja0465247.
- 5.For a review see: Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem. Rev. 2010;110:890. doi: 10.1021/cr900206p.
- 6.Hurst TE, Macklin TK, Becker M, Hartmann E, Kügel W, Salle J-CP-L, Batsanov AS, Marder TB, Snieckus V. Chem. Eur. J. 2010;16:8155. doi: 10.1002/chem.201000401. [DOI] [PubMed] [Google Scholar]
- 7.For aryl iodo- and bromodeboronation reactions, see: Yao M-L, Reddy MS, Yong L, Walfish I, Blevins DW, Kabalka GW. Org. Lett. 2010;12:700. doi: 10.1021/ol9027144. Thompson ALS, Kabalka GW, Akula MR, Huffman JW. Synthesis. 2005:547. Kabalka GW, Mereddy AR. Organometallics. 2004;23:4519. Kabalka GW, Mereddy AR. Tetrahedron Lett. 2004;45:343. Kabalka GW, Akula MR, Zhang J. Nucl. Med. Biol. 2002;29:841. doi: 10.1016/s0969-8051(02)00344-x. Thiebes C, Surya Prakash GK, Petasis NA, Olah GA. Synlett. 1998:141. Kabalka GW, Sastry KAR, Pagni PG. J. Radioanal. Chem. 1982;74:315. Kabalka GW, Gooch EE, Sastry KAR. J. Nucl. Med. 1981;22:908.
- 8.(a) Wu H, Hynes J., Jr Org. Lett. 2010;12:1192. doi: 10.1021/ol9029337. [DOI] [PubMed] [Google Scholar]; (b) Robbins DW, Boebel TA, Hartwig JF. J. Am. Chem. Soc. 2010;132:4068. doi: 10.1021/ja1006405. [DOI] [PubMed] [Google Scholar]; (c) Cordes J, Wessel C, Harms K, Koert U. Synthesis. 2008:2217. [Google Scholar]; (d) Szumigala RH, Devine PN, Gauthier DR, Jr, Volante RP. J. Org. Chem. 2004;69:566. doi: 10.1021/jo035184p. [DOI] [PubMed] [Google Scholar]; (e) Ainley AD, Challenger F. J. Chem. Soc. 1930:2171. [Google Scholar]
- 9.For reviews, see: Darses S, Genêt J-P. Chem. Rev. 2008;108:288. doi: 10.1021/cr0509758. Doucet H. Eur. J. Org. Chem. 2008:2013. Molander GA, Ellis NM. Acc. Chem. Res. 2007;40:275. doi: 10.1021/ar050199q. Stefani HA, Cella R, Vieira AS. Tetrahedron. 2007;63:3623. Molander GA, Figueroa R. Aldrichim. Acta. 2005;38:49. Darses S, Genêt J-P. Eur. J. Org. Chem. 2003:4313.
- 10.For selected examples, see: Molander GA, Raushel J, Ellis NM. J. Org. Chem. 2010;75:4304. doi: 10.1021/jo1004058. Molander GA, Canturk B, Kennedy LE. J. Org. Chem. 2009;74:973. doi: 10.1021/jo802590b. Molander GA, Febo-Ayala W, Ortega-Guerra M. J. Org. Chem. 2008;73:6000. doi: 10.1021/jo800760f. Molander GA, Cooper DJ. J. Org. Chem. 2008;73:3885. doi: 10.1021/jo800383e. Molander GA, Ellis NM. J. Org. Chem. 2006;71:7491. doi: 10.1021/jo061324u. Molander GA, Felix LA. J. Org. Chem. 2005;70:3950. doi: 10.1021/jo050286w.
- 11.(a) Back TG, Chau JH-L, Dyck BP, Gladstone PL. Can. J. Chem. 1991;69:1482. [Google Scholar]; (b) Hiegel GA, Hogenauer TJ, Lewis JC. Synth. Commun. 2005;35:2099. [Google Scholar]
- 12.(a) Alder RW. J. Chem. Soc., Chem. Commun. 1980:1184. [Google Scholar]; (b) Hashimoto S, Kurimoto I, Fujii Y, Noyori R. J. Am. Chem. Soc. 1985;107:1427. [Google Scholar]
- 13.Rates for radical cyclization for this system are on the order of 108 s−1, see: Annunziata A, Galli C, Marinelli M, Pau T. Eur. J. Org. Chem. 2001:1323.
- 14.(a) Salzbrunn S, Simon J, Prakash GKS, Petasis NA, Olah GA. Synlett. 2000:1485. [Google Scholar]; (b) Prakash GKS, Panja C, Mathew T, Surampudi V, Petasis NA, Olah GA. Org. Lett. 2004;6:2205. doi: 10.1021/ol0493249. [DOI] [PubMed] [Google Scholar]
- 15.Molander GA, Colombel V, Braz VA. Org. Lett. 2011;13:1852. doi: 10.1021/ol2003572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo C-Y, Kumar MP, Chang H-K, Lush S-F, Liu R-S. J. Org. Chem. 2005;70:10482. doi: 10.1021/jo0518295. [DOI] [PubMed] [Google Scholar]
- 17.Darweesh WF, Shaaban MR, Farag AM, Metz P, Dawood KM. Synthesis. 2010:3163. [Google Scholar]
- 18.Terao J, Nakamura M, Kambe N. Chem. Commun. 2009:6011. doi: 10.1039/b915620h. [DOI] [PubMed] [Google Scholar]
- 19.Thirumamagal BTS, Narayanasamy S, Venkatesan R. Synth. Commun. 2008;38:2820. [Google Scholar]
- 20.Dai H-L, Liu W-Q, Xu H, Yang L-M, Lv M, Zheng Y-T. Chem. Pharm. Bull. 2009;57:84. doi: 10.1248/cpb.57.84. [DOI] [PubMed] [Google Scholar]
- 21.Hintou T, Kikuchi W, Mukaiyama T. Bull. Chem. Soc. Japan. 2003;76:1645. [Google Scholar]
- 22.Özkan H, Disli A, Yıldırır Y, Türker L. Molecules. 2007;12:2478. doi: 10.3390/12112478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pouchert CJ, Behnke J. The Aldrich library of 13C and 1H FT NMR Spectra. 1st ed. Milwaukee, WI: Aldrich Chemical Company, Inc.; 1993. [Google Scholar]
- 24.Walling C. J. Am. Chem. Soc. 1957;79:4187. [Google Scholar]
- 25.Lin Y-L, Chengy J-Y, Chu Y-H. Tetrahedron. 2007;63:10949. [Google Scholar]
- 26.Kiran YB, Ikeda R, Sakai N, Konakahara T. Synthesis. 2010:276. [Google Scholar]
- 27.Sarkar S, Grimme S, Studer A. J. Am. Chem. Soc. 2010;132:1190. doi: 10.1021/ja910540j. [DOI] [PubMed] [Google Scholar]
- 28.Scheiper B, Bonnekessel M, Krause H, Furstner A. J. Org. Chem. 2004;69:3943. doi: 10.1021/jo0498866. [DOI] [PubMed] [Google Scholar]
- 29.Chesterfield J, McOmie JFW, Sayer ER. J. Chem. Soc. 1955:3478. [Google Scholar]
- 30.Bull JA, Mousseau JJ, Charette AB. Org. Lett. 2008;10:5485. doi: 10.1021/ol802315k. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen C, Ruda GF, Schipani A, Kasinathan G, Leal I, Musso-Buendia A, Kaiser M, Brun R, Ruiz-Pérez LM, Sahlberg B-L, Johansson NG, Gonzalez-Pacanowska D, Gilbert IH. J. Med. Chem. 2006;49:4183. doi: 10.1021/jm060126s. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Li P-H. Chin. J. Chem. 2006;24:770. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.