

Abstract
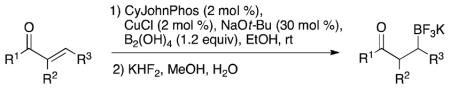
The copper-catalyzed β-boration of α,β-unsaturated carbonyl compounds with tetrahydroxydiborane has been developed. This diboron reagent allows direct, efficient access to boronic acids and their derivatives. Primary, secondary and tertiary α,β-unsaturated amides are converted to the corresponding β-trifluoroboratoamides in good to excellent yields. The β-boration of a variety of α,β-unsaturated esters and ketones is also reported.
Boron homoenolates are versatile compounds that have been developed as synthetic intermediates for a variety of transformations,1 including Suzuki coupling reactions.2 Significantly, secondary β-trifluoroboratoamides have been demonstrated to react stereospecifically with inversion of configuration in palladium catalyzed cross-couplings.2b These developments have greatly expanded the utility of trifluoroborato homoenolates in organic synthesis.
A particularly appealing approach to the synthesis of these compounds is the transition metal-catalyzed β-boration of readily available α,β-unsaturated carbonyl compounds using diboron reagents. The β-boration of α,β-unsaturated carbonyl compounds with bis(pinacolato)diboron is well established.3 Rhodium,4 platinum,5 palladium,6 nickel7 and copper6a–b,8 catalyzed methods have been previously reported. Of these, the copper catalyzed systems have received the most attention. Yun and coworkers were instrumental in expanding the substrate scope from the more traditional unsaturated ketones and esters to include the more challenging α,β-unsaturated amides.9
Although previously reported catalytic systems provide efficient access to pinacol boronate esters, they are less convenient for the syntheses of other boronate derivatives. Conversion of the pinacol esters to the corresponding trifluoroborate can be problematic,10 as pinacol can be difficult to remove from the crude reaction mixtures, requiring tedious purification protocols. Additionally, in the interest of atom economy, use of a borylating reagent that does not require the intermediacy of the pinacol ester would prove beneficial.
Despite the interest in copper catalyzed beta-boration, there have been only a few publications examining alternative diboron reagents,11 each of which provides access to a single boronate ester. Our group has recently established tetrahydroxydiborane (bisboronic acid, BBA)12 as an effective replacement for bis(pinacolato)diboron in palladium catalyzed Miyaura borylation reactions.13 This reagent offers direct access to boronic acids, simplifying purification and providing access to a variety of boronate derivatives through a single intermediate. Herein we report a method for the β-boration of α,β-unsaturated carbonyl compounds using BBA as an effective and atom economical source of boron. To our knowledge this is the first example of the β-boration of α,β-unsaturated carbonyl compounds with BBA, as well as the first copper-catalyzed reaction of this reagent.
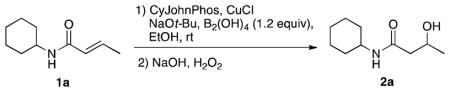
Reaction conditions for the β-borylation were optimized on (E)-N-cyclohexylbut-2-enamide. For ease of analysis, the initially formed boronic acid was converted in situ to the corresponding alcohol by oxidation with NaOH/H2O2. A screen of various solvents, alkoxide bases, copper(I) sources and phosphine ligands established EtOH, NaOt-Bu, CuCl, and 2-(dicyclohexylphosphino)biphenyl (CyJohnPhos) as the preferred conditions. A 1:1 ratio of ligand to copper and 1.2 equivalents of BBA was effective for >90% conversion after 4 hours.
We next sought to reduce the catalyst loading. Interestingly, initial attempts to reduce the catalyst load resulted in incomplete conversion even at long reaction times (Table 1, entries 1–3), despite the relatively short reaction times at high catalyst load. Consequently, attention was focused on the mechanism of the process and specifically the role of the base in these transformations (Scheme 1).9,14
Table 1.
Catalyst Load Optimizationa
 | |||||
|---|---|---|---|---|---|
| entry | CuCl (mol %) | CyJohnPhos (mol %) | NaOtBu (mol %) | reaction time (h) | P:SMb |
| 1 | 10 | 10 | 30 | 18 | >20:1 |
| 2 | 5 | 5 | 15 | 18 | 1.4:1 |
| 3 | 3 | 3 | 9 | 18 | 0.4:1 |
| 4 | 5 | 5 | 30 | 18 | >20:1 |
| 5 | 3 | 3 | 30 | 18 | >20:1 |
| 6 | 1 | 1 | 30 | 18 | >20:1 |
| 7 | 1 | 1 | 30 | 1 | >18:1 |
| 8 | 1 | 1 | 30 | 2 | >19:1 |
| 9 | 1 | 1 | 30 | 3 | >19:1 |
All reactions were carried out on a 0.3 mmol scale in 3 mL of EtOH with 1.2 equiv of BBA.
Ratio of product to starting material calculated by 1H NMR.
Scheme 1.

Proposed Mechanism
Previous studies have established that the base is critical in the formation of an sp2–sp3 species (I), creating a more nucleophilic intermediate that facilitates transmetalation to the active borylcopper species II.11c Insertion of the α,β-unsaturated carbonyl compound into the copper boron bond of II gives a copper enolate (III), which accepts a proton from EtOH to give the borylated product IV, regenerating a copper alkoxide catalyst. We hypothesized that the formation of a borate ester byproduct V sequestered the NaOt-Bu necessary for catalyst regeneration as the reaction proceeded toward completion. Therefore, the incomplete conversion at low catalyst loads could be due to insufficient quantities of base rather than of catalyst.
Encouragingly, we found that keeping 30 mol % of NaOt-Bu allowed a reduction in the amount of CuCl and CyJohnPhos to 1 mol % while retaining efficient conversion to the product (entries 4–6). Reactions run with 2 mol % of CuCl and CyJohnPhos were found to be more reproducible, and thus 2 mol % of both catalyst and ligand were used as the general conditions for subsequent reactions. Increasing the amount of NaOt-Bu beyond 30 mol % provided no advantage.
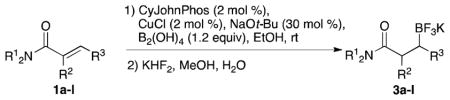
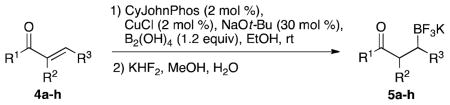
With optimized conditions in hand, we examined the substrate scope of this reaction (Table 2). We first varied the substitution on a series on N-cyclohexyl amides. Methyl substitution at the alpha position was well tolerated, as was mono-alkyl or phenyl substitution at the beta position. Branching at the gamma position was tolerated, but required a longer reaction time, increased catalyst loading, and 2 equiv of BBA for efficient conversion (Table 2, entry 5). Effective conversion could not be obtained, however, for compounds containing multiple substituents at the beta position, presumably for steric reasons (Table 2, entry 6). Next we examined the effect of the amine portion of the amide on the borylation reaction. Primary, secondary and tertiary amides were all tolerated, although primary amides (Table 2, entry 12) required longer reaction times. The reaction could be run on gram scale with only a slight reduction in yield (Table 2, entry 1). Unfortunately, attempts to isolate the boronic acid intermediate synthesized from 1a yielded an inseparable mixture of boronic acid and what appears to be boroxine, along with ligand and traces of starting material.
Table 2.
β-Boration of α,β-Unsaturated Amides with Bisboronic Acida
 | ||||
|---|---|---|---|---|
| entry | time (h) | product | yieldb (%) | |
| 1 | 2 |
|
3a | 90 (82)c |
| 2 | 2 |
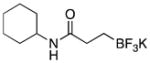
|
3b | 89 |
| 3 | 2 |
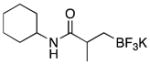
|
3c | 98 |
| 4 | 2 |
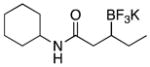
|
3d | 86 |
| 5 | 18 |
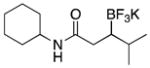
|
3e | 75d |
| 6 | 18 |
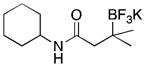
|
3f | −d |
| 7 | 24 |
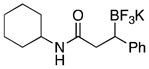
|
3g | 98 |
| 8 | 2 |
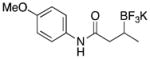
|
3h | 96 |
| 9 | 2 |
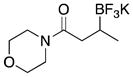
|
3i | 90 |
| 10 | 2 |
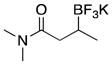
|
3j | 90 |
| 11 | 2 |
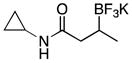
|
3k | 88 |
| 12 | 18 |
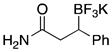
|
3l | 78 |
General conditions: Amide (1.0 equiv), BBA (1.2 equiv), CuCl (2 mol %), CyJohnPhos (2 mol %), NaOt-Bu (30 mol %), EtOH (0.1 M), rt. After filtration and solvent removal, MeOH, KHF2 (8.2 equiv), 0 °C to rt, 20 min.
Yield is given for isolated products.
Gram scale.
Used 2.0 equiv BBA, CuCl (10 mol %), NaOt-Bu (50 mol %) CyJohnPhos (10 mol %).
We were gratified to find that α,β-unsaturated esters and ketones were also efficiently borylated under the standard reaction conditions (Table 3). A variety of substitution patterns were effective for ketones, with cyclic and acyclic substrates proving to be good substrates for the reaction. Even sterically hindered ketones were borylated in good yield, albeit with a longer reaction time (Table 3, entry 3). Esters were equally effective coupling partners (Table 3, entries 5–8), and notably lactone 5h was a competent substrate.
Table 3.
β-Boration of α,β-Unsaturated Carbonyl Compounds with Bisboronic Acida
 | ||||
|---|---|---|---|---|
| entry | time (h) | product | yieldb (%) | |
| 1 | 2 |
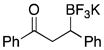
|
5a | 71 |
| 2 | 2 |
|
5b | 93 |
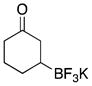
| 3 | 24 |
|
5c | 77 |
| 4 | 2 |
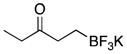
|
5d | 76 |
| 5 | 2 |
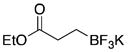
|
5e | 93 |
| 6 | 2 |
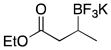
|
5f | 84 |
| 7 | 2 |
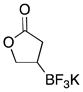
|
5g | 82 |
| 8 | 2 |
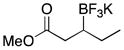
|
5h | 83 |
General conditions: Carbonyl substrate (1.0 equiv), BBA (1.2 equiv), CuCl (2 mol %), CyJohnPhos (2 mol %), NaOt-Bu (30 mol %), EtOH (0.1 M), rt. After filtration and solvent removal, MeOH, KHF2 (8.2 equiv), 0 °C to rt, 20 min.
Yield is given for isolated products.
In summary, we have developed an efficient copper catalyzed method for the synthesis of β-trifluoroborato carbonyl compounds using bisboronic acid as a source of boron – the first copper-catalyzed process for this atom economical reagent. This method uses EtOH as an environmentally benign solvent, avoids purification difficulties encountered in the conversion of pinacol esters to the corresponding trifluoroborates, and provides efficient access to β-amido trifluoroborates. Efforts toward an enantioselective variant are ongoing in our laboratory.
Supplementary Material
Acknowledgments
This research was supported by the NIGMS (R01 GM-035249). Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data. We thank BASF for a generous donation of tetrahydroxydiborane.
Footnotes
Supporting Information Available. Experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bonet A, Sole C, Gulyas H, Fernandéz E. Curr Org Chem. 2010;14:2531. [Google Scholar]
- 2.(a) Molander GA, Petrillo DE. Org Lett. 2008;10:1795. doi: 10.1021/ol800357c. [DOI] [PubMed] [Google Scholar]; (b) Sandrock DL, Jean-Gérard L, Cheng-yi C, Dreher SD, Molander GA. J Am Chem Soc. 2010;132:17108. doi: 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Molander GA, Jean-Gérard L. J Org Chem. 2009;74:1297. doi: 10.1021/jo802453m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Molander GA, Jean-Gérard L. J Org Chem. 2009;74:5446. doi: 10.1021/jo900968h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For a current review see: Schifner JA, Müther K, Oestreich Angew Chem Int Ed. 2010;49:1194. doi: 10.1002/anie.200906521.
- 4.Kabalka GW, Das BC, Das S. Tetrahedron Lett. 2002;43:2323. [Google Scholar]; (b) Shiomi T, Adachi T, Toribatake K, Zhou L, Nishiyama H. Chem Commun. 2009:5987. doi: 10.1039/b915759j. [DOI] [PubMed] [Google Scholar]
- 5.(a) Lawson Y, Lesly G, Marder T, Norman N, Rice C. Chem Commun. 1997:2051. [Google Scholar]; (b) Bell J, Cox A, Cameron N, Evans J, Marder T, Duin M, Elsevier C, Baucherel X, Tulloch A, Tooze R. Chem Commun. 2004:1854. doi: 10.1039/b406052k. [DOI] [PubMed] [Google Scholar]
- 6.(a) Lillo V, Geier MJ, Westcott SA, Férnandez E. Org Biomol Chem. 2009;7:4674. doi: 10.1039/b909341a. [DOI] [PubMed] [Google Scholar]; (b) Bonet A, Gulyás H, Koshevoy IO, Estevan F, Sanaú M, Úbeda MA, Fernandéz E. Chem Eur J. 2010;16:6382. doi: 10.1002/chem.200903095. [DOI] [PubMed] [Google Scholar]
- 7.Hirano K, Yorimitsu H, Oshima K. Org Lett. 2007;9:5031. doi: 10.1021/ol702254g. [DOI] [PubMed] [Google Scholar]
- 8.(a) Takahashi K, Ishiyama T, Miyaura N. Chem Lett. 2000:982. [Google Scholar]; (b) Ito H, Yamanaka H, Tateiwa J, Hosomi A. Tetrahedron Lett. 2000;41:6821. [Google Scholar]; (c) Chea H, Sim HS, Yun J. Adv Synth Catal. 2009;351:855. [Google Scholar]
- 9.Mun S, Lee J, Yun J. Org Lett. 2006;8:4887. doi: 10.1021/ol061955a. [DOI] [PubMed] [Google Scholar]
- 10.Bagutski V, Ros A, Aggarwal V. Tetrahedron. 2009;65:9956. [Google Scholar]
- 11.(a) Gao M, Thorpe SB, Santos WL. Org Lett. 2009;11:1899. doi: 10.1021/ol901359n. [DOI] [PubMed] [Google Scholar]; (b) Thorpe SB, Guo X, Santos WL. Chem Commun. 2011;47:424. doi: 10.1039/c0cc02270e. [DOI] [PubMed] [Google Scholar]; (c) Gao M, Thorpe SB, Kleeberk C, Slebodnick C, Marder TB, Santos WL. J Org Chem. 2011;76:3997. doi: 10.1021/jo2003488. [DOI] [PubMed] [Google Scholar]
- 12.BBA reactions in the literature: Sebelius S, Olsson V, Szabo K. J Am Chem Soc. 2005;127:10478. doi: 10.1021/ja052885q.Olsson V, Sebelius S, Selander N, Szabo K. J Am Chem Soc. 2006;128:4588. doi: 10.1021/ja060468n.Selander N, Kipke A, Sebelius S, Szabo K. J Am Chem Soc. 2007;129:13723. doi: 10.1021/ja074917a.Selander N, Szabo K. Chem Commun. 2008:3420. doi: 10.1039/b804920c.Pilarski LT, Szabo KJ. Angew Chem Int Ed. doi: 10.1002/anie.201102384.
- 13.Molander GA, Trice SLJ, Dreher SD. J Am Chem Soc. 2010;132:17701. doi: 10.1021/ja1089759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dang Li, Lin Z, Marder T. Organometallics. 2008;27:4443. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.