

Abstract
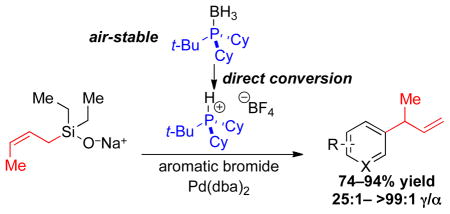
The γ-selective, palladium-catalyzed cross-coupling of sodium (Z)-2-butenyldiethylsilanolate with a variety of aromatic bromides is reported. The protocol provides high yields (73–94%) and site-selectivity (γ/α, 25:1–>99:1) in the coupling of electron-rich, electron-poor, sterically hindered, and heteroaromatic bromides. The use of a configurationally homogeneous (Z)-silanolate, nontransferable ethyl groups, and a sterically bulky trialkylphosphonium tetrafluoroborate salt (t-BuCy2PH+BF4−) prepared directly from the corresponding air-stable phosphine•borane adduct are critical to the success of the method.
The trend toward more active and selective palladium-catalysts has fueled efficient and selective synthetic reactions.1 Specifically, palladium-catalysts incorporating sterically bulky, electron-rich trialkylphosphine ligands for use in C–C bond forming cross-coupling reactions has allowed for milder reaction conditions and the inclusion of previously inaccessible substrates.2,3 However, the synthesis and handling of these highly air-sensitive phosphines poses a technical challenge. The introduction of tetrafluoroborate salts provides bench-stable precursors to air-sensitive trialkylphosphines with no significant change in reactivity when used under basic reaction conditions.4
Previous reports from these laboratories describe the remarkable effect of π-acidic olefin ligands on the site-selectivity of C–C bond formation in the palladium-catalyzed cross-coupling of allylic silanolate salts with aromatic bromides.5 These reaction conditions did however display some sensitivity to the electronic properties of the substrate. Furthermore, stereochemical correlation of products from couplings with an enantio-enriched, α,γ-disubstituted allylic silanolate established that these reagents stereospecifically transfer the allyl group to palladium through a syn SE’ process with complete stereochemical fidelity.6 Taken together, these studies provide a structural model of transmet-alation that invokes a tetracoordinate Pd(II) intermediate bearing a single ancillary ligand, and also highlights the importance of the E vinylic substituent R1 in its placement relative to the palladium ligand periphery (Scheme 1). In addition, recent mechanistic studies with isolated palladium(II) silanolate complexes indicate that nucleo-philic attack at silicon to form hypervalent 10-Si-5 species can have a dramatic effect on the rate of trans-metalation.7 This model prompted our investigation of the effect of silanolate olefin geometry and nontransferable group on reaction generality, rate and selectivity when combined with bulky, monodentate phophine ligands. Our ultimate goal was to identify catalysts that are both more active and less sensitive to the electronic properties of the substrate.
Scheme 1.

Summary of the proposed mechanism illustrating the key transmetalation transition-state structure i.
In a preliminary evaluation of bulky monodentate phosphine ligands, the combination of Pd(dba)2 and t-BuCy2PH+BF4− (3) provided a highly reactive catalyst for the cross-coupling of allylic silanolate salts with aromatic bromides. Therefore, this ligand was used to study the effect of the nontransferable group and olefin geometry of the allylic silanolate on the efficiency and selectivity of the cross-coupling reaction (Table 1). The use of an allylic silanolate with nontransferable methyl groups and (E)-olefin geometry (E)-1a provided a 57% yield of 5a and 42:1 site-selectivity favoring the γ-coupled product (entry 1). Changing the olefin geometry to Z raised the yield of 5a to 92% with slightly lower γ-selectivity (entry 2). Low reactivity was exhibited by the more bulky, diethyl substituted silanolate (E)-1b with only 15% conversion of the aromatic bromide observed (entry 3). Strikingly, the combination of (Z)-olefin geometry and nontransferable ethyl groups (Z)-1b led to higher reaction efficiency (conversion) and exquisite γ-selectivity at a slightly reduced reaction conversion (entry 4). The superior reactivity of (Z)-silanolates relative to (E)-silanolates under these conditions supported our hypothesis that the the disposition of the vinylic methyl group in the transmetalation transition-state structure i is important. The lower conversion observed for diethylsilanolates suggests a slower displacement by the bulkier silanolate at the palladium center to form the requisite Si–O–Pd linkage, however the excellent γselectivity of (Z)-1b warranted further study.
Table 1.
Effect of Silanolate Nontransferable Group and Olefin Geometrya
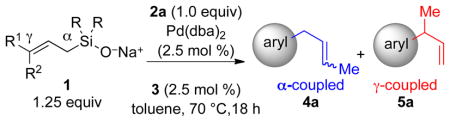 | ||||||
|---|---|---|---|---|---|---|
| entry | 1, R | R1 | R2 | conv,b% | yield γ,b % | γ/αc |
| 1 | 1a, Me | Me | H | 96 | 57 | 42:1 |
| 2 | 1a, Me | H | Me | 100 | 92 | 18:1 |
| 3 | 1b, Et | Me | H | 15 | 4 | – |
| 4 | 1b, Et | H | Me | 69 | 67 | >99:1 |
Reactions performed on 0.1 mmol scale, 2a = 3,5-dimethylbromobenzene, and aryl = 3,5-dimethylbenzene.
Determined by GC analysis.
GC peak area ratio of crude reaction mixtures.
To determine the effect of other bulky, monodentate phosphonium tetrafluoroborate salts required ready access to a variety of ligands. The tetrafluoroborate salts provide a number of technical advantages and similar reactivity (after in situ deprotonation) to the corresponding trialkylphosphines.4 These technical advantages are offset when an air-sensitive phosphine is needed that requires purification or further synthetic elaboration. A significant drawback of these salts is the inability to purify them by silica gel chromatography, or recover them from alkaline or ionic reaction conditions. Conversely, bench-stable phosphine•borane adducts can be carried through multistep synthesis employing, reductive, oxidative, aqueous acidic, or strongly basic conditions, and be easily purified by recrystallization, sublimation or silica-gel chromatography.8 Moreover, trialkylphosphine•borane adducts can be handled without the use of rigorous Schlenk technique or a dry-box.
To achieve the synthesis of trialkylphosphonium tetrafluoroborate salts that bypasses the handling of pyrophoric trialkylphosphines, experimental conditions were developed to transform air-stable phosphine•borane adducts directly into phosphonium tetrafluoroborate salts (Scheme 2). Preparation of 3, began by treatment of Cy2PCl9 with t-BuLi at −78 °C,10 followed by addition of BH3•THF at 0 °C to provide the trialkylphosphine•borane adduct 6 in 78% yield after recrystallization (mp 122–123 °C). Treatment of 6 with HBF4•OEt211 in CH2Cl2 at 0 °C followed by an aqueous fluoroboric acid wash provided a 90% yield of analytically pure 3 (mp 230–231 °C). Likewise, 9 and 10 were prepared from the corresponding trialkylphosphine•borane adducts (7 and 8)12 in high yield under these conditions.
Scheme 2.

Preparation of Trialkylphosphonium Tetrafluoroborate Salts from Phosphine•Borane Adducts
With the trialkylphosphonium salts in hand, the reaction parameters for cross-coupling of (Z)-1b were optimized (Table 2). The use of 9 provided a low yield of the desired product (entry 1). The less bulky ligand 10 provided a similar result to that of 3, however better γselectivity and yield of 5a were achieved with 3. The higher yield and γ-selectivity provided by 3 relative to the other ligands examined suggest this ligand provides an ideal steric environment that allows for transmetalation but retards α-coupling by obstruction of the tetracoordinate π-allyl intermediate. Increasing the reaction concentration provided better conversion and yield (entries 3–5). The use of 1.5 equiv of silanolate provided optimal results (entries 5–7) and Pd(dba)2 was a superior palladium source compared to [allylPdCl]2 (APC) (c.f. entries 5 and 8). The increased γ-selectivity provided by Pd(dba)2 suggests the π-acidic olefin ligand dba continues to serve a beneficial role.13 Reactions were slightly slower at ligand stoichiometries greater than 1:1 with repect to palladium (entries 11 and 12). Only a small decrease in product yield was noted when the reaction temperature was increased to 90 °C (entry 14).
Table 2.
Optimization of Cross-Coupling Reaction Using Trialkylphosphonium Tetrafluoroborate Saltsa
 | ||||||
|---|---|---|---|---|---|---|
| entry | (Z)-1b (equiv) | ligand, mol % | concn (M) | conv,b % | yield γ-5a,b % | γ/αc |
| 1d | 1.5 | 9, 2.5 | 0.5 | 100 | 50 | 38:1 |
| 2d | 1.5 | 10, 2.5 | 0.5 | 100 | 88 | 51:1 |
| 3 | 1.5 | 3, 2.5 | 0.5 | 100 | 94 | >99:1 |
| 4 | 1.5 | 3, 2.5 | 0.25 | 92 | 85 | >99:1 |
| 5 | 1.5 | 3, 2.5 | 1.0 | 100 | 99 | >99:1 |
| 6 | 1.25 | 3, 2.5 | 1.0 | 69 | 67 | >99:1 |
| 7 | 2.0 | 3, 2.5 | 1.0 | 100 | 96 | >99:1 |
| 8e | 1.5 | 3, 2.5 | 1.0 | 97 | 86 | 17:1 |
| 9f | 1.5 | 3, 1.25 | 1.0 | 18 | 18 | >99:1 |
| 10g | 1.5 | 3, 5.0 | 1.0 | 100 | 99 | >99:1 |
| 11 | 1.5 | 3, 5.0 | 1.0 | 93 | 91 | >99:1 |
| 12 | 1.5 | 3, 10 | 1.0 | 89 | 88 | >99:1 |
| 13h | 1.5 | 3, 2.5 | 1.0 | 18 | 18 | >99:1 |
| 14i | 1.5 | 3, 2.5 | 1.0 | 100 | 93 | >99:1 |
Reactions performed on 0.1 mmol scale.
Determined by GC analysis.
GC peak area ratio of crude reaction mixture.
Average of 2 experiments.
APC (1.25 mol %) used as the catalyst.
Pd(dba)2 (1.25 mol %).
Pd(dba)2 (5.0 mol %).
50 °C.
90 °C.
The results from 1.0 mmol scale reactions of a variety of aromatic bromides with (Z)-1b are compiled in Table 3. The model substrate 2a provided the coupling product with excellent γ-selectivity (determined prior to purification) and isolated yield (after aqueous work-up, chromatography and distillation) (entry 1). Likewise, the electron-neutral aromatic bromides 2b–d are excellent substrates in this reaction (entries 2–4). Substrates bearing electron-donating groups provided high yield and γ-selectivity under these conditions (entries 4–6). Noteworthy are the good yields and excellent selectivities observed with substrates containing sterically bulky ortho-substitution, including 2i and the di-ortho-substituted 2h (entries 7 and 8). Substrates bearing electron-withdrawing substituents also reacted smoothly (entries 10 and 11). Heterocyclic bromides, like 2l and 2m are good substrates for allylations under these conditions (entries 12 and 13). Moreover, the racemic precursor14a 5n to the nonsteroidal anti-inflammatory drug naproxen14b was prepared in high yield with excellent γselectivity under these conditions (entry 14).
Table 3.
Preparative Palladium-Catalyzed Allylations of Substituted Aromatic Bromides Using (Z)-1b and 3a
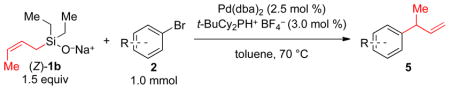 | |||
|---|---|---|---|
| entry | product | yield,b % | γ/αc |
| 1 |
 5a |
90 | >99:1 |
| 2 |
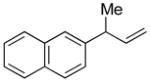 5b |
92 | >99:1 |
| 3 |
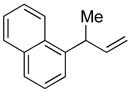 5c |
89 | >99:1 |
| 4 |
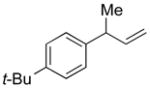 5d |
94 | >99:1 |
| 5 |
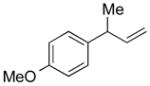 5e |
85 | >99:1 |
| 6 |
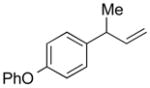 5f |
84 | >99:1 |
| 7 |
5g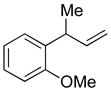
|
85 | >99:1 |
| 8 |
5h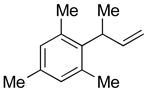
|
73 | >99:1 |
| 9 |
5i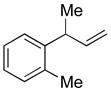
|
74 | >99:1 |
| 10d |
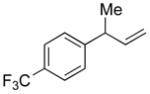 5j |
82 | >99:1 |
| 11 |
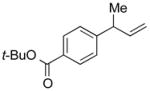 5k |
91 | >99:1 |
| 12 |
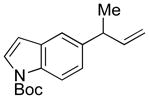 5l |
88 | 99:1e |
| 13d,f |
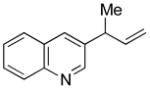 5m |
82 | 25:1 |
| 14 |
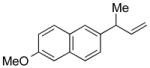 5n |
91 | >99:1 |
Reactions performed on 1.0 mmol scale.
Yield of isolated, purified product.
GC peak area ratio of crude reaction mixture.
5% Pd(dba)2 and 6% t-BuCy2PH+BF4− used.
Determined by 1H NMR analysis (>100:1 S/N).
90 °C reaction temperature used.
The judicious combination of allylic silanolate olefin geometry, nontransferable group, and trialkylphosphonium tetrafluoroborate salt allows for the γ-selective palladium-catalyzed allylation reaction of allylic silanolates with a variety of electronically and sterically differentiated aromatic bromides. Moreover, (Z)-1b is easily prepared from configurationally homogeneous (Z)-crotyltrichlorosilane obtained from the palladium-catalyzed silylation of 1,3-butadiene with trichlorosilane.15 Additionally, the direct conversion of phosphine•borane adducts to useful trialkylphosphonium tetrafluoroborate salts should find widespread utility to chemists interested in the synthesis and application of trialkylphosphine ligands.
Supplementary Material
Acknowledgments
We are grateful for the National Institutes of Health for generous financial support (R01 GM63167). N.S.W. acknowledges the Aldrich Chemical Co. for a Graduate Student Innovation Award and Eli Lilly and Co. for a Graduate Fellowship.
Footnotes
Supporting Information Available. Detailed experimental procedures and spectral characterization of all products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Magano J, Dunetz JR. Chem Rev. 2011;111:2177–2250. doi: 10.1021/cr100346g. [DOI] [PubMed] [Google Scholar]; (b) Torborg C, Beller M. Adv Synth Catal. 2009;351:3027–3043. [Google Scholar]; (c) Fleckenstein CA, Plenio H. Chem Soc Rev. 2010;39:694–711. doi: 10.1039/b903646f. [DOI] [PubMed] [Google Scholar]; (d) Fu G. Acc Chem Res. 2008;41:1555–1564. doi: 10.1021/ar800148f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hartwig JF. Acc Chem Res. 2008;41:1534–1544. doi: 10.1021/ar800098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Shen W. Tetrahedron Lett. 1997;38:5575–5578. [Google Scholar]; (b) Littke AF, Fu GC. Angew Chem Int Ed. 1998;37:3387–3388. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3387::AID-ANIE3387>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (c) Old DW, Wolfe JP, Buchwald SL. J Am Chem Soc. 1998;120:9722–9723. [Google Scholar]
- 3.(a) Fitton P, Rick EA. J Organometal Chem. 1971;28:287–291. [Google Scholar]; (b) Bedford RB, Cazin CSJ, Holder D. Coord Chem Rev. 2004;248:2283–2321. [Google Scholar]
- 4.Netherton MR, Fu GC. Org Lett. 2001;3:4295–4298. doi: 10.1021/ol016971g. [DOI] [PubMed] [Google Scholar]
- 5.Denmark SE, Werner NS. J Am Chem Soc. 2008;130:16382–16393. doi: 10.1021/ja805951j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denmark SE, Werner NS. J Am Chem Soc. 2010;132:3612–3620. doi: 10.1021/ja910804u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denmark SE, Smith RC. J Am Chem Soc. 2010;132:1243–1245. doi: 10.1021/ja907049y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Staubitz A, Robertson APM, Sloan ME, Manners I. Chem Rev. 2010;110:4023–4078. doi: 10.1021/cr100105a. [DOI] [PubMed] [Google Scholar]
- 9.Issleib K, Seidel W. Chem Ber. 1959;92:2681–2694. [Google Scholar]
- 10.Jan D, Delaude L, Simal F, Demonceau A, Noels AF. J Organometal Chem. 2000;606:55–64. [Google Scholar]
- 11.(a) McKinstry L, Livinghouse T. Tetrahedron Lett. 1994;35:9319–9322. [Google Scholar]; (b) McKinstry L, Livinghouse T. Tetrahedron. 1995;51:7655–7666. [Google Scholar]; (c) McKinstry L, Overberg JJ, Soubra-Ghaoui C, Walsh DS, Robins KA, Toto TT, Toto JL. J Org Chem. 2000;65:2261–2263. doi: 10.1021/jo9919456. [DOI] [PubMed] [Google Scholar]
- 12.Overschelde MV, Vervecken E, Modha SG, Cogen S, Eycken EVD, Eycken JVD. Tetrahedron. 2009;65:6410–6415. [Google Scholar]
- 13.Perez-Rodriquez M, Braga AAC, Garcia-Melchor M, Perez-Temprano M, Casares JA, Ujaque G, de Lera AR, Alvarez R, Maseras F, Espinet P. J Am Chem Soc. 2009;131:3650–3657. doi: 10.1021/ja808036j. [DOI] [PubMed] [Google Scholar]
- 14.(a) Smith CR, RajanBabu TV. J Org Chem. 2009;74:3066–3072. doi: 10.1021/jo900198b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Harrington PJ, Lodewijk E. Org Process Res Dev. 1997;1:72–76. [Google Scholar]
- 15.Kira M, Hino T, Sakurai H. Tetrahedron Lett. 1989;30:1099–1102. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.