

Abstract
By differentiating the functional groups on nucleosides, we have designed and developed a one-pot synthesis of deoxyribonucleoside 5′-triphosphates without any protections on the nucleosides. A facile synthesis is achieved by generating an in situ phosphitylating reagent that reacts selectively with the 5′-hydroxyl groups of the unprotected nucleosides. The synthesized triphosphates are of high quality and can be effectively incorporated into DNAs by DNA polymerase. This novel approach is straightforward and cost-effective for triphosphate synthesis.
Nucleoside 5′-triphosphates (dNTPs and NTPs) are the building blocks for the synthesis of nucleic acids (DNA and RNA) and are also utilized in many important biological systems, including DNA replication, RNA transcription, purinergic signaling, neurotransmission, and signal transduction.1 To better understand the roles of triphosphates and meet the needs in nucleic acid research, the first chemical synthesis of nucleoside 5′-triphosphates was achieved over six decades ago.2 Although numerous strategies have been continuously developed in the past decades,3 a convenient synthesis of the nucleoside 5′-triphosphates remains as a long-standing challenge. This is primarily due to the multiple functionalities (hydroxyl and amino groups) of the nucleosides, which generally requires many synthetic steps because of the protection and deprotection of these functionalities. Moreover, the synthesis of nucleoside 5′-triphosphates generates many by-products that are very difficult to remove. Thus, there is an urgent need to develop straightforward strategies for synthesizing nucleoside 5′-triphosphates in order to reduce the cost of triphosphates (especially modified ones) significantly and meet the growing needs in signal regulation research and studies of nucleic acid structure, function, and detection.4
Nucleoside triphosphates are currently synthesized via three major approaches: phosphitylation (or phosphoramiditylation), 3a–c, 5 mono-phosphate isolation/activation,3c–e, 6 and phosphorylation.3b, c, 7 The first strategy employs a highly reactive phosphitylating agent, thereby requiring the protection of both the sugar and nucleobase moieties3c, 5a–c, 5f, g in order to reduce unwanted by-products. Since the 5′-hydroxy groups of the nucleosides are more reactive than the 3′- and 2′-hydroxyl groups, selective protection of the 3′- and 2′-hydroxyl groups, as well as the amino groups on the nucleobases are problematic. Nucleosides undergo lengthy selective protection and deprotection to obtain the partially-protected intermediates containing the free 5′-hydroxyl groups, before the phosphitylation reaction. The protecting groups are normally removed after the phosphite oxidation and the cyclic triphosphate hydrolysis, affording the nucleoside 5′-triphosphates.
The second strategy relies on the nucleoside 5′-monophosphate synthesis, isolation and activation prior to the pyrophosphate treatment.3b, 3d, e, 6 Clearly, this strategy is not ideal for a convenient synthesis. In the third strategy, highly reactive phosphorous oxychloride (POCl3) is employed as the phosphorylating agent, and the trimethylphosphate solvent used in the synthesis aids at reducing the POCl3 reactivity, primarily generating the dichlorophosphate at the 5′-position (equivalent to an activated monophosphate) prior to the pyrophosphate treatment. In theory, the third strategy requires no protection of the nucleosides.7a–c In practice; however, this strategy usually generates numerous by-products (such as the regio-isomers and oligophosphates) and causes purification difficulties, which have even been reported recently.8
To minimize the unwanted by-products and achieve highquality products, we designed a mild and selective phosphitylating reagent in order to differentiate these functionalities. To create a differentiating reagent, we decided to take advantage of the high reactivity of salicyl phosphorochloridite5 and converted it into a weak phosphitylating reagent. After many trials (data not shown), we found that phosphates, such as monophosphate and pyrophosphate, were capable of effectively reducing the reactivity of salicyl phosphorochloridite. Finally, we chose pyrophosphate to both tailor the reactivity and generate the triphosphates after the oxidation and hydrolysis in a one pot synthesis (Scheme 1). Our selective phosphitylating reagent (2) can be generated in situ, and without any purification, it can selectively react with the 5′-hydroxyl groups of nucleosides that contain no protections on the sugar and nucleobases. To demonstrate the proof of principle, we report here a convenient synthesis of the native and modified deoxyribonucleoside triphosphates and demonstrate DNA polymerase extension and PCR reactions using the synthesized native 5′-triphosphates.
Scheme 1.

One-pot synthesis of dNTPs
Nucleoside 5′-triphosphates (4) were synthesized conveniently (Scheme 1). After reacting salicyl phosphorochloridite with pyrophosphate to generate the selective phosphitylating reagent (2), an unprotected nucleoside was added, followed by iodine oxidation and hydrolysis. The developed synthetic conditions are mild, and this strategy was extended to one-pot synthesis of ethano-deoxyadenosine 5′-triphosphate (EdATP, 4e) successfully. 31P-NMR study was also performed to monitor the entire synthesis (Figure 1), where thymidine (d) was used as the model compound. The NMR study of the reactions was conducted by using deuterated DMF (DMF-d7) as the solvent. After the reaction proceeded for 30 min, intermediate 2 was observed as the major product, revealed by an upfield chemical shift from singlet 124.7 ppm (Figure 1b) to 98.9 ppm (td, Figure 1c). Intermediate 2 was formed by two nucleophilic reactions between 1 and pyrophosphate, which displaced the chloride and carboxylate. This mild phosphitylating reagent (2) is also bulky and has the selectivity towards the more reactive 5′-hydroxyl group. After thymidine addition, the other key intermediate (5′-cyclic triphosphite 3d) was observed as the major product 1 hr later, revealed by 105.7 ppm (triplet, Figure 1d). Since nucleosides are much cheaper comparing with the corresponding synthesized triphosphates, we have purposely allowed only a majority of nucleosides to be consumed (approximately 70%) in order to maintain a high regio-selectivity of 5′-cyclic triphosphite intermediates. After the oxidation, hydrolysis and NaCl-ethanol precipitation, the overall reaction yields of dNTPs are 19–46% (determined by HPLC), and the precipitated dNTPs are cable of DNA polymerase and PCR reactions. Furthermore, to confirm their integrity, all synthesized dNTPs were analyzed by NMR and MS, shown in Table 1 and Supporting Information.
Figure 1.

31P-NMR monitoring of 5′-triphosphate 4d formation at room temperature (in DMF-d7; reference: H3PO4 as an external standard). a) Tributylammonium pyrophosphate (−10.2 ppm; b) salicyl phosphorochloridite (124.7 ppm); c) proposed formed phosphitylating reagent 2 in situ, chemical shift observed at 98.9 ppm.; d) the trivalent phosphorous of cyclic triphosphite 3d (105.7 ppm); e) thymidine 5′-triphosphate 4d.
Table 1.
ESI-TOF [M−H]− mass spectrometry analysis of 2′-deoxynucleoside 5′-triphosphates
| entry | dNTP | chemical formula | measured [M−H+]− | (calcd) m/z |
|---|---|---|---|---|
| 4a | dATP | C10H16N5O12P3 | 489.9938 | (489.9936) |
| 4b | dCTP | C9H16N3O13P3 | 465.9814 | (465.9817) |
| 4c | dGTP | C10H16N5O13P3 | 505.9893 | (505.9885) |
| 4d | TTP | C10H17N2O14P3 | 480.9831 | (480.9820) |
| 4e | EdATP | C12H16N5O12P3 | 513.9940 | (513.9936) |
Because of high selectivity of the 5′-triphosphate synthesis, synthesized dNTPs (crude products) can be easily purified by HPLC to offer high purity (typical HPLC shown in Figure 2 and Supporting Information), while ion exchange strategy is not very effective to remove the minor 3′-triphosphates from the 5′-triphosphates. The nucleoside 3′-triphosphate (approximately 5–10%) is the major by-product. The minor peak shown in Figure 2 was isolated via HPLC and characterized via comparison by HPLC and MS with the previously synthesized thymidine 3′-triphosphate. The presence of excess of 1 in the reaction could undergo a nucleophilic attack by the unprotected 3′-hydroxyl group of a deoxynucleoside, producing the undesired 3′-product. We later found that using excess pyrophosphate (such as 2 eq.) to 1 could completely convert 1 to 2, and minimize the by-product formation. In addition, lower temperature (0 or −10 °C), which further reduces reaction rate, can generally minimize formation of the by-products, as observed by RP-HPLC analysis (data not shown). RP-HPLC analysis and co-injection with the dNTP standards revealed that the by-products, commonly observed with the conventional strategies, are significantly reduced when our new approach is used.
Figure 2.
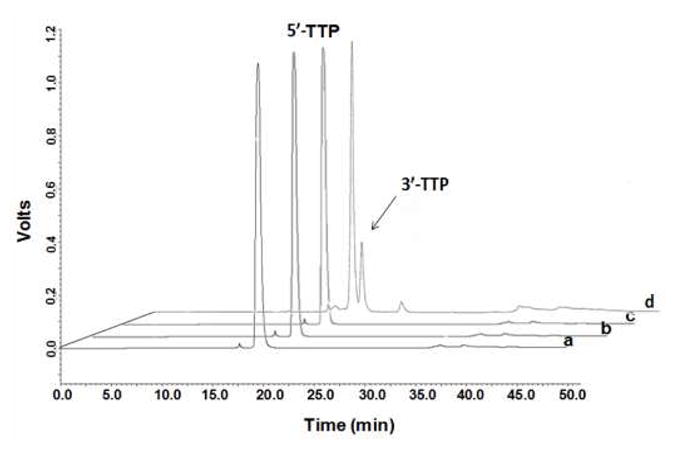
RP-HPLC profiles of chemically synthesized and commercial 2′-deoxynucleoside 5′-triphosphates. a) Synthesized 5′-TTP after NaClethanol precipitation and RP-HPLC purification (retention time: 19.8 min); b) standard 5′-TTP (retention time: 19.4 min); c) co-injection of a and b (retention time: 19.4 min); and d) crude 5′-TTP and 3′-TTP after NaCl-ethanol precipitation (retention time: 19.4 and 20.7 min), respectively.
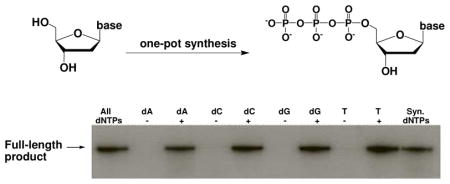
To further confirm the quality of the synthetic dNTPs, we have demonstrated their ability as substrates for DNA polymerization. By using a 32P-labeled primer, we performed the DNA polymerization on a DNA template (Figure 3) with all commercial dNTPs as a positive control, and the HPLC purified or precipitateddNTPs synthesized as the test substrates. Weconducted four polymerization reactions by omitting one dNTP each time to serve as the negative controls. As expected, the positive control gave the full-length DNA product (Lane 2 in Figure 3), while the negative controls with one dNTP missing did not generate any full-length DNA (Lane 3, 5, 7 and 9) and the DNA synthesis stopped after synthesizing short fragments. We then used the HPLC purified individual dNTPs to compensate for the corresponding missing commercial dNTPs in each polymerization reaction, and compared them with the positive control. We observed that DNA polymerase recognized the synthesized dNTPs as well as the standard dNTPs. Polymerase efficiently extended the primer for each reaction containing an individual synthesized dNTP (Lane 4, 6, 8 and 10) to the expected full-length DNA products identical to the DNA synthesized by using the commercial 5′-triphosphates as standard (Lane 2). The incorporation efficiency of the synthesized dNTPs was virtually identical to the standard ones. More convincingly, in the reaction containing all synthetic triphosphates (Lane 11), the full-length DNA was generated similarly to the reaction containing all of the standard dNTPs (Lane 2). Since a DNA polymerase does not recognized 3′-triphosphates as substrates, we conducted polymerase reactions with the crude dNTPs. Our results show that the precipitated synthetic dNTPs can also allow both DNA primer extension and PCR reactions (Figure S16 and 17). Our typical synthesis scale is approximately 0.1 mmole, which can generate sufficient dNTP for 200 PCR reactions (50 μL, 1 mM dNTP each). Furthermore, the synthesis can be easily scaled up; we have done the synthesis at 100-mg scale without any difficulties.
Figure 3.

A. Primer and template sequences used in the polymerization experiment. B. Primer extension reaction of chemically synthesized dNTPs and commercially available dNTPs into DNA by Klenow fragment exo(−) (Kf−). The primer was 5′-end labeled using polynucleotide kinase and [γ-32P] ATP. Polymerization reactions were performed with primer (3.5 μM), template (5 μM), all dNTPs (1.0 mM each), and Kf− (0.05 U/μL) at 37 °C for 1 h. Reactions were analyzed by 19% polyacrylamide gel electrophoresis. Lane 1: primer (P) and all dNTPs, but no Kf−; Lane 2 (positive control): P, template (T), all commercial dNTPs, and Kf−; Lane 3, 5, 7 and 9 (negative controls) omitted dATP, dCTP, dGTP and TTP from Lane 2, respectively; Lane 4, 6, 8 and 10 were compensated with the synthesized dATP, dCTP, dGTP and TTP (respectively) to the corresponding Lane 3, 5, 7 and 9. Lane 11: P, T, all synthesized dNTPs, and Kf−; Lane 12: 5′-32P-labled primer.
In summary, by generating a selective phosphitylating reagent in situ, we have developed a straightforward approach to conveniently synthesize deoxynucleoside 5′-triphosphates without any protection of the nucleosides. The proof of principle was demonstrated by using modified and nonmodified nucleosides. This in situ phosphitylating reagent, generated by reacting salicyl phosphorochloridite with pyrophosphate, is mild and permits high regioselectivity at the 5′-hydroxyl group for the triphosphate synthesis. The quality of the synthesized dNTPs was also confirmed by DNA polymerase primer extension and PCR reactions. Our one-pot synthesis is convenient and cost-effective and eliminates the lengthy protection and deprotection steps associated with most conventional approaches. Moreover, it reduces by-product formation and purification difficulty. This mild chemistry makes synthesis of sensitive modified triphosphates possible. Such a strategy is also applicable to the synthesis of nucleoside α-modified 5′-triphosphates. For instance, intermediate 3 may be oxidized with either sulfur or selenium (data not shown) and may likely be oxidized by borane reagents to afford the corresponding nucleoside 5′-(α-P-thiotriphosphates),5a, 5d, e nucleoside 5′-(α-P-selenotriphosphates),9 or nucleoside 5′-(α-Pboranotriphosphates). 5b, c, 5f, g This general strategy enables scale-up synthesis of a large number of modified triphosphates to meet the emerging needs in nucleic acid structure and function studies and detection research.
Supplementary Material
Acknowledgments
This work was financially supported by NIH (GM095086), NSF (MCB-0824837) and the Georgia Cancer Coalition (GCC) Distinguished Cancer Clinicians and Scientists.
Footnotes
Supporting Information Available: Experimental procedures, 1H, 13C and 31P NMR, HRMS and MALDI-TOF MS analytical data, HPLC profiles. These materials are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Soutourina J, Wydau S, Ambroise Y, Boschiero C, Werner M. Science. 2011;331:1451–4. doi: 10.1126/science.1200188. [DOI] [PubMed] [Google Scholar]; (b) Bogdanov AA, Sumbatyan NV, Shishkina AV, Karpenko VV, Korshunova GA. Biochemistry (Mosc) 2010;75:1501–16. doi: 10.1134/s0006297910130018. [DOI] [PubMed] [Google Scholar]; (c) Storz G. Science. 2002;296:1260–3. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]; (d) McManus MT, Sharp PA. Nat Rev Genet. 2002;3:737–47. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]; (e) Eckstein F, Thomson JB. Methods Enzymol. 1995;262:189–202. doi: 10.1016/0076-6879(95)62018-4. [DOI] [PubMed] [Google Scholar]; (f) Hoffman BL, Ullrich A, Wold WS, Carlin CR. Mol Cell Biol. 1990;10:5521–4. doi: 10.1128/mcb.10.10.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Gish G, Eckstein F. Science. 1988;240:1520–2. doi: 10.1126/science.2453926. [DOI] [PubMed] [Google Scholar]
- 2.Baddiley J, Michelson AM, Todd AR. J Chem Soc (London) 1949:582–586. [Google Scholar]
- 3.(a) Zou K, Horhota A, Yu B, Szostak JW, McLaughlin LW. Org Lett. 2005;7:1485–7. doi: 10.1021/ol050081+. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Horhota AT, Szostak JW, McLaughlin LW. Org Lett. 2006;8:5345–7. doi: 10.1021/ol062232u. [DOI] [PubMed] [Google Scholar]; (c) Burgess K, Cook D. Chemical reviews. 2000;100:2047–60. doi: 10.1021/cr990045m. [DOI] [PubMed] [Google Scholar]; (d) Sun Q, Edathil JP, Wu R, Smidansky ED, Cameron CE, Peterson BR. Org Lett. 2008;10:1703–6. doi: 10.1021/ol8003029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wu W, Freel Meyers CL, Borch RF. Org Lett. 2004;6:2257–60. doi: 10.1021/ol049267j. [DOI] [PubMed] [Google Scholar]; (f) Antonov KV, Esipov RS, Gurevich AI, Chuvikovskii DV, Mikulinskaia GV, Feofanov SA, Miroshnikov AI. Bioorg Khim. 2003;29:616–22. doi: 10.1023/b:rubi.0000008897.08102.ee. [DOI] [PubMed] [Google Scholar]; (g) Schultheisz HL, Szymczyna BR, Scott LG, Williamson JR. J Am Chem Soc. 2010;133:297–304. doi: 10.1021/ja1059685. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Jansen RS, Rosing H, Schellens JH, Beijnen JH. Nucleosides Nucleotides Nucleic Acids. 2010;29:14–26. doi: 10.1080/15257770903451546. [DOI] [PubMed] [Google Scholar]; (i) Warnecke S, Meier C. J Org Chem. 2009;74:3024–30. doi: 10.1021/jo802348h. [DOI] [PubMed] [Google Scholar]; (j) Warnecke S, Meier C. Nucleic Acids Symp Ser (Oxf) 2008:583–4. doi: 10.1093/nass/nrn295. [DOI] [PubMed] [Google Scholar]; (k) Bettendorff L, Nghiem HO, Wins P, Lakaye B. Anal Biochem. 2003;322:190–7. doi: 10.1016/j.ab.2003.08.013. [DOI] [PubMed] [Google Scholar]; (l) Lin N, Yan J, Huang Z, Altier C, Li M, Carrasco N, Suyemoto M, Johnston L, Wang S, Wang Q, Fang H, Caton-Williams J, Wang B. Nucleic Acids Res. 2007;35:1222–9. doi: 10.1093/nar/gkl1091. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Zlatev I, Lavergne T, Debart F, Vasseur JJ, Manoharan M, Morvan F. Org Lett. 2010;12:2190–3. doi: 10.1021/ol1004214. [DOI] [PubMed] [Google Scholar]
- 4.(a) Leclercq TM, Moretti PA, Pitson SM. Oncogene. 2011;30:372–8. doi: 10.1038/onc.2010.420. [DOI] [PubMed] [Google Scholar]; (b) Fausther M, Sevigny J. C R Biol. 2011;334:100–17. doi: 10.1016/j.crvi.2010.12.005. [DOI] [PubMed] [Google Scholar]; (c) Weber G, Shen F, Li W. Adv Exp Med Biol. 1998;431:401–8. doi: 10.1007/978-1-4615-5381-6_80. [DOI] [PubMed] [Google Scholar]; (d) Earnshaw DJGMJ. Biopolymers (Nucleic Acid Sciences) 1998;48:39–55. [Google Scholar]; (e) Earnshaw DJ, Gait MJ. Antisense Nucleic Acid Drug Dev. 1997;7:403–11. doi: 10.1089/oli.1.1997.7.403. [DOI] [PubMed] [Google Scholar]; (f) Watts JK, Deleavey GF, Damha MJ. Drug Discov Today. 2008;13:842–55. doi: 10.1016/j.drudis.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 5.(a) Ludwig J, Eckstein F. J Org Chem. 1989;54:631–635. [Google Scholar]; (b) He K, Porter KW, Hasan A, Briley J, Shaw BR. Nucleic Acids Res. 1999;27:1788–94. doi: 10.1093/nar/27.8.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) He K, Hasan A, Krzyzanowska B, Shaw BR. J Org Chem. 1998;63:5769–5773. doi: 10.1021/jo972002g. [DOI] [PubMed] [Google Scholar]; (d) Lin J, Shaw BR. Chemical Communications. 2000:2115–2116. [Google Scholar]; (e) Lin J, Porter KW, Shaw BR. Nucleosides Nucleotides Nucleic Acids. 2001;20:1019–23. doi: 10.1081/NCN-100002482. [DOI] [PubMed] [Google Scholar]; (f) Krzyzanowska BK, He KZ, Hasan A, Shaw BR. Tetrahedron. 1998;54:5119–5128. [Google Scholar]; (g) Cheek MA, Dobrikov MI, Wennefors CK, Xu Z, Hashmi SN, Shen X, Shaw BR. Nucleic Acids Symp Ser (Oxf) 2008;52:81–2. doi: 10.1093/nass/nrn041. [DOI] [PubMed] [Google Scholar]; (g) Caton-Williams J, Lin L, Smith M, Huang Z. Chemical Communications. 2011;47:8142–8144. doi: 10.1039/c1cc12201k. [DOI] [PubMed] [Google Scholar]
- 6.(a) Maeda M, Patel AD, Hampton A. Nucleic Acids Res. 1977;4:2843–53. doi: 10.1093/nar/4.8.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Simoncsits A, Tomasz J. Nucleic Acids Res. 1975;2:1223–33. doi: 10.1093/nar/2.7.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Moffatt JG. Can J Chem. 1964;42:599–604. [Google Scholar]; (d) Wu W, Bergstrom DE, Davisson VJ. J Org Chem. 2003;68:3860–5. doi: 10.1021/jo020745i. [DOI] [PubMed] [Google Scholar]; (e) Davisson VJ, Davis R, Dixit VM, Poulter CD. J Org Chem. 1987;52:1794. [Google Scholar]; (f) Moffatt JG, Khorana HG. J Am Chem Soc. 1961;83:649–58. [Google Scholar]; (g) Staab HA, Schaller H, Cramer F. Angew Chem. 71:736. [Google Scholar]
- 7.(a) Ludwig J. Acta Biochim Biophys Acad Sci Hung. 1981;16:131–3. [PubMed] [Google Scholar]; (b) Ruth JL, Cheng YC. Mol Pharmacol. 1981;20:415–22. [PubMed] [Google Scholar]; (c) Yoshikawa M, Kato T, Takenishi T. Tetrahedron Lett. 1967;50:5065–8. doi: 10.1016/s0040-4039(01)89915-9. [DOI] [PubMed] [Google Scholar]; (d) Mishra NC, Broom AD. J Chem Soc Chem Commun. 1991:1276–1277. [Google Scholar]
- 8.Gillerman I, Fischer B. Nucleosides Nucleotides Nucleic Acids. 2010;29:245–56. doi: 10.1080/15257771003709569. [DOI] [PubMed] [Google Scholar]
- 9.(a) Brandt G, Carrasco N, Huang Z. Biochemistry. 2006;45:8972–7. doi: 10.1021/bi060455m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Carrasco N, Caton-Williams J, Brandt G, Wang S, Huang Z. Angew Chem Int Ed Engl. 2006;45:94–7. doi: 10.1002/anie.200502215. [DOI] [PubMed] [Google Scholar]; (c) Carrasco N, Huang Z. J Am Chem Soc. 2004;126:448–9. doi: 10.1021/ja0383221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.