

Abstract
Viruses and virus-like particles (VLPs) are useful tools in biomedical research. Their defined structural attributes make them attractive platforms for engineered interactions over large molecular surface areas. In this report, we describe the use of VLPs as multivalent macroinitiators for atom transfer radical polymerization (ATRP). The introduction of chemically reactive monomers during polymerization provides a robust platform for post-synthetic modification via the copper-catalyzed azide-alkyne cycloaddition reaction. These results provide the basis to construct nanoparticle delivery vehicles and imaging agents using protein-polymer conjugates.
Synthetic polymers are chemically diverse in terms of their size and composition, and have long been used for the display of multiple copies of functional units. In contrast to their synthetic counterparts, biopolymers such as viruses and virus-like particles (VLPs) exhibit unique qualities of monodispersity and chemical regularity, allowing for the precise, periodic chemical functionalization of capsid structures.1 VLPs can be both genetically2 and chemically1b modified to alter or introduce functionality and tailor biological functions. The combination of these two types of multivalent structures may prove advantageous in situations requiring organic nanoparticles with defined structural attributes.
Synthetic polymer nanoparticles prepared by emulsion, nanoprecipitation, self-assembly, or layer-by-layer techniques have been widely studied as delivery vehicles.3 Their potential advantages include high drug-loading capacities, ability to improve drug solubility, and the ready introduction of ligands for targeted delivery.4 However, the production of polymeric nanoparticles with precise structural homogeneity remains a challenge to the field.5 The use of biomolecular platforms is an attractive approach toward this end, and many examples exist of the modification of viral and non-viral protein structures with synthetic polymers using ‘grafting to’ approaches.6 In contrast, there are few examples of the use of protein nanoparticles as multivalent macroinitiators for polymerization,7 and none using viruses.8
We describe here the radial polymerization of octa(ethylene glycol)-methacrylate (6, OEGMA) and the azido-functionalized analogue (7, OEGMA-N3) directly from the surface of the bacteriophage Qβ virus-like particle by atom transfer radical polymerization (ATRP). ATRP and other controlled radical polymerization techniques have previously been used to prepare well-defined polymer-protein hybrid structures that maintain their biological functions while gaining improved pharmacokinetic properties.9 We expected ATRP to provide polymers with narrow molecular weight distributions, yielding structurally uniform hybrid protein-polymer nanoparticles from the monodisperse viral platform.
ATRP initiators were installed on the surface of Qβ using a two-step ‘click’ protocol (Figure 1), which would allow other functional units to be installed at the same time if desired. First, N-hydroxysuccinimide 3 was used to attach an azide group to approximately 180 ± 30 of the 720 surface-accessible amine groups of the particle (a value that is controlled by the selection of reaction conditions for this step, and determined experimentally by mass spectrometry analysis of the denatured protein after modification). The resulting azido-particle 3 was then functionalized with a triglyme-based ATRP initiator (4) by copper-catalyzed azide-alkyne cycloadditon (CuAAC) to form Qβ macroinitiator 5.10 Polymers were grown from the multivalent initiator using a 2,2′-bipyridine/CuBr/CuBr2 catalyst system that was previously shown to yield high-molecular weight polymers grafted from protein-based initiators in water.11 A degassed, aqueous copper solution (0.1 mM final concentration) was added to a solution of particle 3 and the commercially available monomer 6 or 7 (final concentrations: 1 mg/mL (0.4 μM) in VLP and 72 mM in monomer). After overnight room-temperature reactions under nitrogen, the polymerizations were terminated by exposure to air and addition of EDTA. The resulting protein:polymer conjugates were purified from the small molecule reactants by sucrose gradient centrifugation and ultrapel-leting.
Figure 1.

Preparation of the Qβ VLP macroinitiator and polymerization from its surface.
Size exclusion chromatography (SEC) and dynamic light scattering (DLS) were used to determine changes in particle size following ATRP. The former (Figure 2A) showed a shift in elution volume from approximately 12 min for unmodified Qβ particles to 9.5 min for the significantly larger poly(OEGMA)-coated product. A similar shift was seen in electrophoretic mobility when the particles were analyzed on a native agarose gel, where polymer coated particles remained near the baseline, a significant retardation compared to particle 5 (Figure 2D). DLS measurements (Figure 2E,F) confirmed the increase in particle hydrodynamic radius from 14 nm (Qβ wild-type) to approximately 24 nm for the polymer-coated particles, with a narrow size distribution (polydispersity = 18%). The virus capsids, when visualized by TEM, were shown to remain intact following ATRP (Figure 2B,C).
Figure 2.
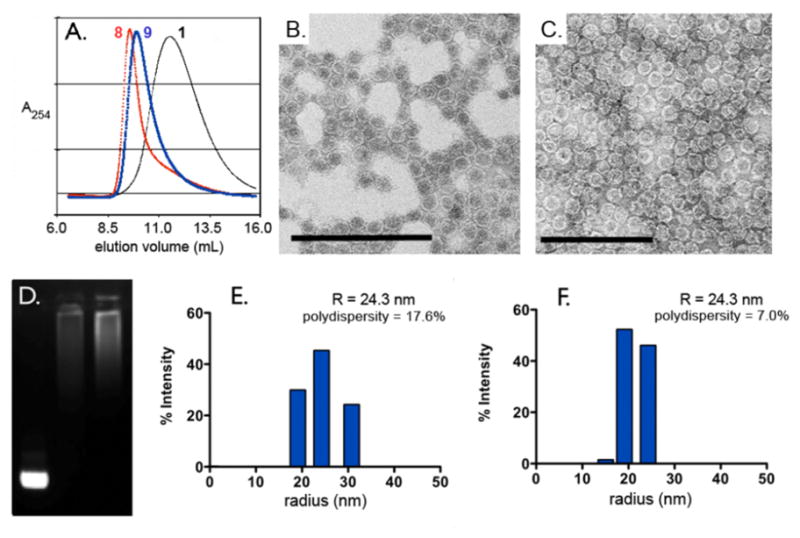
Characterization of Qβ-polymer particles. (A) Size-exclusion chromatographs. (B,C) TEM micrographs of poly(OEGMA) and poly-(OEGMA-N3) coated particles (8 and 9, respectively); scale bar = 200 nm. (D) Non-denaturing agarose gel stained with ethidium bromide: Lane 1= Qβ macroinitiator (5), Lane 2 = Qβ-poly(OEGMA) (8), Lane 3 = Qβ-poly(OEGMA-N3) (9). (E,F) Dynamic light scattering histograms of 8 and 9, respectively.
ATRP provides a tertiary bromide at the terminus of each growing polymer chain, which is amenable to further transformation.12 As shown in Figure 3, we reacted particle 8, which does not contain side-chain azide functionality, with a large molar excess of sodium azide in a water:DMSO mixture to substitute azide for bromide at the particle periphery. The resulting intermediate was then reacted with AlexaFluor488-alkyne under standard CuAAC bioconjugation conditions. Following sucrose gradient purification, the fluorescent particles were found to bear an average of approximately 20 dye molecules per particle by comparing Alexa UV absorbance (495 nm) versus total protein concentration (as measured by Bradford assay). No observable dye labeling occurred when 8 was subjected directly to CuAAC prior to sodium azide treatment, suggesting that no accessible azides remained following CuAAC to particle 3. The labeling of only 20 chains on a particle that started with 180 potential initiation sites suggests that either polymer initiation is strongly inhibited by previously-initiated chains or that azide substitution and click cycloaddition reactions are inhibited by the presence of adjacent chains attached to the particle surface. Given the high efficiency of both azide nucleophilic attack and CuAAC coupling, we suspect that the former explanation is more likely. This issue will be studied further in upcoming experiments. We expect that the method of post-polymerization functionalization and bioconjugation described here will prove especially useful in attaching functional biomolecules such as cell targeting groups, cell penetrating peptides, or diagnostic agents.
Figure 3.

End labeling of Qβ-poly(OEGMA) (8). For the structure of 11, see Figure 4.
The azido-functionalized octa(ethylene glycol)-methacrylate (OEGMA-N3, 7) also served as a competent monomer, allowing for a greater degree of post-polymerization functionalization while retaining PEG-like bio-compatibility.13 The polymerization of 7 proceeded in the same manner as 6 (Figure 1), yielding hybrid Qβ-poly(OEGMA-N3) particle 9 with very similar properties to the poly(OEGMA)-coated 8: an approximate 10 nm increase in radius, narrow size distribution, and dramatically different electrophoretic mobility (Figure 2A,C,D, F).
To demonstrate the reactivity of poly(OEGMA-N3) grafts, 9 was reacted with alkyne-substituted AlexaFluor488 dye 11 under CuAAC conditions (Figure 4A). Sucrose gradient centrifugation revealed an intensely colored green band, indicating a successful ‘click’ reaction (Figure 4B). The potential sensitivity of the molar absorptivity of these dyes to their molecular environment made quantification of the coupling efficiency at high dye loadings impossible by absorbance measurements. The same reaction was therefore performed with gadolinium complex 11, and the coupling result assayed by quantitation of Gd by inductively coupled plasma optical emission spectroscopy (ICP-OES).14 Loading values of 500–650 Gd complexes per particle were routinely found for reactions with different samples of 9.
Figure 4.

Conjugation to polymer side-chains. A) CuAAC reaction to append functionality to 9. B) UV illumination of sucrose gradient following CuAAC between 9 and 11 (upper band = unreacted 11, lower band = particle 14). C) Conjugation of Gd-DOTA complex 12 to azide-functionalized capsid 3.
Nanoparticles bearing Gd complexes have attracted substantial interest as magnetic resonance imaging agents due to benefits anticipated for their large size (slowing rotational relaxation rates) and high Gd loading capacities. Several variations using virus-like particles have been reported.15 We had earlier described the direct attachment of Gd(DOTA)-alkyne complex 12 to azide-derivatized particle 3, and the magnetic resonance behavior of the resulting conjugate 17.14 Examination of the Qβ-polymer-Gd particle 15 found a similar relaxivity on a per Gd basis as 17 at two different Larmor frequencies (Table 1; approximately double the value of the Gd complex alone), suggesting that the polymer backbone does not impart additional conformational rigidity to the complexes or restrict access to water. The per particle relaxivity of 15 (7092 mM−1s−1), however, compares favorably to examples reported in the literature involving the covalent attachment of commercially available Gd reagents to virus particles.15b–d
Table 1.
T1 relaxivities for derivatized virus-like particles.
| Sample | Gd/VLP | Relaxivity per Gd (per VLP)/mM−1s−1 |
|
|---|---|---|---|
| 60 MHz | 90 MHz | ||
| 15 | 610 | 11.63 (7092) | 10.12 (6174) |
| 17 | 350 | 10.7 (3750) | 11.64 (4075) |
The multiple attachment points offered by the azide groups of polymer-decorated particle 9 may also be used to tether a releasable drug cargo. It has been reported that nanoparticles of approximately 25 nm radius are optimal for internalization via endocytotic pathways.16 The relatively acidic environments of endosomal and lysosomal compartments (pH = 4.5–5.5) is most often used as the trigger for drug release, with hydrazones being a popular motif.17 Accordingly, we prepared a ‘clickable’ doxorubicin hydrazone 13 for conjugation to the Qβ-polyazide 9 (Figure 4A). Because of its poorer aqueous solubility than the Gd complex, fewer doxorubicin molecules were attached, an average of 150 per particle (approx. 3 wt-%), determined by measuring doxorubicin concentrations spectrophotometrically (λabs = 490 nm) and comparing these to protein concentrations obtained by Bradford assay.18 The resulting particles 16 remained intact with a narrow size distribution (DLS: radius = 22.7 nm, 14% polydispersity). This stands in contrast to the conjugation of 13 directly to particle 3, which led to degradation of the capsid structure and precipitation of the protein as occasionally happens with hydrophobic or polyaromatic molecules.
The pH sensitivity of the Dox-conjugated particle 16 was assayed by fluorescence spectroscopy. The fluorescence of the Dox chromophores on 16 are known to be quenched at high local concentrations,19 allowing us to monitor the release of the drug (λex = 480 nm; λem = 598 nm) from the conjugate into solution at pH 7.4 and 5.5.20 The maximum fluorescence was reached after approximately 2 h at pH 5.5, while no fluorescence increase was observed after 12 hours for the particles incubated at pH 7.4 (Figure 5A), consistent with the expected properties of the hydrazone linkage.
Figure 5.

(A) In vitro release profile of doxorubicin from particle 16 at pH 7.4 and pH 5.5. (MES = 2(N-Morpholino)ethanesulfonic acid buffer; PBS = phosphate-buffered saline). B) MTT assay for cell viability after treatment with the indicated agents, see text.
The pH-sensitive, doxorubicin-conjugated particle 16 was also found to be cytotoxic to HeLa cells, a cervical cancer cell line that is sensitive to treatment with doxorubicin.21 Cells were incubated with varying concentrations of 16 or appropriate controls for 8 hours in pH 7 buffer (in which no cleavage of Dox from the particle occurs) and were assayed for cell viability the following day using the MTT assay. Polymer conjugate 16 exhibited a similar cytotoxicity profile to free doxorubicin (in terms of overall dox concentrations) under the same assay conditions (Figure 5B), indicating that the particles are internalized and release their payload to effect cell death. The cells showed no significant signs of toxicity when exposed to unloaded particles (9) at through the concentration range used with 16 (Supporting Information).
In summary, we have shown that VLPs derived from the bacteriophage Qβ can serve as a platform for controlled radical polymerization to produce polymer-coated protein nanoparticles. The size and surface properties of Qβ can be significantly altered through ATRP, while still retaining the low polydispersities associated with VLPs. Furthermore, the polymer-coated particles are accessible to bioconjugation at both the chain termini and suitably derivatized polymer sidechains. The platform allows for both conjugation of small-molecule imaging agents and chemotherapeutics. Future studies will focus on varying the nature and properties of the polymers produced by radial polymerization and on further exploration of this platform for development of targeted drug delivery and imaging agents.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (CA112075, RR021886) and by a Pathway to Independence Award (K99EB011530) to JKP. We thank Dr. Lars Liepold and Prof. Trevor Douglas (Montana State University) for relaxivity measurements and Dr. Burkhardt Laufer for the synthesis of Gd complex 12.
Footnotes
Supporting Information. Detailed procedures for all chemical transformations and cell-based assays. The material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Steinmetz NF. Nanomedicine: nanotechnology, biology, and medicine. 2010;6:634–641. doi: 10.1016/j.nano.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Strable E, Finn MG. Curr Top Microbiol Immunol. 2009;327:1–21. doi: 10.1007/978-3-540-69379-6_1. [DOI] [PubMed] [Google Scholar]
- 2.Brown S, Fiedler J, Finn MG. Biochemistry. 2009;48:11155–11157. doi: 10.1021/bi901306p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mora-Huertas CE, Fessi H, Elaissari A. Int J Pharm. 2010;385:113–142. doi: 10.1016/j.ijpharm.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 4.Petros RA, Desimone JM. Nat Rev Drug Discovery. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 5.Good control over size has been achieved in many systems, including polymer nanoparticles, such as: Duan H, Kuang M, Zhang G, Wang D, Kurth DG, Möhwald H. Langmuir. 2005;21:11495–11499. doi: 10.1021/la051638x.Yanagishita T, Fujimura R, Nishio K, Masuda H. Langmuir. 2009;26:1516–1519. doi: 10.1021/la903913h.An SY, Bui MPN, Nam YJ, Han KN, Li CA, Choo J, Lee EK, Katoh S, Kumada Y, Seong GH. J Colloid Interfac Sci. 2009;331:98–103. doi: 10.1016/j.jcis.2008.11.022.
- 6.(a) Kovacs EW, Hooker JM, Romanini DW, Holder PG, Berry KE, Francis MB. Bioconjugate Chem. 2007;18:1140–1147. doi: 10.1021/bc070006e. [DOI] [PubMed] [Google Scholar]; (b) Brunel FM, Lewis JD, Destito G, Steinmetz NF, Manchester M, Stuhlmann H, Dawson PE. Nano Lett. 2010;10:1093–1097. doi: 10.1021/nl1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bruckman MA, Kaur G, Lee LA, Xie F, Sepulveda J, Breitenkamp R, Zhang X, Joralemon M, Russell TP, Emrick T, Wang Q. ChemBioChem. 2008;9:519–523. doi: 10.1002/cbic.200700559. [DOI] [PubMed] [Google Scholar]; (d) Holder PG, Finley DT, Stephanopoulos N, Walton R, Clark DS, Francis MB. Langmuir. 2010;26:17383–8. doi: 10.1021/la1039305. [DOI] [PubMed] [Google Scholar]
- 7.(a) Hu YX, Samanta D, Parelkar SS, Hong SW, Wang QA, Russell TP, Emrick T. Adv Funct Mater. 2010;20:3603–3612. [Google Scholar]; (b) Zeng Q, Li T, Cash B, Li S, Xie F, Wang Q. Chem Commun. 2007:1453–5. doi: 10.1039/b617534a. [DOI] [PubMed] [Google Scholar]; (c) Li H, Li M, Yu X, Bapat AP, Sumerlin BS. Polym Chem. 2011 doi: 10.1039/c1py00031d. Advance Article. [DOI] [Google Scholar]
- 8.Many examples exist showing radial polymerization from polymer and metal nanoparticles. For reviews, see: Barbey R, Lavanant L, Paripovic D, Schuwer N, Sugnaux C, Tugulu S, Klok HA. Chem Rev. 2009;109:5437–5527. doi: 10.1021/cr900045a.Zou H, Wu S, Shen J. Chem Rev. 2008;108:3893–3957. doi: 10.1021/cr068035q.For key examples, see: Ali AMI, Mayes AG. Macromolecules. 2010;43:837–844.Skaff H, Emrick T. Angew Chem, Int Ed. 2004;43:5383–5386. doi: 10.1002/anie.200453822.Perruchot C, Khan MA, Kamitsi A, Armes SP, Von Werne T, Patten TE. Langmuir. 2001;17:4479–4481.
- 9.(a) Heredia KL, Bontempo D, Ly T, Byers JT, Halstenberg S, Maynard HD. J Am Chem Soc. 2005;127:16955–16960. doi: 10.1021/ja054482w. [DOI] [PubMed] [Google Scholar]; (b) Bontempo D, Maynard HD. J Am Chem Soc. 2005;127:6508–6509. doi: 10.1021/ja042230+. [DOI] [PubMed] [Google Scholar]; (c) De P, Li M, Gondi SR, Sumerlin BS. J Am Chem Soc. 2008;130:11288–11289. doi: 10.1021/ja804495v. [DOI] [PubMed] [Google Scholar]; (d) Lele BS, Murata H, Matyjaszewski K, Russell AJ. Biomacromolecules. 2005;6:3380–3387. doi: 10.1021/bm050428w. [DOI] [PubMed] [Google Scholar]
- 10.Hong V, Presolski SI, Ma C, Finn MG. Angew Chem Int Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087.(b) Control experiments showed that no degradation of or insertion into the tertiary bromide initiation site occurs under CuAAC reaction conditions.
- 11.Peeler JC, Woodman BF, Averick S, Miyake-Stoner SJ, Stokes AL, Hess KR, Matyjaszewski K, Mehl RA. J Am Chem Soc. 2010;132:13575–13577. doi: 10.1021/ja104493d. [DOI] [PubMed] [Google Scholar]
- 12.(a) Golas P, Tsarevsky N, Sumerlin B, Walker L, Matyjaszewski K. Aust J Chem. 2007;60:400–404. [Google Scholar]; (b) Tsarevsky NV, Sumerlin BS, Matyjaszewski K. Macromolecules. 2005;38:3558–3561. [Google Scholar]
- 13.(a) Gao W, Liu W, Christensen T, Zalutsky M, Chilkoti A. Proc Natl Acad Sci U S A. 2010;107:16432–16437. doi: 10.1073/pnas.1006044107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao W, Liu W, Mackay JA, Zalutsky MR, Toone EJ, Chilkoti A. Proc Natl Acad Sci U S A. 2009;106:15231–6. doi: 10.1073/pnas.0904378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prasuhn DE, Yeh RM, Obenaus A, Manchester M, Finn MG. Chem Commun. 2007:1269–1271. doi: 10.1039/b615084e. [DOI] [PubMed] [Google Scholar]
- 15.(a) Liepold LO, Abedin MJ, Buckhouse ED, Frank J, Young MJ, Douglas T. Nano Lett. 2009;9:4520–6. doi: 10.1021/nl902884p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liepold L, Anderson S, Willits D, Oltrogge L, Frank J, Douglas T, Young M. Magn Reson Med. 2007;58:871–9. doi: 10.1002/mrm.21307. [DOI] [PubMed] [Google Scholar]; (c) Hooker JM, Datta A, Botta M, Raymond KN, Francis MB. Nano Lett. 2007;7:2207–2210. doi: 10.1021/nl070512c. [DOI] [PubMed] [Google Scholar]; (d) Anderson EA, Isaacman S, Peabody DS, Wang EY, Canary JW, Kirshenbaum K. Nano Letters. 2006;6:1160–1164. doi: 10.1021/nl060378g. [DOI] [PubMed] [Google Scholar]
- 16.Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S. Adv Mater. 2009;21:419–424. doi: 10.1002/adma.200801393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) De Jesus OLP, Ihre HR, Gagne L, Frechet JMJ, Szoka FC. Bioconjug Chem. 2002;13:453–461. doi: 10.1021/bc010103m. [DOI] [PubMed] [Google Scholar]; (b) Bae Y, Fukushima S, Harada A, Kataoka K. Angewandte Chemie-International Edition. 2003;42:4640–4643. doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]; (c) Ulbrich K, Subr V. Adv Drug Deliv Rev. 2004;56:1023–1050. doi: 10.1016/j.addr.2003.10.040. [DOI] [PubMed] [Google Scholar]
- 18.Missirlis D, Kawamura R, Tirelli N, Hubbell J. Eur J Pharm Sci. 2006;29:120–129. doi: 10.1016/j.ejps.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Mohan P, Rapoport N. Mol Pharm. 2010;7:1959–1973. doi: 10.1021/mp100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Ta T, Convertine AJ, Reyes CR, Stayton PS, Porter TM. Biomacromolecules. 2010;11:1915–20. doi: 10.1021/bm1004993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Karukstis KK, Thompson EHZ, Whiles JA, Rosenfeld RJ. Biophys Chem. 1998;73:249–263. doi: 10.1016/s0301-4622(98)00150-1. [DOI] [PubMed] [Google Scholar]
- 21.Eichholtz-Wirth H. Br J Cancer. 1980;41:886–891. doi: 10.1038/bjc.1980.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.