

Abstract
In this paper we describe the phosphine-catalyzed [3 + 2], [3 + 3], [4 + 3], and [3 + 2 + 3] annulations of azomethine imines and allenoates. These processes mark the first use of azomethine imines in nucleophilic phosphine catalysis, producing dinitrogen-fused heterocycles, including tetrahydropyrazolo-pyrazolones, -pyridazinones, -diazepinones, and -diazocinones. Counting the two different reaction modes in the [3 + 3] cyclizations, there are five distinct reaction pathways—the choice of which depends on the structure and chemical properties of the allenoate. All reactions are operationally simple and proceed smoothly under mild reaction conditions, affording a broad range of 1,2-dinitrogen–containing heterocycles in moderate to excellent yields. A zwitterionic intermediate formed from a phosphine and two molecules of ethyl 2,3-butadienoate acted as a 1,5-dipole in the annulations of azomethine imines, leading to the [3 + 2 + 3] tetrahydropyrazolodiazocinone products. The incorporation of two molecules of an allenoate into an eight-membered-ring product represents a new application of this versatile class of molecules in nucleophilic phosphine catalysis. The salient features of this protocol—the facile access to a diverse range of nitrogen-containing heterocycles and the simple preparation of azomethine imine substrates—suggest that it might find extensive applications in heterocycle synthesis.
Introduction
Intermolecular cycloaddition is one of the most powerful tools for the convergent synthesis of a variety of carbo- and heterocycles from simpler precursors.1 Many metallo- and organocatalytic systems have been successfully developed for the cycloadditions of a wide range of starting materials. Among the catalytic systems, nucleophilic phosphine catalysis has been established as a reliable platform for the efficient assembly of a wide array of cyclic products from simple building blocks.2 In particular, activated allenes subjected to nucleophilic phosphine catalysis conditions exhibit superbly diverse reactivity toward electrophilic reagents. These allenes can function as one-, two-, three-, or four-carbon synthons when reacting with a variety of electrophilic coupling partners (including aldehydes, alkenes, imines, and aziridines), undergoing [2 + 1],3 [4 + 1],4 [3 + 2],5 [2 + 2 + 2],6 [3 + 3],7 [4 + 2],8 [4 + 3]9, or [8 + 2]10 annulations.11 The particular reactivity of the allene substrate is often induced by its electrophilic coupling partner. Consequently, the search for new electrophilic substrates exhibiting suitable reactivity for effective use in the synthesis of heterocycles with new skeletons or structural features is a major challenge for the formation of diverse cycloaddition products from the nucleophilic phosphine catalysis of allenes. In this context, we conceived the possibility of introducing a new type of electrophilic coupling reagent, azomethine imines, that might serve as an N–N–C trio-of-atoms synthon in phosphine-catalyzed annulation processes. Herein, we report the phosphine-catalyzed [3 + N] annulations of allenoates and azomethine imines 1 (Scheme 1).
Scheme 1.

Phosphine-Catalyzed [3 + N] Cyclizations of Azomethine Imines with Allenoates
Azomethine imines such as 1 are readily accessible, stable compounds that have been employed recently as efficient 1,3-dipoles in various metal-catalyzed and organocatalytic cycloadditions.12–14 In 2003, Fu reported an efficient Cu-catalyzed protocol for enantioselective [3 + 2] cycloaddition of azomethine imines with alkynes;14a in 2005, he further extended this azomethine imine/alkyne cycloaddition strategy to the kinetic resolution of azomethine imines bearing a stereocenter at the 5-position of the pyrazolidinone core.14b Sibi demonstrated another Cu-catalyzed enantioselective [3 + 2] cycloaddition of azomethine imines with pyrazolidinone acrylates.14i Pale and Sommer presented a heterogeneous Cu(I)-modified zeolite-catalyzed [3 + 2] cycloaddition of azomethine imines with alkynes.14k Hayashi studied Pd-catalyzed [3 + 3] cycloadditions of azomethine imines with trimethylenemethane to produce hexahydropyridazine derivatives under mild conditions,14c and Pd-catalyzed [4 + 3] cycloadditions of azomethine imines with γ-methylidene-δ-valerolactones.14g Toste reported a Au-catalyzed [3 + 3] annulation of azomethine imines with propargyl esters.14j Maruoka developed a Ti-catalyzed enantioselective [3 + 2] cycloaddition of azomethine imines with α,β-unsaturated aldehydes.14m Suga described a Ni-catalyzed enantioselective and diastereoselective [3 + 2] cycloaddition of azomethine imines with 3-acryloyl-2-oxazolidinone.14f Charette also developed a Ni-catalyzed [3 + 3] cycloaddition of aromatic azomethine imines with 1,1-cyclopropane diesters.14l More recently, Scheidt described an N-heterocyclic carbene-catalyzed stereoselective formal [3 + 3] cycloaddition of azomethine imines with enals.14e Chen developed a multifunctional primary amine (derived from cinchona alkaloids)–catalyzed enantioselective [3 + 2] cycloaddition of azomethine imines and cyclic enones,14h and a diarylprolinol salt–catalyzed [3 + 2] cycloaddition of azomethine imines with α,β-unsaturated aldehydes.14d Despite these extensive efforts at using azomethine imines as cyclization partners in heterocycle synthesis, the application of azomethine imines in phosphine-catalyzed cycloaddition has not been reported previously. This situation prompted us to design a cycloaddition of azomethine imines and allenoates using readily available tertiary phosphines as catalysts. It is widely accepted that, in the phosphine-promoted reactions of activated allenoates, the catalysis cycle is typically initiated by the addition of the Lewis basic phosphine to the electrophilic β-carbon atom of the α-allenic ester, leading to the formation of zwitterionic intermediates. We suspected that exposing these intermediates to azomethine imines might allow new [3 + N] cycloadditions, with the pathways followed depending on the number of carbon atoms that the allenoate contributes to the final cyclic products (Scheme 1). Herein, we report the first examples of phosphine-catalyzed cycloadditions of various zwitterionic species to provide functionalized five-, six-, seven-, and eight-membered dinitrogen-containing heterocycles with high efficiency.
The new annulation reactions provide generally applicable routes toward dinitrogen-fused heterocycles, including tetrahydropyrazolo-pyrazolones, -pyridazinones, -diazepinones, and -diazocinones, which are key units in or building blocks of many pharmaceuticals, agrochemicals, biologically active compounds, and other useful chemicals. Among the dinitrogen-containing heterocycles, pyrazolone derivatives are often used as dyes in the food, textile, photography, and cosmetics industries.15 Several pyrazolones also exhibit bioactivity. For example, phenazone has been used as a synthetic drug;16 phenylbutazone has anti-inflammatory activity;16 phenidone and BW755C are inhibitors of lipoxygenase and cyclooxygenase, respectively;17,18 BW357U displays anorectic activity (Figure 1).19 Other dinitrogen-fused heterocycles also exhibit diverse bioactivity and have a variety of applications. Tetrahydropyrazolopyrazolones have been investigated as antibacterial agents,16 herbicides, pesticides,20 antitumor agents,21 calcitonin agonists,22 and potent drugs to relieve Alzheimer’s disease.23 Pyrazolopyridazinone derivatives have been studied extensively as pesticides and, especially, herbicides; more than 60 patents have been filed according to the CAS database. Pyrazolodiazepinone and pyrazolodiazocinone derivatives have also been explored as herbicides, insecticides, acaricides,24 and acetyl-CoA carboxylase inhibitors.25 In addition, the pyrazolidine derivatives obtained through these annulations are readily transformed into 1,3-diamine derivatives via N–N bond cleavage with SmI2;26 this cleavage strategy has been demonstrated in several reports.14j,27 Such 1,3-diamine derivatives serve not only as ligands of metal catalysts but also as biologically important compounds or building blocks for bioactive compounds.28
Figure 1.

Selected examples of biologically active dinitrogen-fused heterocycles.
Results and Discussion
Although the azomethine imines 1 have received much attention,14 including extensive application in 1,3-diploar cycloadditions for the preparation of a plethora of pyrazolidinone derivatives,29 they have never previously been used in nucleophilic phosphine catalysis reactions. Our initial attempts at phosphine-catalyzed annulations of azomethine imines commenced with the reactions of 1-(p-nitrobenzylidene)-3-oxopyrazolidin-1-ium-2-ide (1a). Table 1 presents the results of screening for appropriate reaction conditions and catalysts for the model reaction between 1a and allenoate 2a. Considering the possibility of a background reaction resulting from direct [3 + 2] cycloaddition of the azomethine imine and the allenoate,30 we first attempted the reaction between 1a and the allenoate 2a in the absence of phosphine catalyst in dichloromethane (DCM) at room temperature; TLC analysis revealed no product (Table 1, entry 1). When compound 1a was treated with ethyl 2-methyl-buta-2,3-dienoate (2a) in DCM31 at room temperature in the presence of 20 mol % of PPh3, we isolated a new product in 5% yield (Table 1, entry 2). The efficiency of nucleophilic phosphine catalysis often depends on the nature of the tertiary phosphine catalyst; α-alkyl allenoates, such as 2a, often require more-nucleophilic phosphines for facile reactions.8a,b Indeed, screening of a couple of tertiary phosphines for the catalysis revealed that the reaction efficiency increased as the nucleophilicity of the phosphine increased, reaching 92% product yield for PMe3 (entries 3–6). Using NMR spectroscopy and X-ray crystallography of the homologous tetrahydropyrazolopyrazolone 3ad obtained from the annulation of 1a with the α-isobutyl allenoate 2d, we established the structure of the new annulation product 3aa to be that from a [3 + 2] cycloaddition. Not only did the [3 + 2] annulation exhibit great efficiency, it also provided the tetrasubstituted exocyclic alkylidene as a single E isomer. Hexamethylphosphorous triamide (HMPT), albeit more nucleophilic than PPh3, did not provide any desired product (entry 7).
Table 1.
Phosphine-Catalyzed [3 + 2] Cycloadditions of the Azomethine Imine 1a with the Allenoate 2aa
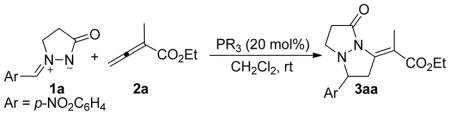 | |||
|---|---|---|---|
| entry | PR3 | time (h) | yield (%)b |
| 1 c | - | 48 | 0 |
| 2 | PPh3 | 24 | 5 |
| 3 | MePPh2 | 24 | 24 |
| 4 | Me2PPh | 24 | 53 |
| 5 | PBu3 | 14 | 88 |
| 6 | PMe3 | 12 | 92 |
| 7 | HMPT | 24 | 0 |
1.2 equiv of allenoate was used.
Isolated yield.
Without phosphine catalyst.
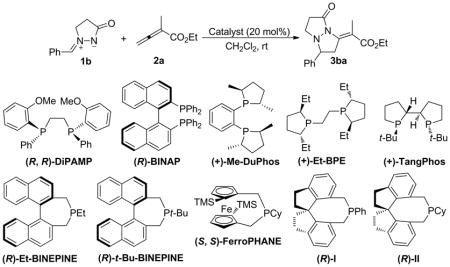
A large number of phosphine-catalyzed annulations have been rendered enantioselective through the use of enantioenriched chiral phosphines.2k To gauge the feasibility of developing enantioselective azomethine imine annulation, we tested the effect of 10 known chiral phosphines (Table 2). DiPAMP, BINAP, Me-DuPhos, TangPhos, Et-BINEPINE, t-Bu-BINEPINE, and FerroPHANE were poor catalysts and provided negligible amounts of products, even after reaction times of five days (entries 1–3 and 5–8). Et-BPE provided the desired tetrahydropyrazolopyrazolone 3ba in 19% yield and 6% ee (entry 4). To our delight, the chiral monophosphines I and II rendered high enantiomeric excesses with a synthetically useful yield in the case of II (entries 9 and 10), demonstrating the feasibility of developing an enantioselective process of synthetic value.
Table 2.
Chiral Phosphine-Catalyzed [3 + 2] Cycloadditions of the Azomethine Imine 1b with the Allenoate 2aa
 | ||||
|---|---|---|---|---|
| entry | catalyst | time (h) | yield (%)b | ee (%)c |
| 1 | (R, R)-DiPAMP | 120 | <5 | n.d.d |
| 2 | (R)-BINAP | 120 | <5 | n.d. |
| 3 | (+)-Me-PuPhos | 120 | <5 | n.d. |
| 4 | (+)-Et-BPE | 120 | 19 | 6 |
| 5 | (+)-TangPhos | 120 | <5 | n.d. |
| 6 | (R)-Et-BINEPHINE | 120 | <5 | n.d. |
| 7 | (R)-t-Bu-BINEPHINE | 120 | <5 | n.d. |
| 8 | (S, S)-FerroPHANE | 120 | <5 | n.d. |
| 9 | (R)-I | 48 | 8 | 93 |
| 10 | (R)-II | 48 | 56e | 89 |
Reactions were conducted with 0.1 mmol of 1b and 1.5 equiv of allenoate in 1 mL of DCM. The absolute configuration of the major enantiomer has not been determined.
Isolated yield.
Enantiomeric excess was determined through HPLC analysis on a chiral stationary phase.
Not determined.
The Z isomer was isolated in 18% yield.
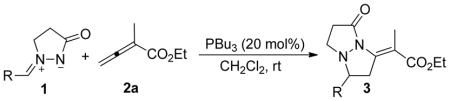
Using 20 mol % of PBu3 as the catalyst, we examined a range of azomethine imines in the [3 + 2] annulations of ethyl 2-methyl-buta-2,3-dienoate (2a) under the optimized reaction conditions (Table 3). Here, we employed PBu3 instead of PMe3 for ease of operation; the reactions of the azomethine imines 1b–1u with the allenoate 2a were also highly efficient (up to 99% yield) when mediated by PBu3. A variety of aryl azomethine imines underwent the [3 + 2] cycloaddition, providing the anticipated tetrahydropyrazolopyrazolones in moderate to excellent yields (entries 1–17). In general, azomethine imines bearing electron-withdrawing groups on the benzene ring provided somewhat higher yields of the cyclization products than did those bearing electron-donating groups (cf. entries 2–4 and 5–11). The reactions employing azomethine imines containing ortho-substituted benzene rings required elevated temperatures and longer times and furnished the tetrahydropyrazolopyrazolones in modest yields (entries 12–14). The azomethine imines bearing 2-naphthyl, 4-pyridyl, and 2-furyl groups, also underwent smooth cyclizations with 2a, readily affording the corresponding pyrazolidinone derivatives in excellent yields (entries 15–17). We were delighted to find that even alkylimines could be employed in [3 + 2] annulations with the allenoate 2a, albeit in moderate yields (entries 18–20).
Table 3.
PBu3-Catalyzed [3 + 2] Cycloadditions of Azomethine Imines 1 with the Allenoate 2aa
 | ||||
|---|---|---|---|---|
| entry | R | time (h) | product | yield (%)b |
| 1 | Ph (1b) | 24 | 3ba | 97 |
| 2 | 4-MeC6H4 (1c) | 24 | 3ca | 85 |
| 3 | 4-i-PrC6H4 (1d) | 24 | 3da | 89 |
| 4 | 4-OMeC6H4 (1e) | 30 | 3ea | 77 |
| 5 | 4-FC6H4 (1f) | 24 | 3fa | 91 |
| 6 | 4-ClC6H4 (1g) | 20 | 3ga | 93 |
| 7 | 4-BrC6H4 (1h) | 20 | 3ha | 97 |
| 8 | 4-CNC6H4 (1i) | 12 | 3ia | 87 |
| 9 | 4-CF3C6H4 (1j) | 24 | 3ja | 98 |
| 10 | 3-NO2C6H4 (1k) | 24 | 3ka | 97 |
| 11 | 2-NO2C6H4 (1l) | 24 | 3la | 94 |
| 12c | 2-MeC6H4 (1m) | 48 | 3ma | 70 |
| 13c | 2-PhC6H4 (1n) | 48 | 3na | 50 |
| 14c | 1-naphthyl (1o) | 48 | 3oa | 57 |
| 15 | 2-naphthyl (1p) | 24 | 3pa | 96 |
| 16 | 4-pyridyl (1q) | 20 | 3qa | 86 |
| 17 | 2-furanyl (1r) | 20 | 3ra | 99 |
| 18 | cyclohexyl (1s) | 36 | 3sa | 55 |
| 19 | n-Bu (1t) | 72 | 3ta | 41 |
| 20 | i-Pr (1u) | 72 | 3ua | 48 |
1.2 equiv of allenoate was used.
Isolated yield.
The reaction was run at 40 °C.
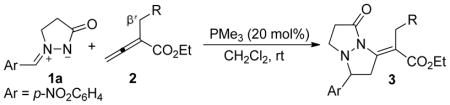
Next, we investigated the reactions of a range of distinctly substituted allenoates 2 with the azomethine imine 1a in the presence of 20 mol % of PMe3 (Table 4). For the sterically more demanding allenoates 2b–2m, we employed the more-reactive PMe3 as the catalyst. Based on the proposed reaction mechanism (vide infra), we did not anticipate the substituent at the β′-carbon atom of the allenoate 2 to exert much influence on the course of the reaction. A variety of β′-alkyl– and β′-aryl–substituted allenoates underwent the cycloaddition at reasonable rates, providing the corresponding tetrahydropyrazolopyrazolones 3 in excellent yields (entries 1–11). In general, alkyl-substituted allenoates required a slightly elevated temperature (40 °C) for the reaction to proceed at a reasonable rate (entries 2–5). Allenoates featuring phenyl groups with either electron-withdrawing or -donating substituents worked very well as substrates at room temperature (entries 6–11). In contrast, the reactions of β′-vinyl– and β′-styryl–substituted allenoates were somewhat sluggish, providing their corresponding products in moderate yields even after prolonged reaction times (entries 12 and 13).
Table 4.
PMe3-Catalyzed [3 + 2] Annulations of the Allenoates 2 with the Azomethine Imine 1aa
 | |||||
|---|---|---|---|---|---|
| entry | R | temp (°C) | time (h) | product | yield (%)b |
| 1 | H (2a) | rt | 12 | 3aa | 92 |
| 2 | Me (2b) | 40 | 20 | 3ab | 96 |
| 3 | Et (2c) | 40 | 20 | 3ac | 92 |
| 4 | i-Pr (2d) | 40 | 20 | 3adc | 95 |
| 5 | t-Bu (2e) | 40 | 24 | 3ae | 98 |
| 6 | Ph (2f) | rt | 24 | 3af | 87 |
| 7 | 2-MeC6H4 (2g) | rt | 20 | 3ag | 95 |
| 8 | 2-FC6H4(2h) | rt | 20 | 3ah | 91 |
| 9 | 4-FC6H4 (2i) | rt | 15 | 3ai | 99 |
| 10 | 4-ClC6H4 (2j) | rt | 36 | 3aj | 88 |
| 11 | 4-BrC6H4 (2k) | rt | 36 | 3ak | 81 |
| 12 | vinyl (2l) | rt | 36 | 3al | 45 |
| 13 | styryl (2m) | rt | 60 | 3am | 30 |
1.2 equiv of allenoate was used.
Isolated yield.
The structure of this compound was verified through single-crystal X-ray analysis.
In contrast to the aforementioned cases wherein excellent selectivity was observed, the reaction involving diethyl 2-vinylidenesuccinate (2n) was more complicated and seemed to follow several competing pathways. Under conditions otherwise identical to those described above, 2n underwent the PBu3-mediated cycloaddition with the azomethine imine 1a in a distinctive manner: a combination of [3 + 2], [3 + 3], and [3 + 4] reactions, affording a mixture of the tetrahydropyrazolopyrazolone 3an, the tetrahydropyrazolopyridazinone 4, and the tetrahydropyrazolodiazepinone 5 in 6, 23, and 63% (trans- and cis-5) yields, respectively (Scheme 2). Intriguingly, the use of PMe3 as the catalyst afforded strikingly different results. The [3 + 2] product 3an and the [3 + 3] product 4 were obtained in 40 and 42% yields, respectively, while the [4 + 3] product 5 was isolated in only 12% yield (trans- and cis-5). We used single-crystal X-ray analyses to unequivocally verify the structures of the annulation products 3an, 4, and trans-5. In an effort to force the reaction to follow one of the cycloaddition modes preferentially, we screened phosphines with various nucleophilicities under a range of reaction conditions (solvents, temperatures, other controllable factors), but we always obtained mixtures of several cycloaddition products. We also investigated the reactions of other azomethine imines with the allenoate 2n, but, in general, they invariably produced more than two products, including those from [3 + 2], [3 + 3], and [4 + 3] cycloadditions. Although the [3 + 3] annulation mode had been observed previously for diethyl 2-vinylidenesuccinate (2n) in its phosphine-mediated annulation with aziridines,7 the [4 + 3] annulation modality had not. The distinctive behavior of the diester allenoate 2n relative to those of the other α-alkyl–substituted allenoates (2a–2m) was due to the enhanced acidity of its β′-protons, thereby facilitating the conversion of the phosphonium enoate to the vinylogous ylide and its subsequent addition at the β′-carbon atom (see the mechanistic discussion below).
Scheme 2.

Phosphine-Catalyzed Annulations of the Azomethine Imine 1a with the Allenoate 2n
On the basis of the reported mechanistic studies of nucleophilic phosphine-catalyzed reactions,2 Scheme 3 presents plausible pathways for the reactions of the azomethine imines 1 and the allenoates 2. Upon conjugate addition of PBu3 to the allenoate 2, the zwitterion A is formed. Because of steric crowding at its α-carbon atom, the β-phosphonium enoate A undergoes addition at its β-carbon atom to form the amide B. A second conjugate addition of the amide to the β-phosphonium enoate motif of intermediate B accomplishes the [3 + 2] cyclization. The thus-formed β-phosphonium enoate C undergoes facile β-elimination to regenerate the catalyst, forming the final tetrahydropyrazolopyrazolone product 3. On the other hand, the carboxylic ester substituent at the β′-carbon atom of the allenoate 2n facilitates the conversion of the phosphonium enoate A to the vinylogous ylide D,32 which then adds to the azomethine imine 1 to form the intermediate E. Unlike the 5-exo cyclization of the intermediate B to form C, the 6-endo cyclization of the amide E to form F is less efficient when PBu3 is the catalyst (23% isolated yield of the tetrahydropyrazolopyridazinone 4); in this case, the intermediate E isomerizes into another amide species H.33 The ylide I is formed from the 7-endo cyclization of H; it then expels the catalyst PBu3 through the ylide-to-enoate conversion to furnish the tetrahydropyrazolodiazepinone 5. When PMe3 is employed as the catalyst, however, conjugate addition of the amide to the β-phosphonium enoates in B and E is the major reaction pathways for the allenoate 2n, due to decreased steric congestion at the β-carbon atom.
Scheme 3.

Mechanisms for the Azomethine Imine–Allenoate [3 + 2], [3 + 3], and [4 + 3] Annulations
With regard to the high E-selectivity for the exocyclic double bond of tetrahydropyrazolopyrazolone 3, Density Functional Calculations at the M06/6-31G** level of theory showed a strong interaction between the phosphorous atom and the carbonyl of the β-phosphonium enoate, most likely of electrostatic nature. This electrostatic interaction is thought to be key for the strereochemical outcome of the reaction (Scheme 4). Structurally, the phosphonium group points away from the bicycle, as a consequence of the relatively puckered heterobicycle and the pyramidalized carbon bearing the phosphonium enoate. This structural motif causes the phosphonium group to depart away from the bicyclic structure, thus favoring rotation of the carbonyl also away from the rest of the molecule. Therefore, it is anticipated that the magnitude of the electrostatic interaction should have an impact on the stereochemistry of the reaction. This lower perceived stereoselectivity (for the E isomer) could be eroded with increasing phosphine bulkiness. To further analyze the dominant stereodirecting effects in the elimination reaction, we performed relaxed coordinate scans for the phosphine-carbon elongation with PMe3 and (R)-II. Our calculations show that it is a combination of factors that lead to the low stereoselectivity when (R)-II is used. The bulkiness provided by the cyclohexyl group, the axial chirality, and the high torque of the benzyl groups in (R)-II, all impose a rigid chiral pocket that changes the orientation of the departure of the leaving phosphine group. In addition, we believe that the bulkier (R)-II may not allow for a P-O(=C) interaction as strong as that with less bulky phosphines, as a consequence of the slightly longer interatomic distance and overall structural rigidity. Both, the weaker electrostatic interaction, and the different departing orientation do not favor the rotation of the enoate in any direction, leading to low stereoselectivities. This can be observed by the differences in the dihedral angle of the enoate group at the transition state for elimination (Scheme 4). The elimination transition state for PMe3 as phosphine is a much later transition state, where the dihedral angle is ~30°, while for (R)-II, it is an early transition state with an enoate dihedral angle of ~10°.
Scheme 4.

Computational study of the stereochemical preference of PMe3 and (R)-II.
Encouraged by the successful incorporation of the azomethine ylides 1 as annulation reaction partners in the phosphine catalysis of α-substituted allenoates, we launched an investigation into the reaction of ethyl 2,3-butadienoate (2o) with the azomethine imine 1a. Our anticipation was that the allenoate 2o would serve as a two- or three-carbon component for [3 + 2] or [3 + 3] annulation, respectively, with the azomethine ylides 1. In the event, treatment of the allenoate 2o with the azomethine imine 1a in DCM at room temperature for 24 h under the influence of 20 mol % of PBu3 afforded the tetrahydropyrazolopyrazolone 3ao and the tetrahydropyrazolopyridazinone 6 in 45 and 36% yields, respectively, along with an unknown product (Table 5, entry 6). We used X-ray crystallography to establish the structures of the [3 + 2] adduct 3ao and the [3 + 3] adduct 6. Although the reaction provided a mixture of products, each compound exhibited exquisite stereoselectivity; 3ao was obtained exclusively in the E isomeric form and 6 as a single trans diastereoisomer. The annulations producing the adducts 3ao and 6 were apparently initiated by the additions at the β-and α-carbon atoms, respectively, of the allenoate 2o (see Scheme 3 and the mechanistic rationale below). In an attempt to improve the yield and chemoselectivity, we tested the effects of other phosphines (Table 5). Using PPh3 instead of PBu3 as the catalyst caused the reaction to proceed much more sluggishly, giving the cyclized products in poor yields, with the [3 + 3] adduct 6 as the major product (entry 2). When we used the more-nucleophilic phosphines EtPPh2, MePPh2, and Me2PPh as catalysts, we obtained the [3 + 2] cycloaddition product 3ao as the major product with improved yields (27, 53, and 69%, respectively) and relatively smaller amounts (5, 2, and 15%, respectively) of the [3 + 3] product (entries 3–5). Trimethylphosphine performed the best in terms of its 3ao/6 selectivity, with a 62% yield of the tetrahydropyrazolopyrazolone 3ao and a negligible amount (3%) of 6 (entry 7). Notably, HMPT facilitated the reaction of the azomethine ylide 1a and the allenoate 2o, albeit in low efficiency (entry 8). One peculiar observation was that an additional byproduct, which we isolated in less than 5% when using either PMe3 or PBu3, was isolated in almost 10% yield when using HMPT as the catalyst (vide infra).
Table 5.
Phosphine-Catalyzed Annulations of the Azomethine Imine 1a with Ethyl 2,3-Butadienoate (2o)a
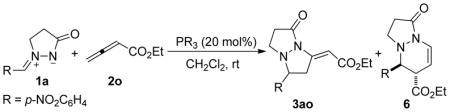 | ||||
|---|---|---|---|---|
| entry | PR3 | time (h) | 3aob yield (%)c |
6b yield (%)c |
| 1 d | – | 48 | 0 | 0 |
| 2 | PPh3 | 24 | 8 | 19 |
| 3 | EtPPh2 | 24 | 27 | 5 |
| 4 | MePPh2 | 24 | 53 | 2 |
| 5 | Me2PPh | 24 | 69 | 15 |
| 6 | PBu3 | 24 | 45 | 36 |
| 7 | PMe3 | 12 | 62 | 3 |
| 8 | HMPT | 24 | 15 | 22 |
1.2 equiv of allenoate was used.
The structure of this compound was verified through single-crystal X-ray analysis.
Isolated yield.
Without phosphine catalyst.
Evidently, the product 3ao was obtained via addition at the γ-carbon atom of the allenoate 2o while 6ao was produced via addition at the α-carbon atom. Consequently, we anticipated that γ-substituted allenoates would provide compounds 6 exclusively. Screening of the reaction conditions and substrates revealed that γ-substituted allenoates, namely γ-methyl, ethyl, isopropyl, tert-butyl, and phenyl allenoates, underwent annulations with the azomethine imine 1a under mild conditions with tolerable conversions. Nevertheless, these annulations appeared to proceed through at least two pathways, providing several spots on the TLC plates. With the γ-methyl and γ-phenyl allenoates as substrates, we could not isolate the pure target products because the reaction mixtures were too messy. From the γethyl, γ-isopropyl, and γ-tert-butyl allenoates 2p–2r, we isolated the [3 + 3] annulation products 6′ as major products in moderate yields (Scheme 5).
Scheme 5.

PBu3-Catalyzed [3 + 3] Annulations of the Azomethine Imine 1a with γ-Substituted Allenoates
Scheme 3 presents a mechanistic rationale for the [3 + 2] annulation of the azomethine imine 1a and the allenoate 2o to form 3ao. The [3 + 3] annulation was initiated through the formation of the β-phosphonium enoate intermediate J (Scheme 6). Addition of the zwitterion J to the iminium unit in 1 produced the phosphonium amide K. The 6-endo cyclization of K furnished the ylide L, which eliminated the phosphine after conversion to the β-phosphonium ester M to yield the tetrahydropyrazolopyridazinone 6′. When the allenoate 2o was the substrate, however, the conjugated enoate 6′ was not isolated; presumably, it isomerized into the enoate 6 because of the more acidic nature of the γ-proton when R1 was a hydrogen atom. This reaction marks the first example of a [3 + 3] annulation in which all three unsaturated carbon atoms of an allene are incorporated into the six-membered-ring product and suggests the future development of other [3 + 3] annulations between 1,3-dipoles and allenoates.
Scheme 6.

Mechanistic Rationale for the Formation of Compounds 6 and 6′
Next, we studied in further detail the unidentified byproduct generated from the above-mentioned reaction of ethyl 2,3-butadienoate (2o) and the azomethine imine 1a. To our delight, using a combination of NMR spectroscopy and X-ray crystallography, we determined the structures of these unknown products to be an equilibrating mixture of unexpected [3 + 2 + 3] products: the 1-oxo-2,3,5,6-tetrahydro-1H-pyrazolo[1,2-a][1,2]diazocine derivative 7 and the 1-oxo-2,3,5,10-tetrahydro-1H-pyrazolo[1,2-a][1,2]diazocine derivative 8 (Scheme 7). In this particular reaction, a trimeric zwitterionic intermediate, formed from two molecules of ethyl 2,3-butadienoate (2o) and a phosphine, presumably acted as a 1,5-dipole34 synthon to react with the azomethine imine, providing the [3 + 2 + 3] cycloaddition products. To the best of our knowledge, this reaction is the first example of this trimeric 1,5-dipole being captured in a phosphine-catalyzed intermolecular annulation of allenoates and electrophiles.5a, 35 This intriguing finding prompted us to further investigate this new reaction modality of the allenoate 2o under nucleophilic phosphine catalysis conditions. Although the literature is replete with examples of the incorporation of two molecules of alkenes, acetylenes, and allenes under transition-metal catalysis conditions36, these incidents are relatively rare in the realm of organocatalysis.37
Scheme 7.

Phosphine-Catalyzed [3 + 2 + 3] Annulation of the Azomethine Imine 1 and the Allenoate 2o
We varied the reaction conditions in an attempt to improve the yields of the [3 + 2 + 3] adducts. An initial survey of the reaction parameters revealed that the highest [3 + 2 + 3] product yield resulted when we conducted the reaction in DCM/benzene (4:1) at 0 °C. We employed this mixed solvent because DCM aided the dissolution of the azomethine imine and benzene favored the formation of the trimeric zwitterionic intermediate from a phosphine and two ethyl 2,3-butadienoate (2o) units, as observed by Lu.5a Varying the solvent or increasing the reaction temperature was deleterious to the reaction selectivity—specifically, it increased the amounts of the [3 + 2] and [3 + 3] cycloaddition products. In the mixed solvent (4:1 DCM/benzene) at 0 °C, the reaction between the azomethine imine 1a and the allenoate 2o displayed behavior somewhat distinct from that in DCM at room temperature (cf. Tables 5 and 6). For instance, the use of PPh3 as the catalyst at 0 °C in 4:1 DCM/benzene produced the [3 + 2] adduct as the major product (Table 6, entry 1). The trialkylphosphines PBu3 and PMe3 exhibited similar reaction profiles (entries 2 and 3). Dimethylphenylphosphine consistently provided the highest efficiency for the formation of the tetrahydropyrazolopyrazolone 3ao (entry 4); the overall reaction yield increased to 99%, of which 29% corresponded to the tetrahydropyrazolodiazocinone products 7a/8a. The use of HMPT as catalyst produced even more of the [3 + 2 + 3] product, but the overall mass recovery was lower (entry 5). This observation prompted us to test the effects of tris(sec-alkyl)phosphines (entries 6 and 7). We found that PCy3 was the most effective catalyst, affording the [3 + 2 + 3] adduct 7a/8a as the major product in 65% yield after 120 h (entry 8). Triisopropylphosphine also produced the tetrahydrodiazocine 7a/8a as the major product, but with lower efficiency. Tris(tert-butyl)phosphine displayed barely any reactivity at this low temperature, presumably because of steric hindrance; TLC revealed almost no cycloaddition products.
Table 6.
Phosphine-Catalyzed [3 + 2 + 3] Annulations of the Azomethine Imine 1a and the Allenoate 2oa
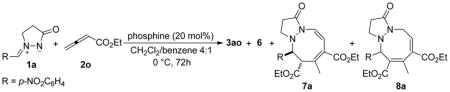 | ||||
|---|---|---|---|---|
| entry | PR3 | 3ao yield (%)b |
6 yield (%)b |
7a/8ac,d yield (%)b |
| 1 | PPh3 | 29 | 19 | <2 |
| 2 | PBu3 | 26 | 12 | 4 |
| 3 | PMe3 | 51 | 3 | 6 |
| 4 | PMe2Ph | 65 | 5 | 29 |
| 5 | HMPT | 50 | 4 | 32 |
| 6 | P(i-Pr)3 | 24 | 10 | 26 |
| 7 | PCy3 | 5 | 2 | 61 |
| 8e | PCy3 | 7 | 2 | 65f |
2.4 equiv of allenoate was used.
Isolated yield.
The structure of this compound was verified through single-crystal X-ray analysis.
The 7a:8a ratios were not determined, except the case in entry 8.
The reaction was run for 120 h.
The 7a:8a ratio was 19:81, based on integration of signals in the 1H NMR spectrum recorded in CDCl3.
Under these optimized reaction conditions (in 4:1 DCM/benzene at 0 °C with 20 mol % of PCy3), we investigated the reactions of a variety of azomethine imines 1 with the allenoate 2o (Table 7). When the azomethine imines 1 were derived from benzaldehydes featuring electron-donating or -withdrawing substituents at the para position, the reactions proceeded smoothly to afford the tetrahydropyrazolodiazocinones 7/8 in moderate to good yields (Table 7, entries 1–8); thus, electron-withdrawing and -donating groups at the para position provided similar reaction efficiencies. In contrast, meta and ortho substitution favored electron-withdrawing substituents (entries 9–16). In particular, ortho-halogen–substituted aryl groups produced the bicyclic products 7/8 in excellent yields (88–92%; entries 13–15). The 2-naphthaldehyde–derived azomethine imine 1m was also a viable substrate (entry 17).
Table 7.
Phosphine-Catalyzed [3 + 2 + 3] Cycloadditions of the Azomethine Imines 1 with the Allenoate 2oa
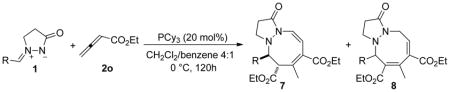 | ||||
|---|---|---|---|---|
| entry | R | product | yield (%)b | ratio of 7:8c |
| 1 | Ph (1b) | 7b + 8b | 81 | 34:66 |
| 2 | 4-MeC6H4 (1c) | 7c + 8c | 64 | 39:61 |
| 3 | 4-i-PrC6H4 (1d) | 7d + 8d | 67 | 42:58 |
| 4 | 4-OMeC6H4 (1e) | 7e + 8e | 76 | 44:56 |
| 5 | 4-FC6H4 (1f) | 7f + 8f | 68 | 27:73 |
| 6 | 4-ClC6H4 (1g) | 7g + 8g | 63 | 37:63 |
| 7 | 4-BrC6H4 (1h) | 7hd + 8h | 77 | 29:71 |
| 8 | 4-CNC6H4 (1i) | 7id + 8i | 51 | 21:79 |
| 9 | 3-NO2C6H4 (1k) | 7k + 8k | 77 | 17:83 |
| 10 | 3-FC6H4 (1v) | 7v + 8v | 79 | 25:75 |
| 11 | 3-ClC6H4 (1w) | 7w + 8w | 82 | 24:76 |
| 12 | 3-BrC6H4 (1x) | 7x + 8x | 75 | 22:78 |
| 13 | 2-BrC6H4 (1y) | 7y + 8y | 92 | 28:72 |
| 14 | 2-ClC6H4 (1z) | 7z + 8z | 90 | 24:76 |
| 15 | 2-FC6H4 (1zz) | 7zz + 8zz | 88 | 25:75 |
| 16 | 2-NO2C6H4 (1l) | 7l + 8l | 73 | 9:91 |
| 17 | 2-naphthyl (1p) | 7p + 8p | 76 | 32:68 |
2.4 equiv of allenoate was used.
Isolated yield.
Based on integration of signals in the 1H NMR spectrum recorded in CDCl3; the solvent had a very strong effect on the ratio.
The structure of this compound was verified through single-crystal X-ray analysis.
Scheme 8 provides a mechanistic proposal for this unprecedented cyclization reaction. The key event is the formation of the trimeric zwitterionic intermediate N, which is then captured in situ by the azomethine imine 1. The trimeric zwitterionic intermediate N is generated via the conjugate addition of the β-phosphonium dienloate J to another molecule of the allenoate 2o. The self-cycloaddition of the allenoate 2o to form P under phosphine catalysis has been reported previously, by Lu in 1995.5a Here, for the first time, we have realized the intermolecular cycloaddition of the trimeric intermediate N with the azomethine imine. In a scenario where the fewest zwitterionic intermediates are involved,38 the intermediate N adds to the azomethine imine 1 to form the intermediate Q. The 8-endo cyclization of Q produces the ylide R. Well-established proton transfers and subsequent β-elimination of the phosphine leads to formation of the tetrahydropyrazolodiazocinone product S. It appears that the cross-conjugated exomethylene group in compound S isomerizes into the endocyclic double bond to give the tetrahydropyrazolodiazocinone 8. In turn, we isolated compound 8 as an equilibrating mixture with its tautomer 7.
Scheme 8.

Mechanism for the [3 + 2 + 3] Cycloadditions of the Azomethine Imines 1 and the Allenoate 2o
To compare the relative ground state energies of the tetrahydropyrazolodiazocinones S (R = phenyl), 7b, and 8b, we studied these three molecules computationally using density functional theory (DFT) with the M06 functional, a hybrid meta-GGA functional that can be a good choice for main-group thermochemistry.39 Given the conformer-rich landscape of the our tetrahydropyrazolodiazocinones, we used the M06/6-31G** level of theory to perform a conformational study, focusing on the positioning of the ring substituents and inversion of the sp3-hybridized nitrogen atom. We further minimized at least two of the lowest-energy conformers for each isomer (S, 7b, 8b) using finer grids and calculated the analytic Hessian for each minimum. Electronic energies were calculated using the M06 functional and the correlation-consistent polarized triple-ζ basis set cc-pVTZ++40 as implemented in Jaguar 7.6.110.41 For S, 7b, and 8b, the computed relative ground state free energies (ΔG; including zero-point, solvation for DCM, and thermodynamic corrections to the free energy at 298 K) at the M06/cc-pVTZ++ level were 4.1, 0.8, and 0.0 kcal/mol, respectively; thus, isomerization of the intermediate S to the product 8b is thermodynamically favored and compounds 8b and 7b would be in equilibrium at ambient temperature, as observed experimentally (Table 7, entry 1).
In determining the structures of the [3 + 2 + 3] annulation products, all of our 1H and 13C NMR spectra, recorded in CDCl3 at room temperature, indicated that the products existed as mixtures of the tautomers 7 and 8 in solution, with 8 as the major isomer. To further study the interconversion of compound 7 and its tautomer 8 in solution, we recorded 1H NMR spectra of the 7a/8a and 7h/8h pairs in various deuterated solvents and at various temperatures—making some interesting observations. For the 7a/8a pair, 8a was the major isomer in either CDCl3 or CD2Cl2 at room temperature; the ratio of 7a to 8a was approximately 1:4. Even in CD2Cl2 at −80 °C, the ratio of 7a to 8a remained unchanged, although the two signals of the aromatic protons broadened as the temperature increased.42 Relative to the behavior of the 7a/8a pair, the 7h/8h equilibrium was very sensitive to the solvent and temperature. In CDCl3 at room temperature, 8h was the major isomer and the ratio of 8h to 7h was 71:29. In contrast, in CD2Cl2 at room temperature, 7h existed as the sole isomer. When we decreased the temperature from room temperature to −20 °C, the two signals of the aromatic protons each resolved from one broad peak to two distinct sharp peaks. More interestingly, in CDCl3 at 0 or −20 °C, the mixture of the two tautomers 7h and 8h completely converged into 7h.42 These observations are consistent with the crystal growth behavior. When we left the 7h/8h compound pair in DCM/MeOH, we isolated single crystals of 7h exclusively. From the mixture of compounds 7a and 8a, we grew single crystals of the single tautomer 8a from DCM/hexane or DCM/MeOH.
The fused pyrazolidinone heterocycles, arising from the phosphine-catalyzed annulations of azomethine imines with allenoates, can be transformed into other useful compounds. For example, SmI2 selectively reduced the exocyclic double bond to provide pyrazolopyrazolone 9 as a single diastereoisomer43,44 while treatment with NaBH4 in EtOH furnished the ring-opened product 10 as a mixture of diastereoisomers (Scheme 9). The dihydropyrazolopyrazolone 11, obtained through the CuBr/TBHP oxidation,45 should be a useful precursor for further functionalization through cross-coupling reactions. Treatment of the tetrahydrodiazocine 7b/8b with SmI2 reduced the enamine double bond selectively to provide the dihydrodiazocine 12 in 73% isolated yield.43,44
Scheme 9.

Reactions of the Tetrahydropyrazolopyrazolone 3ba and the Tetrahydropyrazolodiazocinone 7b
Conclusion
We have investigated the phosphine-catalyzed [3 + 2], [3 + 3], [4 + 3], and [3 + 2 + 3] annulations of azomethine imines with allenoates. These cyclization reactions are operationally simple and proceed smoothly under very mild reaction conditions, providing a broad range of fused pyrazolidinone heterocycles in moderate to excellent yields. The positive characteristics of this protocol—including the production of a diverse range of dinitrogen-fused bicycles and the ready preparation of azomethine imine substrates—suggest that these reactions might have extensive applicability in organic synthesis. Notably, we captured a trimeric zwitterionic intermediate, formed from two molecules of ethyl 2,3-butadienoate and a molecule of a phosphine, that acted as a 1,5-dipole in the cycloaddition reaction, leading to [3 + 2 + 3] adducts. The incorporation of two molecules of allenoates into cycloaddition products is a new application for this versatile class of molecules under phosphine catalysis conditions. In addition, we investigated, both spectroscopically and computationally, the unique tautomerization behavior of the tetrahydropyrazolodiazocinone products. Studies to address the further applications of azomethine imines in nucleophilic phosphine catalysis and the development of asymmetric variants of these annulations, as well as their use in the synthesis of biologically relevant molecules, are ongoing in our laboratories.
Supplementary Material
Acknowledgments
This study was supported by the National Natural Science Foundation of China (H.G.), the startup research funding from China Agricultural University (H.G.), Chinese Universities Scientific Fund (Project No. 2011JS029 and 2011JS031) (H.G.), the National Scientific and Technology Supporting Program of China (2011BAE06B05-5) (H.G.), Nutriechem Company (H.G.), and the U.S. National Institutes of Health (NIH; O.K.: R01GM071779 and P41GM081282). Computational facilities were funded by grants from ARO-DURIP and ONR-DURIP.
Footnotes
Supporting Information Available: Detailed experimental procedures and characterization data for all new compounds (PDF). Crystallographic data of compounds 3ad, 3an, 3ao, 4, trans-5, 6, 7h, 7i, and 8a (CIF files). This material is available free of charge via the Internet at http://pubs.acs.org.
References and Footnotes
- 1.(a) Gothelf KV. In: Cycloaddition Reactions in Organic Synthesis. Kobayashi S, Jørgensen KA, editors. Wiley-VCH; Weinheim, Germany: 2002. pp. 211–245. [Google Scholar]; (b) Padwa A, Pearson WH, editors. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry—Toward Heterocycles and Natural Products; John Wiley and Sons; Hoboken, NJ: 2003. [Google Scholar]; (c) Hashimoto T, Maruoka K, Ma S, editors. Handbook of Cyclization Reactions. Vol. 3. Wiley-VCH; Weinheim, Germany: 2009. pp. 87–168. [Google Scholar]
- 2.For reviews on phosphine-promoted cycloadditions, see: Lu X, Zhang C, Xu Z. Acc Chem Res. 2001;34:535–544. doi: 10.1021/ar000253x.Valentine DH, Hillhouse JH. Synthesis. 2003:317–334.Methot JL, Roush WR. Adv Synth Catal. 2004;346:1035–1050.Lu X, Du Y, Lu C. Pure Appl Chem. 2005;77:1985–1990.Nair V, Menon RS, Sreekanth AR, Abhilash N, Biju AT. Acc Chem Res. 2006;39:520–530. doi: 10.1021/ar0502026.Ye LW, Zhou J, Tang Y. Chem Soc Rev. 2008;37:1140–1152. doi: 10.1039/b717758e.Denmark SE, Beutner GL. Angew Chem, Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943.Kwong CKW, Fu MY, Lam CSL, Toy PH. Synthesis. 2008:2307–2317.Aroyan CE, Dermenci A, Miller SJ. Tetrahedron. 2009;65:4069–4084.Cowen BJ, Miller SJ. Chem Soc Rev. 2009;38:3102–3116. doi: 10.1039/b816700c.Marinetti A, Voituriez A. Synlett. 2010:174–194.Kolesinska B. Cent Eur J Chem. 2010:1147–1171.
- 3.Xu S, Zhou L, Ma R, Song H, He Z. Org Lett. 2010;12:544–547. doi: 10.1021/ol902747c. [DOI] [PubMed] [Google Scholar]
- 4.(a) Meng X, Huang Y, Chen R. Org Lett. 2009;11:137–140. doi: 10.1021/ol802453c. [DOI] [PubMed] [Google Scholar]; (b) Zhang Q, Yang L, Tong X. J Am Chem Soc. 2010;132:2550–2551. doi: 10.1021/ja100432m. [DOI] [PubMed] [Google Scholar]
- 5.For examples of [3 + 2] cycloadditions, see: Zhang C, Lu X. J Org Chem. 1995;60:2906–2908.Xu Z, Lu X. Tetrahedron Lett. 1997;38:3461–3464.Zhu G, Chen Z, Jiang Q, Xiao D, Cao P, Zhang X. J Am Chem Soc. 1997;119:3836–3837.Pyne SG, Schafer K, Skelton BW, White AH. Chem Commun. 1997;67:2267–2268.Wang JC, Ng SS, Krische MJ. J Am Chem Soc. 2003;125:3682–3683. doi: 10.1021/ja030022w.Zhu XF, Henry CE, Kwon O. Tetrahedron. 2005;61:6276.Wilson JE, Fu GC. Angew Chem, Int Ed. 2006;45:1426–1429. doi: 10.1002/anie.200503312.Cowen BJ, Miller SJ. J Am Chem Soc. 2007;129:10988–10989. doi: 10.1021/ja0734243.Xia Y, Liang Y, Chen Y, Wang M, Jiao L, Huang F, Liu S, Li Y, Yu ZX. J Am Chem Soc. 2007;129:3470–3471. doi: 10.1021/ja068215h.Mercier E, Fonovic B, Henry C, Kwon O, Dudding T. Tetrahedron Lett. 2007;48:3617–3620.Henry CE, Kwon O. Org Lett. 2007;9:3069–3072. doi: 10.1021/ol071181d.Fang YQ, Jacobsen EN. J Am Chem Soc. 2008;130:5660–5661. doi: 10.1021/ja801344w.Voituriez A, Panossian A, Fleury-Bregeot N, Retailleau P, Marinetti A. J Am Chem Soc. 2008;130:14030–14031. doi: 10.1021/ja806060a.
- 6.Zhu X, Henry CE, Wang J, Dudding T, Kwon O. Org Lett. 2005;7:1387–1390. doi: 10.1021/ol050203y. [DOI] [PubMed] [Google Scholar]
- 7.For an example of a [3 + 3] cycloaddition, see: Guo H, Xu Q, Kwon O. J Am Chem Soc. 2009;131:6318–6319. doi: 10.1021/ja8097349.
- 8.For examples of [4 + 2] cycloadditions of α-alkyl allenoates, see: Zhu XF, Lan J, Kwon O. J Am Chem Soc. 2003;125:4716–4717. doi: 10.1021/ja0344009.Wurz RP, Fu GC. J Am Chem Soc. 2005;127:12234–12235. doi: 10.1021/ja053277d.Tran YS, Kwon O. Org Lett. 2005;7:4289–4291. doi: 10.1021/ol051799s.Tran YS, Kwon O. J Am Chem Soc. 2007;129:12632–12633. doi: 10.1021/ja0752181.Castellano S, Fiji HDG, Kinderman SS, Watanabe M, de Leon P, Tamanoi F, Kwon O. J Am Chem Soc. 2007;129:5843–5845. doi: 10.1021/ja070274n.For [4 + 2] reactions of simple allenoates, see: Zhu XF, Schaffner AP, Li RC, Kwon O. Org Lett. 2005;7:2977–2980. doi: 10.1021/ol050946j.For examples of a [4 + 2] reaction in which the allenoate acts as a two-atom contributor, see: Ishar MPS, Kumar K, Kaur S, Kumar S, Girdhar NK, Sachar S, Marwaha A, Kapoor A. Org Lett. 2001;3:2133–2136. doi: 10.1021/ol010026a.Meng X, Huang Y, Zhao H, Xie P, Ma J, Chen R. Org Lett. 2009;11:991–994. doi: 10.1021/ol802917d.
- 9.For an example of a [4 + 3] cycloaddition, see: Kumar K, Kapoor R, Kapur A, Ishar MPS. Org Lett. 2000;2:2023–2025. doi: 10.1021/ol0000713.
- 10.Kumar K, Kapur A, Ishar MPS. Org Lett. 2000;2:787–789. doi: 10.1021/ol000007l. [DOI] [PubMed] [Google Scholar]
- 11.For other selected examples of phosphine-catalyzed annulations, see: Trost BM, Li CJ. J Am Chem Soc. 1994;116:10819–10820.Lu C, Lu X. Org Lett. 2002;4:4677–4679. doi: 10.1021/ol0270733.Liu B, Davis R, Joshi B, Reynolds DW. J Org Chem. 2002;67:4595–4598. doi: 10.1021/jo016154u.Kuroda H, Tomita I, Endo T. Org Lett. 2003;5:129–131. doi: 10.1021/ol020198n.Du Y, Lu X, Zhang C. Angew Chem, Int Ed. 2003;42:1035–1037. doi: 10.1002/anie.200390266.Jung CK, Wang JC, Krische MJ. J Am Chem Soc. 2004;126:4118–4119. doi: 10.1021/ja049377l.Thalji RK, Roush WR. J Am Chem Soc. 2005;127:16778–16779. doi: 10.1021/ja054085l.Kamijo S, Kanazawa C, Yamamoto Y. Tetrahedron Lett. 2005;46:2563–2566.Zhao GL, Shi M. Org Biomol Chem. 2005;3:3686–3694. doi: 10.1039/b510572b.Silva F, Sawaki M, Gouverneur V. Org Lett. 2006;8:5417–5419. doi: 10.1021/ol0624225.Virieux D, Guillouzic AF, Cristau HJ. Tetrahedron. 2006;62:3710–3720.Gabillet S, Lecerclé D, Loreau O, Carboni M, Dézard S, Gomis JM, Taran F. Org Lett. 2007;9:3925–3927. doi: 10.1021/ol701563e.Nair V, Mathew SC, Biju AT, Suresh E. Angew Chem, Int Ed. 2007;46:2070–2073. doi: 10.1002/anie.200604025.Ye LW, Sun XL, Wang QG, Tang Y. Angew Chem, Int Ed. 2007;46:5951–5954. doi: 10.1002/anie.200701460.Sriramurthy V, Barcan GA, Kwon O. J Am Chem Soc. 2007;129:12928–12929. doi: 10.1021/ja073754n.Creech GS, Zhu XF, Fonovic B, Dudding T, Kwon O. Tetrahedron. 2008;64:6935–6942. doi: 10.1016/j.tet.2008.04.075.
- 12.For early works on azomethine imines, see: Godtfredsen WO, Vangedal S. Acta Chem Scand. 1955;9:1498–1509.Huisgen R, Grashey R, Laur P, Leitermann H. Angew Chem. 1960;72:416–417.
- 13.For early reports demonstrating that azomethine imines 1 are stable and easily handled compounds, see: Dorn H, Otto A. Chem Ber. 1968;101:3287–3301. doi: 10.1002/cber.19681010934.Dorn H, Otto A. Angew Chem, Int Ed Engl. 1968;7:214–215.
- 14.For recent examples of the cycloadditions of azomethine imines, see: Shintani R, Fu GC. J Am Chem Soc. 2003;125:10778–10779. doi: 10.1021/ja036922u.Suárez A, Downey CW, Fu GC. J Am Chem Soc. 2005;127:11244–11245. doi: 10.1021/ja052876h.Shintani R, Hayashi T. J Am Chem Soc. 2006;128:6330–6331. doi: 10.1021/ja061662c.Chen W, Yuan XH, Li R, Du W, Wu Y, Ding LS, Chen YC. Adv Synth Catal. 2006;348:1818–1822.Chan A, Scheidt KA. J Am Chem Soc. 2007;129:5334–5335. doi: 10.1021/ja0709167.Suga H, Funyu A, Kakehi A. Org Lett. 2007;9:97–100. doi: 10.1021/ol062675y.Shintani R, Murakami M, Hayashi T. J Am Chem Soc. 2007;129:12356–12357. doi: 10.1021/ja073997f.Chen W, Du W, Duan YZ, Wu Y, Yang SY, Chen YC. Angew Chem, Int Ed. 2007;46:7667–7670. doi: 10.1002/anie.200702618.Sibi MP, Rane D, Stanley LM, Soeta T. Org Lett. 2008;10:2971–2974. doi: 10.1021/ol800904t.Shapiro ND, Shi Y, Toste FD. J Am Chem Soc. 2009;131:11654–11655. doi: 10.1021/ja903863b.Keller M, Sido ASS, Pale P, Sommer J. Chem Eur J. 2009;15:2810–2817. doi: 10.1002/chem.200802191.Perreault C, Goudreau SR, Zimmer LE, Charette AB. Org Lett. 2008;10:689–692. doi: 10.1021/ol702414e.Hashimoto T, Maeda Y, Omote M, Nakatsu H, Maruoka K. J Am Chem Soc. 2010;132:4076–4077. doi: 10.1021/ja100787a.
- 15.Elguero J. In: Pyrazoles: Comprehensive Heterocyclic Chemistry II. Katritzky AR, Rees CW, Scriven EFV, editors. Vol. 3. Elsevier; Oxford: 1996. pp. 1–75. and references cited therein Varvounis G, Fiamegos Y, Pilidis G. Adv Heterocycl Chem. 2001;80:75–165.Eicher T, Hauptmann S. The Chemistry of Heterocycles. 2. Wiley-VCH; Weinheim: 2003.
- 16.For reviews, see: Marchand-Brynaert J, Ghosez L. In: Recent Progress in the Chemical Synthesis of Antibiotics. Lukacs G, Ohno M, editors. Springer; Berlin: 1990. Hanessian S, McNaughton-Smith G, Lombart HG, Lubell WD. Tetrahedron. 1997;53:12789–12854.Konaklieva MI, Plotkin BJ. Curr Med Chem Anti-infect Agents. 2003;2:287–302.For related research, see: Jungheim LN, Sigmund SK, Holmes RE, Barnett CJ, Ternansky RJ. 202046 Eur Pat Appl EP. Chem Abstr. 1986;106:119880.Ternansky RJ, Draheim SE. In: In Recent Advances in the Chemistry of β-Lactam Antibiotics. Bentley PH, Southgate RH, editors. Royal Society of Chemistry; London: 1989. pp. 139–156.Ternansky RJ, Holmes RA. Drugs Future. 1990;15:149–157.Cusan C, Spalluto G, Prato M, Adams M, Bodensieck A, Bauer R, Tubaro A, Bernardi P, Da Ros T. Il Farmaco. 2005;60:327–332. doi: 10.1016/j.farmac.2004.12.009.
- 17.Cucurou C, Battioni JP, Thang DC, Nam NH, Mansuy D. Biochemistry. 1991;30:8964–8970. doi: 10.1021/bi00101a008. [DOI] [PubMed] [Google Scholar]
- 18.Kawai K, Shiojiri H, Hasegawa J, Nozaki M, Tsurumi K, Nozawa Y, Fujimura H. Prostaglandins Cardiovasc Dis, [Int Symp] 1986:61–67. [Google Scholar]
- 19.White HL, Howard JL, Cooper BR, Soroko FE, McDermed JD, Ingold KJ, Maxwell RA. J Neurochem. 1982;39:271–273. doi: 10.1111/j.1471-4159.1982.tb04733.x. [DOI] [PubMed] [Google Scholar]
- 20.(a) Fischer R, Bretschneider T, Gesing ERF, Feucht D, Kuck K-H, Loesel P, Malsam O, Arnold C, Auler T, Hills MJ, Kehne H. 2005016873 PCT Int Appl WO. ; Chem Abstr. 2005;142:261530. [Google Scholar]; (b) Kosower EM, Radkowsky AE, Fairlamb AH, Croft SL, Nea RA. Eur J Med Chem. 1995;30:659–671. [Google Scholar]
- 21.Hatayama S, Hayashi K, Honma M, Takahashi I. 2002220338 Jpn Kokai Tokkyo Koho JP. ; Chem Abstr. 2002;137:150215. [Google Scholar]
- 22.Bhandari A, Boros EE, Cowan DJ, Handlon AL, Hyman CE, Oplinger JA, Rabinowitz MH, Turnbull PSPCT. 2003076440 IntApplWO. ; Chem Abstr. 2003;139:261291. [Google Scholar]
- 23.Kosower EM, Hershkowitz E. 94658 Isr Patent ISXXAQ IL. ; Chem Abstr. 1994;122:214077. [Google Scholar]
- 24.(a) Maetzke T, Stoller A, Wendeborn S, Szczepanski H. 2001017972 PCT Int Appl WO. ; Chem Abstr. 2001;134:237482. [Google Scholar]; (b) Krueger B-W, Fischer R, Bertram H-J, Bretschneider T, Boehm S, Krebs A, Schenke T, Santel H-J, Lurssen K, Schmidt RR, Erdelen C, Wachendorff-Neumann U, Stendel W. 5358924 US. ; Chem Abstr. 1994;122:25904. [Google Scholar]; (c) Kunz W, Nebel K, Wenger J. 9952892 PCT Int Appl WO. ; Chem Abstr. 1999;131:359759. [Google Scholar]
- 25.Kamata M, Yamashita T. 2009196966 Jpn Kokai Tokkyo Koho JP. ; Chem Abstr. 2009;151:337200. [Google Scholar]
- 26.(a) Burk MJ, Feaster JE. J Am Chem Soc. 1992;114:6266–6267. [Google Scholar]; (b) Burk MJ, Martinez JP, Feaster JE, Cosford N. Tetrahedron. 1994;50:4399–4428. [Google Scholar]
- 27.(a) Kobayashi S, Shimizu H, Yamashita Y, Ishitani H, Kobayashi J. J Am Chem Soc. 2002;124:13678–13679. doi: 10.1021/ja027681d. [DOI] [PubMed] [Google Scholar]; (b) Shirakawa S, Lombardi PJ, Leighton JL. J Am Chem Soc. 2005;127:9974–9975. doi: 10.1021/ja052307+. [DOI] [PubMed] [Google Scholar]; (c) Yamashita Y, Kobayashi S. J Am Chem Soc. 2004;126:11279–11282. doi: 10.1021/ja049498l. [DOI] [PubMed] [Google Scholar]
- 28.(a) Tashiro T. Nippon Kagaku Kaishi. 1988:684–690. [Google Scholar]; (b) Shurig JE, Brandner WT, Huflutalen JB, Doyle GJ, Gylys JA. Cisplatin. Academic Press; New York: 1980. p. 227. [Google Scholar]; (c) Armarego WLF, Kobayashi T. J Chem Soc C. 1970:1597–1600. [Google Scholar]
- 29.For a review of the pyrazolidinones, see: Claramunt RM, Elguero J. Org Prep Proced Int. 1991;23:273–320.
- 30.For a review of direct [3 + 2] cycloaddition of allenes with a variety of 1,3-dipoles in the absence of catalyst, see: Pinho e Melo TMVD. Curr Org Chem. 2009;13:1406–1431.
- 31.A survey of reaction solvents revealed that haloalkanes (DCM, CHCl3, 1,2-dichloroethane) were superior to other common organic solvents. See the Supporting Information for a full list of solvents for which we measured the reaction yields.
- 32.Khong SN, Tran YS, Kwon O. Tetrahedron. 2010;66:4760–4768. doi: 10.1016/j.tet.2010.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baldwin JE. J Chem Soc, Chem Commun. 1976:734–736. [Google Scholar]
- 34.For reviews on 1,5-dipoles, see: Taylor EC, Turchi IJ. Chem Rev. 1979;79:181–231.Grigg R. Chem Soc Rev. 1987;16:89–121.Pinho e Melo TMVD. Eur J Org Chem. 2006:2873–2888.
- 35.For the self-cycloaddition of a trimeric zwitterionic intermediate, formed from two molecules of ethyl 2,3-butadienoate (2o) and a phosphine, acting as a 1,5-dipole, see ref. 5a. For an example of Rh(I)-catalyzed [3 + 2 + 2] cyclization of methyl buta-2,3-dienoate, in which two allene units, each acting as a two-membered synthon, were incorporated into the product, see: Barluenga J, Vicente R, López LA, Tomás M. Tetrahedron. 2010;66:6335–6339.
- 36.For some examples of the incorporation of two molecules of alkenes, acetylenes, and allenes under transition-metal catalysis conditions, see: Schwiebert KE, Stryker JM. J Am Chem Soc. 1995;117:8275–8276.Heller B, Sundermann B, Buschmann H, Drexler HJ, You JS, Holzgrabe U, Heller E, Oehme G. J Org Chem. 2002;67:4414–4422. doi: 10.1021/jo011032n.Barluenga J, Barrio P, López LA, Tomás M, García-Granda S, Alvarez-Rúa C. Angew Chem Int Ed. 2003;42:3008–3011. doi: 10.1002/anie.200351216.Saito S, Masuda M, Komagawa S. J Am Chem Soc. 2004;126:10540–10541. doi: 10.1021/ja0494306.Heller B, Sundermann B, Buschmann H, Drexler HJ, You JS, Holzgrabe U, Heller E, Oehme G. J Org Chem. 2002;67:4414–4422. doi: 10.1021/jo011032n.Hirabayashi T, Sakaguchi S, Ishii Y. Adv Synth Catal. 2005;347:872–876.Hilt G, Paul A, Harms K. J Org Chem. 2008;73:5187–5190. doi: 10.1021/jo800735x.Yamasaki R, Terashima N, Sotome I, Komagawa S, Saito S. J Org Chem. 2010;75:480–483. doi: 10.1021/jo902251m.
- 37.For L-proline–catalyzed self-aldolization of propionaldehyde and acetaldehyde, see: Chowdari NS, Ramachary DB, Córdova A, Barbas CF., III Tetrahedron Lett. 2002;43:9591–9595.Córdova A, Notz W, Barbas CF., III J Org Chem. 2002;67:301–303. doi: 10.1021/jo015881m.
- 38.An alternative route to the tetrahydropyrazolodiazocine 8 may be proceed via several zwitterionic intermediates; in this case, the tetrahydropyrazolodiazocine 8 would be the immediate product from the β-elimination of phosphine from the β-phosphonium enoate intermediate V. See the Supporting Information for such a path.
- 39.(a) Zhao Y, Truhlar DG. Theo Chem Acc. 2008;120:215–241. [Google Scholar]; (b) Zhao Y, Truhlar DG. Acc Chem Res. 2008;41:157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 40.Dunning TH. J Chem Phys. 1989;90:1007–1023. [Google Scholar]
- 41.Jaguar . version 7.6. Schrödinger; New York, NY: 2009. [Google Scholar]
- 42.See the 1H NMR spectra in the Supporting Information
- 43.The relative stereochemistry of the ring substituents of 9 and 12 was assigned according to the NOESY-NMR experiments. See the Supporting Information for details.
- 44.Davies SG, Rodriguez-Solla H, Tamayo JA, Cowley AR, Concellon C, Garner AC, Pakes AL, Smith AD. Org Biomol Chem. 2005:1435–1447. doi: 10.1039/b500566c. [DOI] [PubMed] [Google Scholar]
- 45.Glazier ER. J Org Chem. 1962;27:4397–4399. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.