

Abstract
HIV-1 integrase (IN) is a validated therapeutic target for antiviral agents. However, the emergence of viral strains resistant to clinically studied IN inhibitors demands new structure and new mechanism IN inhibitors. Herein, we describe the design and discovery of novel IN inhibitors targeting the catalytic domain as well as its interaction with LEDGF/p75, which is essential for the HIV-1 integration as an IN cofactor. By merging the pharmacophores of salicylate and catechol, the 2,3-dihydroxybenzamide (5a) was identified as a new scaffold to inhibit the strand transfer reaction efficiently. Further structural modifications on the 2,3-dihydroxybenzamide scaffold revealed that the heteroaromatic functionality attached on the carboxamide portion and the piperidin-1-ylsulfonyl substituted at the phenyl ring are beneficial for the activity, resulting in a low micromolar IN inhibitor (5p, IC50 = 5 μM) with more than 40-fold selectivity for the strand transfer over the 3′-processing reaction. More significantly, this active scaffold remarkably inhibited the interaction between IN and LEDGF/p75 cofactor. The prototype example, N-(cyclohexylmethyl)-2,3-dihydroxy-5-(piperidin-1-ylsulfonyl) benzamide (5u) inhibited the IN-LEDGF/p75 interaction with an IC50 value of 8 μM. Based on the molecular modeling, the mechanism of action was hypothesized to involve the chelation of the divalent metal ions inside the IN active site. And the inhibitor of IN-LEDGF/p75 interaction was properly bound to the LEDGF/p75 binding site in IN protein. This work provided a new and efficient approach to evolve novel HIV-1 IN inhibitors from rational integration and optimization of previously reported inhibitors.
Keywords: HIV-1 integrase inhibitors; 2,3-dihydroxybenzamide; strand transfer; 3′-process; LEDGF/p75; chelation
Introduction
The virally encoded IN is a critical enzyme for viral replication. The protein catalyzes the insertion of the reverse-transcribed proviral cDNA into the host cell genome through a multistep process that includes two catalytic reactions: 3′-endonucleolytic processing of the proviral DNA ends (termed 3′-processing) and the integration of the 3′-processed viral DNA into the cellular DNA (referred to as strand transfer).1 Divalent metals, such as Mg2+ or Mn2+, are required for not only the 3′-processing and strand transfer steps, but also for the assembly of IN onto specific viral donor DNA to form a complex that is competent to carry out either function.2 It is generally accepted that Mg2+ is a more reasonable cofactor for integration because of its 1 000 000-fold abundance over Mn2+ in cells.2,3 Thus the chelation of the critical metal cofactor can cause the functional impairment of IN, providing an alternative opportunity for the design and development of highly efficient IN inhibitors.4 In fact, all of the small molecule HIV-1 IN inhibitors that exhibit potent antiviral activity by the inhibition of viral DNA integration, commonly contain a structural motif that coordinates the two divalent magnesium ions in the enzyme’s active site.4 Typically, the most developed and promising β-diketoacid, naphthyridine carboxamide, pyrimidinone and quinolone carboxylic acid classes of IN inhibitors belong to this category.5 Among these chelation inhibitors, the pyrimidinone MK-0518 has been approved by the FDA and currently as the first and only HIV-1 IN inhibitor for the treatment of HIV infection.6
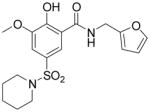
Although great strides have been achieved in the design and discovery of IN inhibitors as antiviral agents,7,8 the emergence of viral strains resistant to clinically studied IN inhibitors and the dynamic nature of the HIV-1 genome demand a continued effort toward the discovery of novel inhibitors to keep a therapeutic advantage over the virus. One approach to discover structurally novel classes of IN inhibitors is to revive previously identified IN inhibitor chemical classes, which displayed potent IN inhibition, but were developmentally halted due to unwanted pharmacokinetic, pharmacodynamic, or toxicological properties. The salicylhydrazide and polyhydroxyl aromatics (as exemplified by the structures in Fig. 1) were previously reported as potent IN inhibitors,9,10 but the inherently high cytotoxicity in these compounds limited their therapeutic application as antiretroviral agents. Recent structural modifications on the salicylhydrazide family resulted in a significant decrease in the cytotoxicity while the IN inhibitory activity was retained,11,12 thereby confirming the feasibility of reviving old drugs by structural optimization.
Figure 1.

Representative examples of salicyl and catechol-containing IN inhibitors
In this study, we were interested in designing new IN inhibitors by merging the pharmacophores of the salicylic acid and catechol to generate novel chemical scaffolds. Actually, the adjacent carboxylic and hydroxyl groups on salicylic acid could serve as the metal binding pharmacophore. On the other hand, the polyhydroxylated aromatic inhibitors are usually active against both 3′-processing and strand transfer reactions13 that might imply a different mechanism targeting both steps. Since HIV-1 resistant strains exhibited cross-resistance to different strand transfer specific chemical classes in preclinical and clinical development studies,5 the optimal integration of salicylic acid and catechol pharmacophores is expected to produce novel inhibitors by chelating a divalent metal on the IN active site. These inhibitors are likely to be effective against viral strains that display resistance to strand transfer specific inhibitors.
Consequently, by combining the privileged fragments of salicyl and catechol-containing IN inhibitors, we designed four classes of 2-hydroxybenzoic acid derivatives with various substitution patterns on the phenyl ring to determine an optimal scaffold (as shown in Fig. 2). The catechol moiety is susceptible to oxidation into a quinone species that have a propensity to cross-link with cellular proteins, thereby leading to cytotoxicity,14 an outcome that we tried to mitigate by incorporating alkyoxy groups such as benzyloxy, 4′-fluorobenzyloxy and naphthalenylmethoxy into 3-, 4-, 5-, and 6-postion of the 2-hydroxybenzoic acid, respectively (Fig. 2, compounds 3a–j). Furthermore, the aryl group might serve as the pharmacophore to interact with the hydrophobic binding surface of the IN, as revealed by the aryl diketoacid IN inhibitors. On the other hand, we tried to apply hydroxyamic acid (Fig. 2, 4a–d) or 2,3-dihydroxybenzoyl amide (Fig. 2, 5a–x, 6a–d) as an isostere of the 3-substituted-2-hydroxybenzoic acid to the design of IN chelation class inhibitor. This idea was also inspired by the recent discovery of the dihydroxybenzoylnaphthylhydrazone (DHBNH, Fig. 1) as a novel HIV-1 reverse transcriptase inhibitor that exerts its inhibitory effect through a metal-chelating mechanism.15,16 We propose that the neighboring carbonyl and two free hydroxyl groups on the 2,3-dihydroxybenzoyl amide might sufficiently bind to the two metal cofactors in the IN active site, and the substituent on the amide portion could provide the interactions with the hydrophobic pocket of the enzyme. Herein, we report the synthesis, evaluation and SAR study of these salicylic acid-based IN inhibitors and establish their binding mode. We further tested their ability to inhibit the interaction between IN and LEDGF/p75 with the idea that some of these compounds may act as an allosteric inhibitors of IN.17
Figure 2.

Scaffold hopping approach for design of new IN inhibitors by combining the privileged fragments of salicylate and catechol
Chemistry
The 3/4/5/6-substituted salicylic acid derivatives were synthesized readily from the corresponding precursors. As outlined in Scheme 1, the direct O-alkylation of the 3/4/5-hydroxy-2-hydroxybenzoic acids by the corresponding bromide in the NaH/DMF solution afforded the desired products (3a–g). The intramolecular hydrogen bonding between the 1-carboxy group and 2-hydroxy group secured the selective alkylation on the 3/4/5-hydroxy site. Similarly, the 6-subtituted analogs (3h–j) were prepared from methyl 2,6-dihydroxybenzoate by reacting with different bromide in the presence of NaI and K2CO3 followed by the hydrolysis in 1N NaOH/THF solution. However, because the 1-carboxy group can form intramolecular hydrogen bonds with neighboring 2- and 6-hydroxyl groups which lowered the reactivity, the methyl 2,6-dihydroxybenzoate was employed as the starting material for the synthesis of 6-alkyloxy analogs (3h–j). The resulting 3/4/5/6-substituted salicylic acids were conveniently converted into the corresponding hydroxamic acids (4a–d) by reacting with hydroxylamine in the presence of activating agent and base.
Scheme 1.

Preparation of the 3/4/5/6-substituted salicylic acid derivatives. Reagents and conditions: (i) NaH, DMF, bromide, 7h, rt; (ii) ClCO2iBu, NMM, NH2OHHCl, THF, 0°C, 2h; (iii) K2CO3, NaI, bromide, acetonitrile, DMF, 14h, rt; (iv) THF, 1N NaOH, 80°C, 24h.
For the 2,3-dihydroxybenzamide series (5a, 5c–m), the synthesis was generally achieved by the condensation of 2,3-dihydroxybenzoic acid with the corresponding amine, as depicted in Scheme 2. And the preparation of 1-benzyloxy derivative (5b) needed one more alkylation prior to the condensation.
Scheme 2.

Synthesis of 2,3-dihydroxybenzamide derivatives. R is as indicated in Table 2. Reagents andcondition: (i) EDCI, DIPEA, HOBt, THF or DCM, various amine, 6~8h, rt; (ii) BnBr, NaH, DMF.
In this series, further structural modifications were conducted on the phenyl ring by incorporating an acidic functionality into the benzylamino portion or the benzoyl moiety. These analogs required a separate preparation of the coupling components. For example, the substituted benzylamine was first synthesized as the building block for the N-(2,4-substitutedphenyl methyl) 2,3-dihydroxybenzamide derivatives (5n–o). As indicated in Scheme 3, the ring-opening of isochroman-3-one by methyl amine produced the 2-(2-(hydroxymethyl)phenyl)-N-methylacetamide 9, which was treated with methanesulfonyl chloride to yield the chloride 10 instead of the expected methanesulfonate. The conversion of the chloride into the amine 12n was achieved by the nucleophilic attack of azide followed by reduction. The resulting 2-(2-(aminomethyl)phenyl)-N-methylacetamide was coupled with 2,3-dihydroxybenzoic acid afforded the desired product 5n. Another methylcarbamoyl-substituted benzylamine 12o was synthesized from 5-fluoro-2-methylbenzoic acid via the sequential bromination, azido replacement and the reduction. The condensation of the 2,3-dihydroxybenzoic acid with the resultant 2-(aminomethyl)-5-fluoro-N-methylbenzamide generated the final product N-(4-fluoro-2-(methylcarbamoyl)benzyl)-2,3-dihydroxybenzamide 5o.
Scheme 3.

Synthesis of N-(2,4-substitutedphenyl methyl) 2,3-dihydroxybenzamide derivatives. Reagents and conditions: i) NH2Me, THF; ii) MsCl, TEA, DCM; iii) NaN3, DMF, 80°C; iv) Ph3P, THF; v) 2,3-dihydroxy-benzoic acid, EDCI, HOBt, TEA or NMM, DCM; vi) NH2Me, EDCI, HOBt, DCM; vii) NBS, AIBN, CCl4, reflux.
For the synthesis of 5-substituted 2,3-dihydroxybenzamide derivatives, the 5-sulfamoyl substituted 2,3-dihydroxybenzoic acid was first synthesized as the component for the condensation reaction. As shown in Scheme 4, the 2,3-dimethoxybenzoic acid was treated with chlorosulfonic acid to provide the sulfonylated compound 15. The sulfonamidation of 15 with piperidine, methylamine, or tertbutyl amine afforded compounds 16p–r. Then the condensation of 16p–r with 4-fluorobenzyl amine in the presence of HOBt and EDCI provided compounds 17p–r. The sequential de-methylation of the 2,3-dimethoxyl groups by boron tribromide afforded the desired products 5p–r. More derivatives of 2,3-dihydroxy-5-(piperidin-1-ylsulfonyl)benzamide with variation on the amide portion (5s–v, 6c) were synthesized in a similar fashion, from the common intermediate of 16p, which underwent the demethylation before the coupling with various amines. The demethylation of 16p by boron tribromide yielded the mixture of 3-demethylated and 2,3-demethylated products, which were separated by column chromatography.
Scheme 4.

Synthesis of 5-sulfoxamide substituted 2,3-dihydroxybenzamide derivatives. Reagents and conditions: (i) chlorosulfonic acid, 15h, rt; (ii) alkyl amine, Et3N, CH2Cl2, reflux, 22h; (iii) EDCI, DIPEA, HOBt, THF, 4-fluorobenzylamine, 6.5h, rt.; (iv) 15eq BBr3, CH2Cl2, −20°C, 10h or LiCl, DMF, 110°C, 8h; (v) 15 eq BBr3, CH2Cl2, −20°C, 10h; (vi) EDCI, DIPEA, HOBt, DMF, amine, 6.5h, rt.
The amino or cyano group substituted at 5-position of the 2,3-dihydroxybenzamide was introduced via nitration on the 2,3-dimethoxybenzoic acid. As shown in Scheme 5, after the condensation with 4-fluorobenzyl amine, the 5-nitro- 2,3-dimethoxybenzoic acid 20 was reduced into the corresponding 5-amino derivative 21 by stannous chloride in hydrochloric acid. Then the 5-amino derivatives were further converted into amide (5w–x) or cyano analogs (5y) by amidation and diazo-reaction followed by nucleophilic replacement, respectively.
Scheme 5.

Synthesis of 5-substituted 2,3-dihydroxybenzamide derivatives. Reagents and conditions: (i) conc.H2SO4, HNO3, 0°C; (ii) 4-fluorobenzylamine, EDCI, DIPEA, HOBt, DCM,, 6.5h, rt; (iii) SnCl2, HCl, THF, rt; (iv) MsCl for 22w (or AcCl for 22x), TEA, DCM, rt; (v) LiCl, DMF, 110°C, 8h then 15 eq BBr3, CH2Cl2, −20°C, 10h (for 5w–x) or AlBr3, MeCN, 0°C, 8h (for 5y); (vi) NaNO2, EtOH, H2O, −15°C, then CuCN, H2O, 50°C,1h, rt overnight.
The substitution at 4-position was examined with alkylamino group. The synthesis of 4-alkyamino substituted 2,3-dihydroxybenzamide derivatives (6a–b) was achieved via a formylation on the phenyl ring followed by reductive amination, as shown in Scheme 6.
Scheme 6.
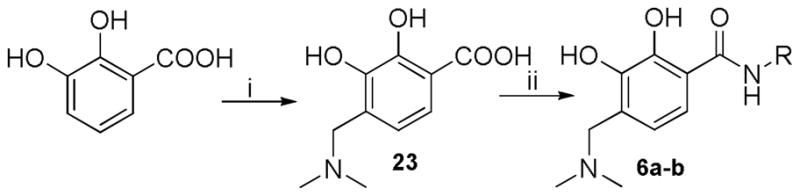
Synthesis of 4-alkylamino substituted 2,3-dihydroxybenzamide derivatives. Reagents and conditions: (i) 40% HCHO (aq), dimethylamine hydrochloride, Et3N, EtOH; (ii) EDCI, DIPEA, HOBt, DMF, amine, 6.5h, rt.
Results and Discussion
Identification of the 2,3-dihydroxybenzamide as the active scaffold of HIV-1 IN inhibitors
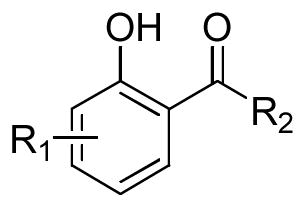
As depicted in Table 1, the 3/4/5/6-alkyloxy substituted salicylic acid derivatives (3a–j) generally displayed weak inhibition against IN regardless of the substituent structure and position. Even the incorporation of the chelation-advancing hydroxylamino group into the 3/4/5/6-alkyloxy-salicylic acid scaffold just slightly improved the binding (4a–d), although hydroxamic acids were reported to facilitate the binding of two Mg2+ ions by the azaindole-based IN inhibitors,18 which implied the ineffectiveness of the alkyloxy substituted salicylic acid scaffold as IN inhibitor. However, the evolved 2,3-dihydroxybenzamide (5a–b) displayed moderate inhibition against strand transfer reaction. The N-(4-fluorobenzyl)-2,3-dihydroxybenzamide 5a exhibited IC50 values of 100μM and 35μM in inhibiting 3′-processing and strand transfer, respectively. The elimination of the phenolic hydroxy at the 3-position by conversion to a benzyl ether reduced the inhibitory potency (5b, IC50 = 111 μM for strand transfer) by 2.5-fold relative to the 3-hydroxy analog 5a, which might result from the reduction of the metal-binding region. In addition, the 2,3-dihydroxybenzamide derivatives (5a and 5b) were not cytotoxic in H630 cells at the concentration of up to 40 μM.
Table 1.
Scaffold screening: inhibition of HIV-1 integrase catalytic activities and the cytotoxicityby salicylic acid derivatives with various substitution pattern.
 | |||||
|---|---|---|---|---|---|
| Compd | R1 | R2 | IC50 (μM) | CC50 (μM) H630 cells | |
| 3′-Processing | Strand transfer | ||||
| 3a |
|
OH | > 100 | > 100 | > 40 |
| 3b |
|
OH | > 100 | > 100 | > 40 |
| 3c |
|
OH | > 100 | 140±46 | > 40 |
| 3d |
|
OH | > 100 | > 100 | > 40 |
| 3e |
|
OH | > 100 | > 100 | > 40 |
| 3f |
|
OH | > 100 | > 100 | > 40 |
| 3g |
|
OH | > 100 | > 100 | > 40 |
| 3h |
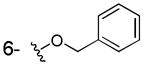
|
OH | > 100 | > 100 | > 40 |
| 3i |
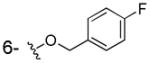
|
OH | > 100 | > 100 | > 40 |
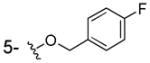
| 3j |
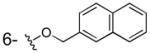
|
OH | > 100 | 186±15 | > 40 |
| 4a |
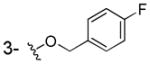
|
NHOH | > 100 | > 100 | NDa |
| 4b |
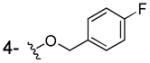
|
NHOH | > 100 | 100 | ND |
| 4c |
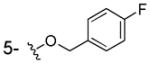
|
NHOH | > 100 | 85±7 | ND |
| 4d |
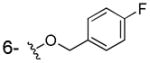
|
NHOH | > 100 | 97±6 | ND |
| 5a | 3-OH |
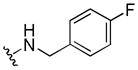
|
100 | 35±25 | > 40 |
| 5b | 3-OBn |
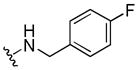
|
> 333 | 111 | > 40 |
ND: not determined.
Consequently, the 2,3-dihydroxybenzamide was chosen as the template for further structural modification to increase potency. The SAR study on the 2,3-dihydroxybenzamide-based IN inhibitors included structural variation on the left side catechol group and the right side benzamide moiety. The substitution on the phenyl ring of the catechol core was investigated, and the structural variation on the “right side” carboxamide group was explored with heterocycle (aliphatic and aromatic) and substituted phenyl ring separately. The activity data is summarized in Table 2 and rationalized by molecular modeling.
Table 2.
Inhibition of HIV-1 integrase catalytic activities, integrase-LEDGF/p75 interaction, and cytotoxicity of the 2,3-dihydroxybenzamide series with variations on the phenyl ring and the carboxamide moiety.
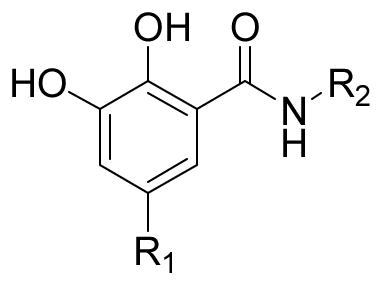 | ||||||
|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | IC50 (μM) | IC50 (μM) LEDGF/p75-IN | CC50 (μM) H630 cells | |
| 3′-Processing | Strand transfer | |||||
| 5a | H |
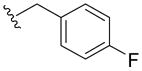
|
>100 | 35 ± 25 | > 100 | > 40 |
| 5c | H |
|
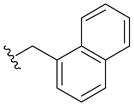
>100 | 26± 4 | 34± 9 | 21 |
| 5d | H |
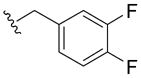
|
>100 | 23 ± 7 | 56±8 | > 40 |
| 5e | H |
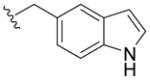
|
>100 | 24 ± 2 | 45± 9 | > 40 |
| 5f | H |
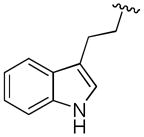
|
100 | 100 | 63± 20 | 15 |
| 5g | H |
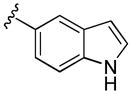
|
>100 | 58± 12 | 88± 19 | > 40 |
| 5h | H |
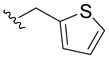
|
83±21 | 12±4 | 95± 11 | > 40 |
| 5i | H |
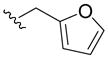
|
90±14 | 15±5 | 89±8 | > 40 |
| 5j | H |
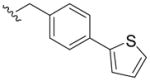
|
> 100 | 21± 5 | > 100 | 15 |
| 5k | H |
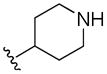
|
> 100 | > 100 | > 100 | > 10 |
| 5l | H |
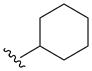
|
> 100 | > 100 | > 100 | > 40 |
| 5m | H |
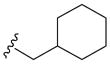
|
> 100 | 18±9 | > 100 | 21 |
| 5n | H |
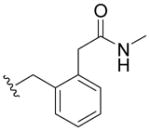
|
>100 | 56 ± 19 | > 100 | > 40 |
| 5o | H |
|
> 100 | 100 | > 100 | > 40 |
| 5p |
|
|
200 | 5 | ND | 13 |
| 5q | -SO2NHMe |
|
>100 | 44 ± 7 | > 100 | > 40 |
| 5r | -SO2NHtBu |
|
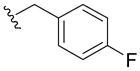
70 | 8±5 | 100 | > 10 |
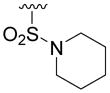
| 5s |
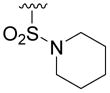
|
|
> 100 | 13 | > 100 | 33 |
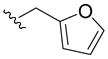
| 5t |
|
|
89 | 11 | > 100 | 31 |
| 5u |
|
|
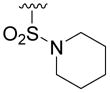
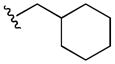
53±4 | 19±3 | 8± 1 | >40 |
| 5v |
|
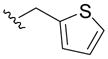
|
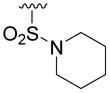
> 100 | 44±19 | > 50 | >40 |
| 5w |
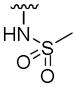
|
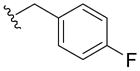
|
> 100 | 34±10 | 100 | |
| 5x |
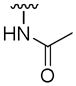
|
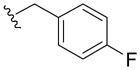
|
55±7 | 14±4 | > 100 | < 10 |
| 5y | CN |
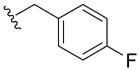
|
75±7 | 22±8 | ND | > 10 |
| 6a |
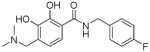
|
> 100 | 20±4 | > 50 | > 10 | |
| 6b |
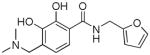
|
> 100 | 39±8 | > 50 | 7 | |
| 6c |
|
>100 | > 100 | 13±4 | > 25 | |
| 6d |
|
> 100 | > 100 | 51±4 | > 25 | |
SAR study with respect to the structural variation on the carboxamide portion and phenyl ring of the 2,3-dihydroxybenzamide scaffold
We prepared compounds with modification on the “right side” of the core structure (Table 2, 5a–o). A range of aryl- or alkyl-substituted amines were investigated, whereby the heteroaromatic amine and the amide collectively caused an increase in the 3′-processing inhibitory activity (5h–i, 5r, 5t–u, 5x–y) compared to the parent compound 5a. The lipophilic substituent such as naphthalenyl and 3,4-difluorophenyl groups were beneficial for the strand transfer inhibition (5c, IC50 = 26 μM; 5d, IC50 = 23 μM). In particular, the thiophenyl, furanyl and (thiophen-2-yl)phenyl substitutions markedly enhanced the potency of strand transfer inhibition (5h, IC50 = 12 μM; 5i, IC50 = 15 μM; 5j, IC50 = 21 μM). But the effect of the indolyl substitution varied according to the linker length and substituted position (5e–g), in which the best substituent was (1H-indol-5-yl) methyl group (5e, IC50 = 24 μM). Conversely, the N-methyl carbamoyl substitution at the 2-position of the 4-fluorophenyl ring (5o) resulted in a loss of IN inhibitory potency. Interestingly, the replacement of the phenylmethyl functionality with the aliphatic counterpart, cyclohexylmethyl group improved the activity (5m, IC50 = 18 μM vs. 5a, IC50 = 35 μM), while the direct cyclohexyl or 4-piperidinyl group was deemed unfavorable at this carboxamide position (5k and 5l, IC50 > 100 μM). This result indicated the importance of the hydrophobic substituent in the inhibition of the catalytic activities of IN.
As for the substitution effect of the 2,3-dihydroxyphenyl group on the potency, the introduction of a piperidin-1-ylsulfonyl group at 5-position of the phenyl ring led to a 7-fold improvement in the strand transfer inhibitory potency (5p, IC50 = 5 μM) relative to the parent compound 5a. However, a more polar substituent such as N-methyl or butyl sulfonyl group at the 5-position of 2,3-dihydroxyphenyl ring caused a decrease in the inhibitory activity (5q, IC50 = 44 μM; 5r, IC50 = 8 μM), in accordance with the reduction of the hydrophobicity. More electron-withdrawing groups were examined with respect to the effect on the potency. The methylsulfonamide, acetylamide or cyano group at 5-postion produced moderate potent inhibitors against the strand transfer (5w, IC50 = 34 μM; 5x, IC50 = 14 μM; 5y, IC50 = 22 μM). Further structural modification with the alkylamino group substituted at 4-position displayed slight influence on the potency (6a, IC50 = 20 μM; 6b, IC50 = 39 μM). As a comparison, the replacement of the 3-hydroxyl group with a methoxy or an amino group suffered a severe loss of potency (6c, 6d), suggesting that the 3-hydroxy group was involved in the two metal-binding interaction.
With the privileged structures disclosed above, we designed new analogs with simultaneous substitution on both sides (5s–v). However, the combination of the favorable substitution on both sides didn’t result in a synergistic effect on the inhibitory activity as we expected. The resulting active compounds exhibited low micromolar inhibitory potency against the strand transfer reaction (5s, IC50 = 13μM; 5t, IC50 = 11μM; 5u, IC50 = 19μM; 5v, IC50 = 44μM).
These salicylate and catechol-merged IN inhibitors were hypothesized to function by the chelation of the divalent metal ions in the active site of IN. According to the SAR study, the 2,3-dihydroxybenzamide core might serve as the metal-binding motif, and the substituents on the phenyl ring possibly afforded an additional interaction with the key residues in the binding pocket. The aryl or aliphatic cyclic substituent on the right side carboxamide portion might be responsible for the interaction with the hydrophobic surface of the enzyme. These observations can be reasonably rationalized from the docking studies by molecular modeling.
The 2,3-dihydroxybenzamide derivatives inhibit the interaction of HIV-1 IN and LEDGF/p75 cofactor
Lens epithelium-derived growth factor (LEDGF/p75) is a cellular cofactor of IN that promotes viral integration by tethering the pre-integration complex to the chromatin.19,20 Instead of targeting the catalytic activity of IN, the disruption of the integrase-LEDGF/p75 interaction, and the consequent inhibition of HIV replication, represents a new frontier for the design and development of novel anti-HIV agents for AIDS therapy.[21] However, very few small molecule inhibitors of the IN-LEDGF/p75 interaction have been reported to date.17,22–25
Considering that the specific protein-protein interaction between IN catalytic core domain (CCD) and the LEDGF/p75 IN-binding domain (IBD) was characterized by IBD residues Ile365, Asp366, Phe406 and Val408,26 we were interested in testing these salicylate and catechol-merged compounds in the inhibition of IN-LEDGF/p75 interaction, since our chemical frame contained both the aromatic moiety and the carboxylic functionality. Interestingly, most of our 2,3-dihydroxybenzamide derivatives were able to inhibit the IN-LEDGF/p75 interaction at micromolar concentrations in the AlphaScreen assay. As shown in Table 2, the active IN strand transfer inhibitors displayed consistent potency in inhibiting the interaction of IN-LEDGF/p75, among which the highest potency was exhibited by 5u (IC50 = 8μM) bearing the privileged structures on both sides. The preliminary SAR study indicated that the substituent attached on the carboxamide portion played an important role in the disruption of IN-LEDGF/p75 interaction. In this position, the lipophilic structure as well as the heteroaromatics was favored (Table 2, 5c–i). However, the catechol structure was not essential for inhibiting IN-LEDGF/p75 interaction, exemplified by 6c and 6d which were almost inactive against the strand transfer but moderately active in preventing IN-LEDGF/p75 interaction (6c, IC50 = 13 μM; 6d, IC50 = 51 μM). Furthermore, the piperidin-1-ylsulfonyl substituent at 5-position of 2,3-dihydroxyphenyl ring might potentiate the interaction with the LEDGF-binding site of IN when properly oriented (5u, IC50 = 8μM; 6c, IC50 = 13 μM), which was depicted in the following molecular modeling.
Our study provides new important chemical frames to disrupt the protein-protein interaction between IN and its cellular cofactor LEDGF/p75, which could advance the design and discovery of anti-HIV agents possessing a novel mechanism of action.
Molecular Modeling Studies
We selected 5p, 5t and 5u as active and 6c as inactive representative structures to dock against an HIV-1 IN model which has been built from a crystal structure of prototype foamy virus (PFV) intasome.27 Compounds were then docked into the binding site using Standard Precision method of Glide from Schrodinger, Inc.28,29 The binding modes of compounds 5p, 5t, 5u and 6c in the IN active site are shown in Figures 3 and 4. The docking modes in PVF IN of all active compounds are similar to that of raltegravir. Both hydroxyl groups of 5p form metal chelating interactions with the two Mg2+ ions along with the amino acid residues Asp64, Asp116 and Glu152. The fluoro-phenyl moiety of 5p packed into the tight binding site formed by viral DNA bases DA17, DC16 and amino acid residues Pro145 and Gln146. Phenyl ring forms stacking interaction with DC16. Thus, the compound displaces reactive DNA 3′OH group from the active site and causes the deactivation of the intasome. On the other hand, the piperidine moiety of 5p occupies the hydrophobic pocket formed by Pro142, Tyr143 and Asn117. Compounds 5t and 5u exhibited a binding mode that is similar to 5p, while the inactive compound 6c, has the disadvantage of possessing a single hydroxyl group to form metal chelating interaction. In addition, its piperidine and furanyl moieties are not properly positioned into hydrophobic pockets. As a result, 6c forms weak interactions with the active site residues thereby potentially contributing to its weak strand transfer activity.
Figure 3.


The proposed binding modes of 5p, 5t, 5u and 6c in the modeled HIV-1 intasome. The close contact residues are shown in surface model. Inhibitors are shown as pink sticks (colored by atom types) and the two Mg+2 are depicted as green spheres.
Figure 4.


A close-up view of the proposed binding modes of 5p, 5t, 5u and 6c in the modeled HIV-1 IN active site. The close interacting residues are depicted as cyan sticks. 5p, 5t, 5u and 6c are shown in pink sticks (colored by atom types) while green sphere represent the Mg2+ ions.
Active LEDGF/p75 inhibitor, 5u, was also docked into the LEDGF/p75 binding site of the HIV IN crystal structure 3LPU. Figure 5 shows the predictive binding mode of 5u into the LEDGF/p75 binding site of HIV IN. The inhibitor develops strong interactions with IN residues in the LEDGF/p75 binding site. One hydroxyl group from central phenyl ring forms hydrogen bond with the backbone carbonyl oxygen of Thr125. Cyclohexyl moiety formulates hydrophobic interaction with Tyr99 and Gln95, while piperidine moiety creates hydrophobic interactions with Trp132 and Leu102.
Figure 5.
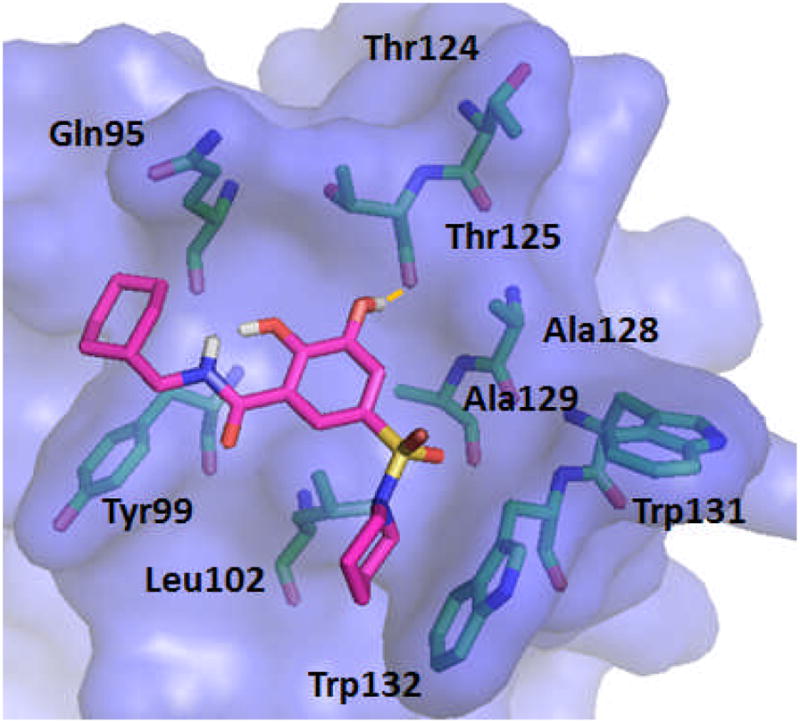
The proposed binding mode of 5u in to the LEDGF binding site of HIV IN
Conclusions
In this study, through the optimal combination of the active structures of salicylic acid and catechol, we successfully identified a novel class of salicylic acid-based IN inhibitors targeting both the catalytic domain of IN and the IN-LEDGF/p75 interaction. The hit compound, 2,3-dihydroxybenzamide, showed low micromolar IN inhibitory activity without exerting significant cytotoxicity. These inhibitors are predicted to function by chelating the metal cofactor in the active site of IN. Moreover, the substituents on the phenyl ring and the carboxamide portion are responsible for the binding with the key residues in the drug-binding pocket. Molecular modeling revealed the binding mode of the active and inactive compounds, confirming the chelation mechanism. Our study also supports a potential for allosteric inhibition by some of these compounds due to their similar potency in inhibiting the interaction between IN and LEDGF/p75.
Experimental Section
General Synthetic Methods
The 1H NMR spectra were recorded on a Varian 300-MHz spectrometer. The data are reported in parts per million relative to TMS and referenced to the solvent. Elemental analyses were obtained using a Vario EL spectrometer. Melting points (uncorrected) were determined on a Buchi-510 capillary apparatus. The MS spectra were obtained on an APEXIII 7.0 T FTMS mass spectrometer. The flash column chromatography was performed on silica gel H (10-40μm). Anhydrous solvents were obtained by standard procedure. For the target compounds the purity was determined by analytical HPLC using a Dionex P680 equipped with UV detector. HPLC conditions: Global chromatography column (250 mm×4.6 mm); solvent gradient, A, 0.05% TFA in water; B, 0.05% TFA in 90% acetonitrile in water (solvent system 1); or A, 0.05% TFA in water; C, methanol (solvent system 2), with gradient indicated below; flow rate, 1.0 mL/min; UV detector, 254 nm.
Methods
Method A: 10% B for 2 min, 10% B to 90% B in 13 min, 90% B for 5 min, 90% B to 10% B in 5 min.
Method B: 10% B for 2 min, 10% B to 90% B in 8 min, 90% B for 10 min, 90% B to 10% B in 5 min.
Method C: 10% B for 2 min, 10% B to 90% B in 13 min, 90% B for 5 min, 90% B to 10% B in 3 min.
Method D: 10% B for 2 min, 10% B to 50% B in 8 min, 50% B to 90% B in 5 min, 90% B to 10% B in 3 min.
Method E: 10% B for 2 min, 10% B to 20% B in 3 min, 20% B to 90% B in 5 min, 90% B for 5 min, 90% B to 10% B in 5 min.
Method F: 10% C for 2 min, 10% C to 90% C in 8 min, 90% C for 10 min, 90% C to 10% C in 5 min.
Method G: 10% C for 2 min, 10% to 95% C in 3 min, 95% C for 15 min, 95% to 10% C in 5 min, 10% C for 3 min.
Method H: 10% C for 2 min, 10% C to 20% C in 15 min, 10% C to 90% C in 5 min, 90% C to 10% C in 6 min.
3-(Benzyloxy)-2-hydroxybenzoic acid (3a)
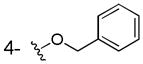
To the solution 60% NaH (100mg, 2.2mmol) in DMF (5mL) was added 2,3-dihydroxy benzoic acid at room temperature and stirred for 1h, then benzyl bromide (0.119mL, 1mmol) was added to the above solution and stirred for 7h. Then the reaction was quenched by adding H2O (20 mL) and the reaction mixture was acidified with 1N HCl to pH = 1–3. The solution was extracted with EtOAc (20 mL), washed by brine (3 × 20 mL) and dried over anhydrous Na2SO4. The solvent was removed under vacuum. The residue was purified by flash chromatography using petroleum ether/ethyl acetate (1:1) to give compound 3a as a white solid (54 mg, 22% yield). Mp: 151–153°C; 1H NMR (300 MHz, CDCl3): δ 7.55-7.31 (m, 6H), 7.11 (m, 1H), 6.81 (m, 1H), 5.19(s, 2H). 13C NMR (100MHz, CD3OD): δ 174.3, 149.0, 139.0, 130.0, 129.5, 129.2, 124.1, 121.6, 120.0, 72.8. EI-MS m/z: 244 (M)+. Purity: system 1, 97.6% (method A, tR = 15.63 min); system 2, 96.8% (method F, tR = 14.61 min).
3-(4-Fluorobenzyloxy)-2-hydroxybenzoic acid (3b)
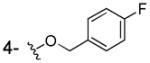
3b was prepared as a white solid according to the same procedure described for 3a, (42 mg, 28% yield). 1H NMR (300MHz, d6-DMSO): δ 7.49 (dd, J = 5.7 Hz, 8.7Hz 2H), 7.36 (dd, J = 1.5 Hz, 8.1 Hz, 1H), 7.22 (m, 3H), 6.81 (t, J = 8.1 Hz, 1H), 5.09 (s, 2H). 13C NMR (100MHz, CD3OD): δ 174.3, 164.4 (d, 1JCF=242.6Hz), 154.7, 148.9, 135.0, 131.4 (d, 3JCF=8.2Hz), 124.3, 121.7, 120.0, 116.6 (d, 2JCF=21.5Hz), 115.0, 72.2. EI-MS m/z: 262 (M)+. Purity: system 1, 99.2% (method A, tR = 15.71 min); system 2, 98.1% (method F, tR = 14.63 min).
3-((Naphthalen-2-yl)methoxy)-2-hydroxybenzoic acid (3c)
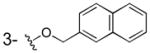
3c was prepared as a white solid according to the same procedure described for 3a (58mg, 26% yield). 1H NMR (300MHz, d6-DMSO): δ 7.96-7.89 (m, 4H), 7.58 (m, 1H), 7.50 (m, 2H), 7.36 (m, 1H), 7.26 (m, 1H), 6.78 (m, 1H), 5.28 (s, 2H). 13C NMR (100MHz, CD3OD): δ 174.3, 165.3, 154.6, 136.5, 135.2, 135.1, 129.7, 129.5, 129.2, 128.0, 127.7, 127.6, 127.0, 124.2, 121.8, 120.0, 115.0, 73.0. EI-MS m/z: 294 (M)+. Purity: system 1, 96.2% (method A, tR = 17.22 min); system 2, 97.8% (method F, tR = 16.44 min).
4-(Benzyloxy)-2-hydroxybenzoic acid (3d)
3d was prepared as a white solid according to the same procedure described for 3a, (62mg, 39% yield). Mp: 173–175°C; 1H NMR (300MHz, CDCl3): δ 7.81 (m, 1H), 7.37 (m, 5H), 6.55 (m, 2H), 5.10 (s, 2H). 13C NMR (100MHz, CD3OD): δ 164.5, 137.4, 132.5, 128.9, 128.4, 128.1, 107.2, 101.8, 70.1. EI-MS m/z: 244 (M)+. Purity: system 1, 95.4% (method A, tR = 16.26 min); system 2, 99.3% (method F, tR = 15.43 min).
4-(4-Fluorobenzyloxy)-2-hydroxybenzoic acid (3e)
3e was prepared as a white solid according to the same procedure described for 3a, (72mg, 53% yield). 1H NMR (300MHz, d6-DMSO): δ 7.68 (m, 1H), 7.48 (m, 2H), 7.21 (m, 2H), 6.54 (m, 2H), 5.11 (s, 2H). 13C NMR (100MHz, CD3OD): δ 173.9, 166.5 (d, 1JCF=244.1Hz), 165.8, 163.3, 134.5, 133.5, 131.3 (d, 3JCF=8.2Hz), 116.8 (d, 2JCF=21.6Hz), 109.0, 107.6, 103.1, 70.9. EI-MS m/z: 262 (M)+. Purity: system 1, 95.1% (method A, tR = 16.33 min); system 2, 94.7% (method F, tR = 15.34 min).
5-(Benzyloxy)-2-hydroxybenzoic acid (3f)
3f was prepared as a white solid according to the same procedure described for 3a, (66mg, 46% yield). Mp: 162–164°C; 1H NMR (300MHz, CDCl3): δ 7.45-7.33 (m, 6H), 7.21 (m, 1H), 6.95 (m, 1H), 5.04 (s, 2H). 13C NMR (100MHz, CD3OD): δ 158.2, 152.9, 139.1, 130.0, 129.4, 129.2, 125.9, 119.5, 115.9, 72.3. EI-MS m/z: 244 (M)+. Purity: system 1, 95.1% (method A, tR = 16.01 min); system 2, 95.1% (method F, tR = 14.60 min).
5-(4-Fluorobenzyloxy)-2-hydroxybenzoic acid (3g)
3g was prepared as a white solid according to the same procedure described for 3a (49mg, 40% yield). 1H NMR (300MHz, CDCl3): δ 10.08 (brs, 1H), 7.41 (m, 3H), 7.20 (m, 1H), 7.08 (m, 2H), 6.96 (m, 1H), 5.00 (s, 2H). 13C NMR (100MHz, CD3OD): δ 173.6, 164.4 (d, 1JCF=243.3Hz), 158.3, 152.8, 135.2, 131.2 (d, 3JCF=8.2Hz), 126.0, 119.6, 116.7 (d, 2JCF=21.5Hz), 115.9, 114.0, 71.6. EI-MS m/z: 262 (M)+. Purity: system 1, 94.7% (method A, tR = 16.10 min); system 2, 95.4% (method F, tR = 15.16 min).
Methyl 2-(benzyloxy)-6-hydroxybenzoate (7h)
To a suspension of methyl 2,6-dihydroxybenzoate (84mg, 0.5mmol), K2CO3(83mg, 0.6mmol), and NaI (22mg, 0.15mmol) in CH3CN (5mL) and DMF (5mL) was added benzyl bromide, the mixture was stirred for 14h at room temperature. Evaporated to remove most of the CH3CN, then the mixture was acidified with 1N HCl. H2O (20 mL) and EtOAc (20mL) were added. The organic layer was washed with brine (3×20mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was chromatographied on silica gel (PE/EtOAc=20:1) to provide methyl 2-(benzyloxy)-6-hydroxybenzoate 7h (51mg, 40% yield) as white solid, with starting material being recovered (44 mg, 52%). 1H NMR (300MHz, CDCl3): δ 11.54 (s, 1H), 7.50-7.30 (m, 6H), 6.64 (m, 1H), 6.48 (m, 1H), 5.13 (s, 2H), 3.96 (s, 3H).
2-(Benzyloxy)-6-hydroxybenzoic acid (3h)
The reaction mixture of 2-(benzyloxy)-6-hydroxybenzoate 7h (39mg, 0.15mmol) in 3mL THF and 3mL 1N NaOH was heated to 80°C for 24h, then the solution was acidified with 1 N HCl to pH 1–2 and extracted with ethyl acetate (2*15mL). Washed with brine and dried over Na2SO4, the solution was evaporated under vacuum to give 2-(benzyloxy)-6-hydroxybenzoic acid 3h (34mg, 92% yield) as a white solid. Mp: 123–126°C; 1H NMR (300MHz, CDCl3): δ 12.10 (s, 1H), 11.38 (brs, 1H), 7.41 (m, 6H), 6.75 (m, 1H), 6.59 (m, 1H), 5.46(s, 2H). 13C NMR (100MHz, CD3OD): δ 173.5, 164.6, 160.6, 138.0, 136.5, 130.2, 129.8, 129.2, 112.1, 105.3, 73.0. EI-MS m/z: 244 (M)+. Purity: system 1, 97.5% (method A, tR = 16.57 min); system 2, 95.5% (method F, tR = 14.29 min).
Methyl 2-(4-fluorobenzyloxy)-6-hydroxybenzoate (7i)
7i was prepared as a white solid according to the same procedure described for 7h, (42mg, 22% yield). 1H NMR (300MHz, CDCl3): δ 11.49 (s, 1H), 7.45-7.35 (m, 3H), 7.09 (m, 2H), 6.63 (m, 1H), 6.47 (m, 1H), 5.08(s, 2H), 3.93(s, 3H).
2-(4-Fluorobenzyloxy)-6-hydroxybenzoic acid (3i)
3i was prepared as a white solid according to the same procedure described for 3h (55mg, 90% yield). 1H NMR (300MHz, CDCl3): δ 12.18 (s, 1H), 11.30 (brs, 1H), 7.45-7.38 (m, 3H), 7.17-7.11 (m, 2H), 6.75 (d, J = 8.4 Hz, 1H), 6.56 (d, 1H, J = 8.4 Hz), 5.22(s, 2H). 13C NMR (100MHz, CD3OD): δ173.5, 165.8 (d, 1JCF=243.3Hz), 164.4, 160.6, 136.3, 134.3, 131.2 (d, 3JCF=8.9Hz), 116.8 (d, 2JCF=21.6Hz), 112.0, 105.4, 72.2. EI-MS m/z: 262 (M)+. Purity: system 1, 99.2% (method A, tR = 16.48 min); system 2, 99.6% (method F, tR = 14.27 min).
Methyl 2-((naphthalen-3-yl)methoxy)-6-hydroxybenzoate (7j)
7j was prepared as a white solid according to the same procedure described for 7h (39mg, 20% yield). 1H NMR (300MHz, CDCl3): δ 11.55 (s, 1H), 7.97 (s, 1H), 7.90-7.84 (m, 3H), 7.59-7.49 (m, 3H), 7.34 (m, 1H), 6.64 (d, J = 8.4 Hz, 1H), 6.54 (d, J = 8.4 Hz, 1H), 5.28 (s, 2H), 3.99 (s, 2H).
2-((Naphthalen-2-yl)methoxy)-6-hydroxybenzoic acid (3j)
3j was prepared as a white solid according to the same procedure described for 3h (47mg, 30% yield). 1H NMR (300MHz, CDCl3): δ 12.21 (s, 1H), 7.95-7.87 (m, 4H), 7.55 (m, 3H), 7.41 (m, 1H), 6.75 (d, J=8.4 Hz, 1H), 6.63 (d, J =8.4 Hz, 1H), 5.43 (s, 2H). 13C NMR (100MHz, CD3OD): δ 173.6, 164.5, 160.7, 137.5, 135.6, 135.3, 130.0, 129.9, 129.7, 129.5, 129.3, 129.1, 128.3, 127.9, 126.8, 126.6, 112.0, 73.4. EI-MS m/z: 294 (M)+. Purity: system 1, 98.4% (method A, tR = 18.13 min); system 2, 98.9% (method F, tR = 15.89 min).
3-(4-fluorobenzyloxy)-N,2-dihydroxybenzamide (4a)
To a solution of 3b (1.31g, 5.0 mmol) in dry THF (25.0 ml) was added NMM (0.58 ml, 5.25 mmol) at −15°C, stirred for 15 min, then added ClCO2iBu (0.69 ml, 5.25 mmol) during 15 min, and the mixture was stirred for 1h at −15°C. Then hydroxylamine hydrochloride (0.52 g, 7.5mmol) which was dissolved in H2O (10 ml) was added to above system during 30 min at 0°C. The reaction mixture was stirred for 1.5h, then poured into ice-water, washed with ether 3 times. The organic phases were combined and washed with sat. NaHCO3 and brine, dried with anhydrous Na2SO4, filtered, and concentrated under vacuum to give 4a as a white solid. 1HNMR (CD3OD, 300MHz) δ: 3.95 (s, 1H), 3.97 (s, 1H), 5.09 (s, 2H), 7.08 (t, J =8.7Hz, 2H), 7.22–7.30 (m, 2H), 7.41–7.49 (m, 3H).
4-(4-fluorobenzyloxy)-N,2-dihydroxybenzamide (4b)
Compound 4b was prepared from 3d according to the same procedure as 4a. 4b was purified as a white solid. 1HNMR (CDCl3, 300MHz) δ: 5.01 (s, 2H), 5.32–5.36 (m, 1H), 5.40 (br, 1H), 5.57 (br, 1H), 6.48–6.53 (m, 2H), 7.04–7.11 (m, 3H), 7.35–7.40 (m, 2H).
5-(4-fluorobenzyloxy)-N,2-dihydroxybenzamide (4c)
Compound 4c was prepared from 3g according to the same procedure as 4a. 4c was purified as a white solid. 1HNMR (CDCl3, 300MHz) δ: 5.00 (s, 2H), 6.84 (d, J =8.7Hz, 1H), 7.04–7.16 (m, 4H), 7.32 (d, J =2.4Hz, 1H), 7.42–7.47 (m, 3H).
2-(4-fluorobenzyloxy)-N,6-dihydroxybenzamide (4d)
Compound 4d was prepared from 3i according to the same procedure as 4a. 4d was purified as a white solid. 1HNMR(CDCl3, 300MHz) δ: 5.12 (s, 2H), 6.47 (d, J =8.7Hz, 1H), 6.67 (d, J =8.4Hz, 1H), 7.00–7.11 (m, 2H), 7.23–7.33 (m, 1H), 7.39–7.44 (m, 2H), 10.36 (br, 1H).
N-(4-Fluorobenzyl)-2,3-dihydroxybenzamide (5a)
A solution of 2,3-dihydroxybenzoic acid (154mg, 1mmol), EDCI (380mg, 2mmol), DIPEA (2mmol), and HOBt (290mg, 2mmol) in dry THF (15mL) was stirred at rt under N2. To this solution was added 90% (4-fluorophenyl)methanamine (0.152 mL, 1.2 mmol). The reaction mixture was stirred for 6.5 h at rt. After removal of most of THF, 20 mL of EtOAc was added to the residue. The solution was washed by 1N HCl, sat. NH4Cl and dried over Na2SO4. The concentration provided the residue which was purified by chromatography using petroleum ether/ethyl acetate (1:1) as eluent to give compound 5a as a white solid (51mg, 57% yield). 1H NMR (300MHz, d6-DMSO): δ 12.43 (brs, 1H), 8.33 (m, 1H), 7.40-7.32 (m, 3H), 7.20-7.14 (m, 2H), 6.94 (m, 1H), 6.71 (m, 1H), 4.48 (s, 2H). 13C NMR (100MHz, CD3OD): δ 171.9, 163.9 (d, 1JCF=242.6Hz), 150.8, 147.9, 136.6, 130.9 (d, 3JCF=8.2Hz), 120.2, 119.2, 119.1, 117.1, 116.6 (d, 2JCF=21.6Hz), 43.7. EI-MS m/z: 261 (M)+. Purity: system 1, 96.3% (method A, tR = 14.44 min); system 2, 95.0% (method F, tR = 13.13 min).
3-(Benzyloxy)-2-hydroxybenzoic acid (8b)
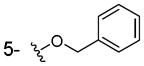
To a stirred solution of 2,3-dihydroxybenzoic acid (154mg, 1mmol) in DMF (5mL) was added NaH (48mg, 2mmol) and BnBr (0.125mL, 1.05mmol). The above mixture was stirred for 8h and the excess DMF was removed under reduced pressure. The residue was diluted with DCM (10mL) and the organic phase was washed with brine (5mL), dried with anhydrous Na2SO4, filtered, concentrated under vacuum to give the 8b (61mg, 25% yield) as colorless oil. 1HNMR (300 MHz, CDCl3): δ 7.41-7.20 (m, 6H), 6.79 (br, 1H), 6.50 (br, 1H), 4.99 (s, 2H).
3-(Benzyloxy)-N-(4-fluorobenzyl)-2-hydroxybenzamide (5b)
Compound 5b was prepared from 8b and (4-fluorophenyl)methanamine according to the same procedure described for 5a. Purification via column chromatography using PE/EA=5:1 afforded 5b (31 mg, 35% yield) as a white solid. 1H NMR (300MHz, CDCl3): δ 11.20 (s, 1H), 7.45-7.28 (m, 6H), 7.18 (dd, J = 1.5Hz, 8.1Hz, 1H), 7.14-7.12 (m, 1H), 7.04-6.99 (m, 3H), 6.74 (t, J =7.8Hz, 1H), 5.16 (s, 2H), 4.59 (d, J =5.7Hz, 2H). 13C NMR (100MHz, CDCl3): δ 168.7, 162.2 (d, 1JCF=245.0Hz), 150.6, 147.6, 136.5, 133.5, 129.5 (d, 3JCF=7.7Hz), 128.6, 128.1, 127.5, 118.8, 118.4, 117.2, 115.6 (d, 2JCF=21.4Hz), 71.2, 42.9. EI-MS: m/z 351 (M)+. HRMS (EI): calcd for C21H18FNO3 (M)+ 351.1271, found 351.1278. Purity: system 1, 99.3% (method B, tR = 14.20 min); system 2, 96.5% (method F, tR = 15.59 min).
2,3-Dihydroxy-N-(naphthalen-1-ylmethyl)benzamide (5c)
Compound 5c was prepared from 2,3-dihydroxybenzoic acid and naphthalen-1-ylmethanamine according to the same procedure described for 5a. 5c was purified as a white solid. (78mg, 45% yield) 1H NMR (300MHz, CDCl3): δ 8.03 (d, 1H, J =8.4Hz), 7.92-7.85 (m, 2H), 7.51-7.43 (m, 5H), 6.79 (d, J =7.8Hz, 1H), 6.68 (t, J =8.1Hz, 1H), 6.52 (br, 1H), 5.82 (s, 1H), 5.08 (d, J =4.5Hz, 2H). 13 C NMR (100MHz, CDCl3): δ 169.5, 148.7, 145.5, 133.6, 132.6, 131.1, 128.6, 128.3, 126.4, 126.0, 125.8, 125.2, 123.0, 118.5, 118.4, 116.9, 114.6, 41.3. EI-MS: m/z 293 (M)+. HRMS (EI): calcd for C18H15NO3 (M)+ 293.1052, found 293.1058. Purity: system 1, 98.0% (method A, tR = 15.96 min); system 2, 99.1% (method F, tR = 14.32 min).
N-(3,4-Difluorobenzyl)-2,3-dihydroxybenzamide (5d)
Compound 5d was prepared from 2,3-dihydroxybenzoic acid and (3,4-difluorophenyl)methanamine as a white solid according to the same procedure described for 5a (52mg, 42% yield). 1H NMR (300MHz, CDCl3): δ 7.29-7.13 (m, 4H), 6.94 (dd, J =1.5Hz, 7.8Hz, 1H), 6.73 (t, J =8.1Hz, 1H), 5.49 (s, 1H), 4.53 (s, 2H). 13C NMR (100MHz, CDCl3): δ 169.7, 148.5 (d, 1JCF=266.4Hz), 147.9 (d, 1JCF=268.2Hz), 145.3, 135.3, 123.0, 118.2 (d, 3JCF=8.1Hz), 117.1, 116.7 (d, 2JCF=17.2Hz), 115.9 (d, 2JCF=17.8Hz), 114.7, 41.6. EI-MS: m/z 279 (M)+, 280 (M+1)+. HRMS (EI): calcd for C14H11F2NO3 279.0707 (M)+, found 279.0704. Purity: system 1, 97.9% (method A, tR = 14.78 min); system 2, 95.0% (method F, tR = 13.43 min).
N-((1H-indol-5-yl)methyl)-2,3-dihydroxybenzamide (5e)
Compound 5e was prepared from 2,3-dihydroxybenzoic acid and (1H-indol-5-yl)methanamine according to the same procedure described for 5a. 5e was purified as a brown solid. (31mg, 36% yield) 1H NMR (300MHz, CDCl3): δ 8.23 (br, 1H), 7.63 (s, 1H), 7.41 (d, J =8.4Hz, 1H), 7.19 (d, J =8.4Hz, 1H), 7.04 (d, J =7.8Hz, 1H), 6.85 (d, J =7.8Hz, 1H), 6.72 (t, J =8.1Hz, 1H), 6.56-6.52 (m, 2H), 5.79 (br, 1H), 4.72 (d, J =5.1Hz, 2H). 13C NMR (100MHz, CDCl3): δ 169.5, 148.8, 145.6, 135.3, 128.2, 127.7, 124.9, 121.6, 119.7, 118.5, 118.3, 116.6, 114.6, 111.4, 101.6, 44.0. EI-MS: m/z 282 (M)+. HRMS (EI): calcd for C16H14N2O3 282.1004 (M)+, found 282.1006. Purity: system 1, 95.0% (method A, tR = 13.58 min); system 2, 96.0% (method F, tR = 12.33 min).
N-(2-(1H-indol-3-yl)ethyl)-2,3-dihydroxybenzamide (5f)
Compound 5f was prepared from 2,3-dihydroxybenzoic acid and 2-(1H-indol-3-yl)ethanamine according to the same procedure described for 5a. 5f was purified as a yellow solid. (36mg, 42% yield) 1H NMR (300MHz, CDCl3): δ 8.08 (br, 1H), 7.64 (d, J =8.1Hz, 1H), 7.41 (d, J =8.1Hz, 1H), 7.25-7.22 (m, 1H), 7.18-7.13 (m, 1H), 7.09-7.08 (m, 1H), 7.01 (dd, J =2.7Hz, 6.6Hz, 1H), 6.67-6.64 (m, 2H), 6.40 (br, 1H), 5.77 (s, 1H), 3.82-3.76 (m, 2H), 3.11 (t, J =6.3Hz, 2H). 13C NMR (100MHz, CDCl3): δ 169.7, 148.8, 145.6, 136.5, 136.3, 127.0, 122.3, 121.8, 119.1, 118.4, 118.3, 116.5, 114.5, 112.0, 111.3, 39.8, 24.9. EI-MS: m/z 296 (M)+. HRMS (EI): calcd for C17H16N2O3 (M)+ 296.1161, found 296.1159. Purity: system 1, 95.4% (method A, tR = 14.43 min); system 2, 95.1% (method F, tR = 13.09 min).
2,3-Dihydroxy-N-(1H-indol-5-yl)benzamide (5g)
Compound 5g was prepared from 2,3-dihydroxybenzoic acid and 1H-indol-5-amine according to the same procedure described for 5a. 5g was purified as a white solid. (40mg, 40% yield) 1H NMR (300MHz, CDCl3): δ 8.35 (br, 1H), 8.11 (br, 1H), 7.87 (d, J =1.8Hz, 1H), 7.41 (d, J =8.4Hz, 1H), 7.31-7.26 (m, 2H), 7.10 (dt, J =1.2Hz, 7.8Hz, 2H), 6.84 (t, J =7.8Hz, 1H), 6.57-6.56 (m, 1H), 5.41 (s, 1H). 13 C NMR (100MHz, CDCl3): δ 168.4, 148.8, 145.6, 133.8, 128.5, 127.7, 125.3, 118.6, 118.5, 117.3, 117.1, 115.3, 114.3, 111.2, 101.9. EI-MS: m/z 268 (M)+, 269 (M+1)+. HRMS (EI): calcd for C15H12N2O3 (M)+ 268.0848, found 268.0846. Purity: system 1, 95.2% (method A, tR = 13.15 min); system 2, 99.5% (method F, tR = 12.22 min).
2,3-Dihydroxy-N-(thiophen-2-ylmethyl)benzamide (5h)
Compound 5h was prepared from 2,3-dihydroxybenzoic acid and thiophen-2-ylmethanamine according to the same procedure described for 5a. 5h (190mg, 19% yield) was purified as a white solid. 1H NMR (300MHz, CDCl3): δ 12.55 (s, 1H), 7.27 (dd, J =1.5Hz, 5.1Hz, 1H), 7.06-7.03 (m, 2H), 6.98 (t, J = 2.7Hz, 1H), 6.87 (dd, J =1.5Hz, 8.4Hz, 2H), 6.74 (t, J =8.4Hz, 1H), 5.91 (brs, 1H), 5.81 (s, 1H), 4.80 (d, J =5.7Hz, 2H). 13C NMR (100MHz, CDCl3): δ 169.5, 148.7, 145.5, 140.4, 126.6, 125.9, 125.0, 118.5, 118.4, 116.9, 114.6, 37.9. EI-MS: m/z 249 (M)+. HRMS (EI): calcd for C12H11NSO3 249.0456 (M)+, found 249.0460. Purity: system 1, 96.1% (method C, tR = 13.71 min); system 2, 96.8% (method G, tR = 8.96 min).
N-(Furan-2-ylmethyl)-2,3-dihydroxybenzamide (5i)
Compound 5i was prepared from 2,3-dihydroxybenzoic acid and furan-2-ylmethanamine according to the same procedure described for 5a. 5i was purified as an off-white solid (102 mg, 33% yield). 1H NMR (300MHz, CDCl3): δ 7.39 (q, 1H), 7.04 (dd, J =1.5Hz, 7.8Hz, 1H), 6.89 (dd, J =1.5Hz, 8.1Hz, 1H), 6.75 (t, J =8.1Hz, 1H), 6.62 (brs, 1H), 6.36 (brs, 1H), 6.32 (d, J =3.0Hz, 1H), 5.80 (brs, 1H), 4.63 (d, J =6.0Hz, 2H). 13C NMR (100MHz, CDCl3): δ 169.7, 150.5, 148.9, 145.7, 142.4, 118.6, 118.4, 116.4, 114.0, 110.5, 107.9, 36.4. EI-MS: m/z 234.1 [M+H]+. HRMS (EI): calcd for C12H11NO4 (M)+ 233.0688, found 233.0689. Purity: system 1, 97.2% (method C, tR = 12.84 min); system 2, 95.9% (method G, tR = 8.73 min).
2,3-Dihydroxy-N-(4-(thiophen-2-yl)benzyl)benzamide (5j)
Compound 5j was prepared from 2,3-dihydroxybenzoic acid and (4-(thiophen-2-yl)phenyl)methanamine according to the same procedure described for 5a. 5j (78mg, 31% yield) was purified as a pale-yellow solid. 1H NMR (300MHz, CDCl3): δ 7.61 (d, J =8.4Hz, 2H), 7.35 (d, J =8.7Hz, 2H), 7.30 (t, J = 6.0Hz, 2H,), 7.04–7.10 (m, 2H), 6.89 (dd, J =1.8Hz, 8.1Hz, 1H), 6.76 (t, J =8.1Hz, 1H), 6.62 (brs, 1H), 5.81 (brs, 1H), 4.65 (d, J =5.7Hz, 2H). 13C NMR (100MHz, CDCl3): δ 169.8, 149.2, 145.9, 143.7, 128.4, 128.1, 126.4, 125.0, 123.3, 118.7, 118.2, 115.9, 113.7, 43.4. ESI-MS: m/z 326.1 [M+H]+. HRMS (ESI): calcd for C18H15NO3Na (M+Na)+ 348.0675, found 348.0670. Purity: system 1, 99.0% (method C, tR = 16.59 min); system 2, 99.1% (method G, tR = 9.58 min).
2,3-Dihydroxy-N-(piperidin-4-yl)benzamide (5k)
To a stirred solution of tert-butyl 4-oxopiperidine-1-carboxylate (184 mg, 1.86 mmol) and triethylamine (1.29 mL, 9.29 mmol) in CH2Cl2 (200 mL, anhydrous) was added the solution of di-tert-butyl dicarbonate (810 mg, 3.72 mmol) in CH2Cl2 (20 mL, anhydrous) dropwise. After stirring at rt for 16 h, the solvent was evaporated and the residue was purified by chromatography using petroleum ether/ethyl acetate (5:1) as eluent to give compound tert-butyl 4-oxopiperidine-1-carboxylate as white solid (318 mg, 86% yield).
To the solution of tert-butyl 4-oxopiperidine-1-carboxylate (360mg) in NH3/MeOH (15ml) was added Pd/C (15mg). The mixture was stirred under H2 atmosphere at rt overnight. The Pd/C was filtered and the filtrate was concentrated under vacuum. The concentration provided the residue which was purified by chromatography using DCM/MeOH (12:1) as eluent to give compound 4-aminopiperidine-1-carboxylate as white solid (226mg, 63% yield). 1H NMR (CDCl3, 300MHz): δ 4.07 (m, 2H), 2.81-2.65 (m, 3H), 1.84-1.79 (m, 2H), 1.48 (s, 9H), 1.38 (m, 2H). EI-MS: m/z 200 (M)+.
A solution of 2,3-dihydroxybenzoic acid (339mg, 2.2mmol), EDCI (422mg, 2.2mmol), DIPEA (2.2mmol), and HOBt (297mg, 2.2mmol) in dry CH2Cl2 (11mL) was stirred at room temperature. To this solution was added 4-aminopiperidine-1-carboxylate (220mg, 1.1mmol). The reaction mixture was stirred for 6.5 h at room temperature. After removal of most of CH2Cl2, 20 mL of EtOAc was added to the residue. The solution was washed by 1N HCl, saturated NH4Cl and dried over Na2SO4. The concentration provided the residue that was purified by chromatography using petroleum ether/ethyl acetate (5:1) as eluent to give compound tert-butyl 4-(2,3-dihydroxybenzamido)piperidine-1-carboxylate as white solid (190 mg, 51% yield). 1H NMR (CDCl3, 300MHz): δ 7.04 (d, J =8.1Hz, 1H), 6.85 (d, J =7.8Hz, 1H), 6.76 (t, J =8.1Hz, 1H), 6.16 (s, 1H), 5.79 (brs, 1H), 4.15-4.10 (m, 4H), 2.90 (t, J =9.9Hz, 2H), 2.04-1.98 (m, 3H), 1.49 (s, 9H).
To the solution of 4-(2,3-dihydroxybenzoyl)piperadine-1-carboxylate (67mg, 0.2mmol) in anhydrous DCM (5ml) on ice bath was added TFA(0.578 ml) dropwise. The solution was allowed to warm to r.t for 2h. The solvent was evaporated and the residue was purified by chromatography using methylene chloride/acetone (2:1) as eluent to give 5k as white solid (47mg, 100% yield). 1H NMR (300MHz, CD3OD): δ 7.11 (d, J =8.4Hz, 1H), 6.86 (d, J = 8.1Hz, 1H), 6.62 (t, J =7.8Hz, 1H), 4.08-4.06 (m, 1H), 3.34 (d, J =6.3Hz, 2H), 2.88 (t, J =12.3Hz, 2H), 2.00 (d, J =6.6Hz, 2H), 1.82 (q, 2H). 13C NMR (100MHz, CDCl3): δ 171.6, 150.7, 147.8, 120.3, 120.2, 119.4, 117.2, 48.9, 46.4, 44.8, 30.0. ESI-MS: m/z 237.1 [M+H]+. HRMS (ESI): calcd for C12H17N2O3 (M+H)+ 237.1239, found 237.1225. Purity: system 1, 97.0% (method D, tR = 3.38 min); system 2, 96.8% (method H, tR = 3.79 min).
N-Cyclohexyl-2,3-dihydroxybenzamide (5l)
Compound 5l was prepared from 2,3-dihydroxybenzoic acid and cyclohexanamine according to the same procedure as 5a. 5l (570mg, 38% yield) was purified as a white solid. 1H NMR (300MHz, CDCl3): δ 7.03 (d, J =7.5Hz, 1H), 6.86 (d, J =8.1Hz, 1H), 6.75 (t, J =8.1Hz, 1H), 6.09 (brs, 1H), 5.91 (brs, 1H), 4.02-3.90 (m, 1H), 2.05-2.00 (m, 2H), 1.82-1.74 (m, 2H), 1.71-1.32 (m, 4H), 1.28-1.20 (m, 2H). 13C NMR (100MHz, CD3OD): δ 169.0, 149.2, 146.0, 118.5, 117.9, 115.7, 114.1, 48.6, 33.0, 25.4, 24.8. EI-MS: m/z 235 (M)+. HRMS (EI): calcd for C13H17NO3 (M)+ 235.1205, found 235.1208. Purity: system 1, 97.8% (method C, tR = 15.27 min); system 2, 97.9% (method G, tR = 9.48 min).
N-(Cyclohexylmethyl)-2,3-dihydroxybenzamide (5m)
Compound 5m was prepared from 2,3-dihydroxybenzoic acid and cyclohexylmethanamine according to the same procedure as 5a. 5m (222mg, 31% yield) was purified as a white solid. 1H NMR (300MHz, CDCl3): δ 7.04 (d, J =7.8Hz, 1H), 6.87 (d, J =8.1Hz, 1H), 6.75 (t, J =8.1Hz, 1H), 6.38 (brs, 1H), 5.81 (brs, 1H), 3.29 (t, J =6.6Hz, 2H), 1.77 (m, 5H), 1.35-1.12 (m, 4H), 1.01 (m, 2H). EI-MS: m/z 249 (M)+. HRMS (EI): calcd for C14H19NO3 (M)+ 249.1365, found 249.1365. Anal. Calcd (found): C, 67.45 (67.29); H, 7.68 (7.68); N, 5.62 (5.67). Purity: system 1, 99.1% (method C, tR = 16.40 min); system 2, 96.0% (method G, tR = 9.88 min).
2-(2-(Hydroxymethyl)phenyl)-N-methylacetamide (9)
To a stirred solution of isochroman-3-one (316 mg, 2.13 mmol) in THF (10 mL) was added NH2Me (662 mg, 21.35 mmol). The above mixture was stirred at rt for 8 hours and excess THF was removed under reduced pressure. The residue was diluted with DCM (10 mL) and the organic phase was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, concentrated under vacuum. The residue was chromatographied using PE/EA=1:1 to provide 9 (377 mg, 99% yield) as colorless oil. 1H NMR (300MHz, CDCl3): δ 7.39-7.37 (m, 1H), 7.29-7.24 (m, 3H), 6.37 (br, 1H), 4.66 (s, 2H), 4.38 (br, 1H), 3.62 (s, 2H), 2.74 (d, J =4.8Hz, 3H).
2-(2-(Chloromethyl)phenyl)-N-methylacetamide (10)
To a stirred solution of 9 (0.45 mL, 5.9 mmol) in DCM (10 mL) was added MsCl (0.45 mL, 5.9 mmol) and TEA (1mL, 7.4mmol). The above mixture was stirred at rt for 8 hours and the residue was diluted with DCM (10 mL) and the organic phase was washed with brine (20 mL), dried with anhydrous Na2SO4, filtered, concentrated under vacuum. The residue was chromatographied using PE/EA=1:1 to give 10 (267mg, 28% yield) as colorless oil. 1H NMR (300MHz, CDCl3): δ 7.42-7.29 (m, 4H), 5.42 (br, 1H), 4.65 (s, 2H), 3.73 (s, 2H), 2.77 (d, J =5.1Hz, 3H).
2-(2-(Azidomethyl)phenyl)-N-methylacetamide (11n)
To a stirred solution of 10 (287 mg, 1.36 mmol) in DMF (5 mL) was added NaN3 (106 mg, 1.63 mmol), the mixture was stirred at 80 °C for 8 hours. The residue was diluted with DCM (10 mL) and the organic phase was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, concentrated under vacuum to give 11n which was used directly for the next step without further purification.
2-(2-(Aminomethyl)phenyl)-N-methylacetamide (12n)
To a stirred solution of 11n in THF (5mL) was added Ph3P (713mg, 2.72mmol), the mixture was stirred at rt for 8 hours and evaporated to remove THF. The residue was diluted with DCM (10mL) and the organic phase was washed with brine (5mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was chromatographed using DCM/MeOH=10:1 to afford 12n (94 mg, 39% yield in 2 steps) as yellow oil.
2,3-Dihydroxy-N-(2-(2-(methylamino)-2-oxoethyl)benzyl)benzamide (5n)
Compound 5n was prepared from 2,3-dihydroxybenzoic acid and 12q according to the same procedure described for 5a. 5n was purified as a white solid. (28 mg, 39% yield) 1H NMR (300MHz, CDCl3): δ 7.49-7.46 (m, 1H), 7.32-7.29 (m, 2H), 7.23-7.17 (m, 4H), 4.64 (d, J =3.6Hz, 2H), 3.64 (s, 2H), 2.78 (d, J =4.5Hz, 3H). 13C NMR (100MHz, CDCl3): δ 172.4, 169.7, 148.9, 145.4, 136.1, 133.7, 130.6, 130.1, 128.2, 127.7, 118.5, 118.3, 117.2, 114.4, 41.5, 40.0, 26.1. EI-MS: m/z 314 (M)+, 316 (M+2)+. HRMS (EI): calcd for C17H18N2O4 (M)+ 314.1267, found 314.1265. Purity: system 1, 98.9% (method A, tR = 11.82 min); system 2, 98.7% (method F, tR = 11.95 min).
5-Fluoro-N,2-dimethylbenzamide (13)
Compound 13 w a s p r epared from 5-fluoro-2-methylbenzoic acid and methanamine according to the same procedure as 5a. 13 was purified as a white solid. (173 mg, 82% yield) 1H NMR (300MHz, CDCl3): δ 7.20-7.15 (m, 1H), 7.07-6.98 (m, 2H), 3.00 (d, J =4.2Hz, 3H), 2.39 (s, 3H).
2-(Bromomethyl)-5-fluoro-N-methylbenzamide (14)
To a solution of 13 (581 mg, 3.48 mmol) in CCl4 (10 mL) was added NBS (743 mg, 4.17 mmol) and AIBN (171 mg, 1.04 mmol). The above mixture was stirred at reflux for 8h and evaporated to remove CCl4 under vacuum. The residue was diluted with DCM (10mL) and the organic phase was washed with brine (10mL), dried with anhydrous Na2SO4, filtered, concentrated under vacuum, the residue was chromatographed using DCM/MeOH=10:1 to provide 14 (467mg, 55% yield) as a yellow solid. 1H NMR (300MHz, CDCl3): δ 7.59-7.41 (m, 3H), 5.75 (s, 2H), 3.32 (s, 3H).
2-(Azidomethyl)-5-fluoro-N-methylbenzamide (11o)
Compound 11o was prepared from 14 according to the same procedure described for 11n. The crude product 11o was used directly for next step without further purification.
2-(Aminomethyl)-5-fluoro-N-methylbenzamide (12o)
Compound 12o was prepared from 11o according to the same procedure described for 12n. 12o was purified as a yellow oil. (92mg, 68% yield) 1H NMR (300MHz, CD3OD): δ 7.60-7.55 (m, 1H), 7.47-7.43 (m, 1H), 7.37-7.30 (m, 1H), 4.12 (s, 2H), 2.93 (s, 3H).
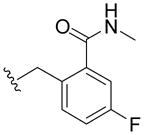
N-(4-Fluoro-2-(methylcarbamoyl)benzyl)-2,3-dihydroxybenzamide (5o)
Compound 5o was prepared from 2,3-dihydroxybenzoic acid and 12o as a white solid according to the same procedure described for 5a (31mg, 61% yield) 1H NMR (300MHz, CDCl3): δ 7.57-7.53 (m, 1H), 7.47-7.43 (m, 1H), 7.19-7.01 (m, 3H), 6.91-6.86 (m, 1H), 6.16 (br, 1H), 5.73 (br, 1H), 4.58 (d, J =6.3Hz, 2H), 3.06 (s, 3H). 13C NMR (100MHz, CDCl3): δ 169.7, 169.4, 161.3 (d, 1JCF=247.0Hz), 148.7, 145.4, 137.0, 131.9 (d, 4JCF=8.2Hz), 131.7 (d, 3JCF=10.5Hz), 128.5 (d, 3 JCF=12.7Hz), 120.3, 120.1, 118.8, 117.1 (d, 2JCF=21.6Hz), 114.1 (d, 2JCF=26.0Hz), 40.5, 26.2. EI-MS: m/z 318 (M)+, 319 (M+1)+. HRMS (EI): calcd for C16H15FN2O4 (M)+ 318.1016, found 318.1020. Purity: system 1, 95.3% (method A, tR = 14.49 min); system 2, 97.7% (method F, tR = 13.15 min).
5-Chlorosulfonyl-2,3-dimethoxy-benzoic acid (15)
2,3-dimethoxybenzoic acid (10 mmol, 1.82 g) was added in several portions to chlorosulfonic acid (10 mL) and stirred at rt. for 15h. The solution was diluted with CH2Cl2 (50 mL) and poured onto a mixture of CH2Cl2 and ice on a chilled salt bath. The organic phase was washed with water (3 × 20mL), then with brine (3 × 20mL), dried over Na2SO4 and concentrated to give a white solid 15 (2.246g, 80% yield). 1H NMR (300MHz, CDCl3): δ 8.34 (d, J = 2.4 Hz, 1H), 7.67 (d, J = 2.4 Hz, 1H), 4.17 (s, 3H), 4.04 (s, 3H).
2,3-Dimethoxy-5-(piperidine-1-sulfonyl)-benzoic acid (16p)
Piperidine (0.356 mL, 3.6 mmol) was added to a stirred solution of 15 (0.84 g, 3.0 mmol) and Et3N (0.5 mL, 3.6 mmol) in CH2Cl2 (20 mL). The solution was heated to reflux for 22h. The reaction mixture was washed with dilute HCl (2× 30 mL), then brine, dried over anhydrous Na2SO4 and concentrated in vacuo. Flash chromatography (hexane/EtOAc=2:1) provide 16p as a white solid (0.83g, 84% yield). 1H NMR (300MHz, CDCl3): δ 8.01 (d, J = 1.2 Hz, 1H), 7.46 (d, J = 1.2 Hz, 1H), 4.13 (s, 3H), 3.97 (s, 3H), 3.03 (m, 4H), 1.65 (m, 4H), 1.45 (m, 2H).
N-(4-Fluoro-benzyl)-2,3-dimethoxy-5-(piperidine-1-sulfonyl)-benzamide (17p)
17p was prepared as a white solid according to the same procedure described for 5a (85mg, 83% yield). 1H NMR (300MHz, CDCl3): δ 8.51 (m, 1H), 8.07 (s, 1H), 7.38 (s, 1H), 7.31 (m, 2H), 7.02 (m, 2H), 4.61 (d, J = 4.8 Hz, 2H), 3.92 (s, 3H), 3.89 (s, 3H), 3.02 (m, 4H), 1.63 (m, 4H), 1.42 (m, 2H).
N-(4-Fluorobenzyl)-2-methoxy-3-hydroxy-5-(piperidin-1-ylsulfonyl)benzamide (18p)
The solution of 1M BBr3 (2.61 mmol) in CH2Cl2 was added dropwise to a stirred solution of 17p (76 mg, 0.174 mmol) in anhydrous CH2Cl2 (20 mL) at −40°C. The mixture was stirred at −20°C for 10h. Methanol (2 mL) was added to quench the reaction and the solution was washed with H2O and brine, dried over Na2SO4, and concentrated. The residue was purified by chromatography (hexane/EtOAc=2:1) to give 18p as a white solid (60mg, 82% yield). 1H NMR (300MHz, CDCl3): δ 12.27 (s, 1H), 7.71 (s, 1H), 7.67 (m, 1H), 7.39 (m, 2H), 7.23 (s, 1H), 7.03 (m, 2H), 4.61 (d, J = 6.0 Hz, 2H), 3.94 (s, 3H), 2.96 (m, 4H), 1.59 (m, 4H), 1.43 (m, 2H).
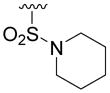
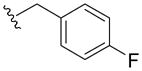
N-(4-Fluoro-benzyl)-2,3-dihydroxy-5-(piperidine-1-sulfonyl)-benzamide (5p)
The solution of 1M BBr3 (2.13 mmol) in CH2Cl2 was added dropwise to a stirred solution of 18p (60 mg, 0.142 mmol) in anhydrous CH2Cl2 (20 mL) at −40°C. The mixture was stirred at −20°C for 10h. Methanol (2 mL) was added to quench the reaction and the solution was washed with H2O and brine, dried over Na2SO4, and concentrated. The residue was purified by chromatography (hexane/EtOAc=2:1) to give 5p as a white solid (25mg, 42% yield). 1H NMR (300MHz, CDCl3): δ 13.65 (s, 1H), 7.55 (m, 1H), 7.38 (m, 4H), 7.04 (m, 2H), 6.02 (m, 1H), 4.60 (d, J = 5.7 Hz, 2H), 2.97 (m, 4H), 1.62 (m, 4H), 1.41 (m, 2H). 13C NMR (100MHz, CD3OD): δ 170.5, 164.0 (d, 1JCF=243.3Hz), 154.8, 148.7, 136.6, 131.2 (d, 3JCF=8.2Hz), 127.6, 120.0, 118.0, 117.1, 116.7 (d, 2JCF=21.6Hz), 48.7, 44.0, 26.9, 25.0. EI-MS m/z: 408 (M)+. Purity: system 1, 99.8% (method A, tR = 16.34 min); system 2, 97.6% (method F, tR = 14.31 min).
2,3-Dimethoxy-5-(N-methylsulfamoyl)benzoic acid (16q)
To the solution of 15 (349 mg, 1.24 mmol) in DCM (5 mL) was added MeNH2 (0.2 mL, 1.87 mmol) and TEA (0.35 mL, 2.49 mmol). The above mixture was stirred at rt for 8h and the residue was diluted with DCM (10 mL) and the organic phase was washed with brine (10 mL), dried over anhydrous Na2SO4, filtered, concentrated under vacuum, the residue was chromatographed using DCM/MeOH=50:1 to separate the 16q (303mg, 89% yield) as a white solid. 1HNMR (300MHz, CD3OD): δ 7.68 (d, J =2.1Hz, 1H), 7.43 (d, J =2.1Hz, 1H), 3.92 (s, 3H), 3.91 (s, 3H), 2.52 (s, 3H).
N-(4-Fluorobenzyl)-2,3-dimethoxy-5-(N-methylsulfamoyl)benzamide (17q)
Compound 17q was prepared from 16q and (4-fluorophenyl)methanamine according to the same procedure described for 5a. 17q was purified as a white solid. (94mg, 62% yield) 1H NMR (300MHz, CDCl3): δ 8.21 (s, 1H), 7.52 (s, 1H), 7.35-7.31 (m, 2H), 7.07-7.01 (m, 2H), 4.63 (d, J =5.1Hz, 2H), 4.52 (br, 1H), 3.96 (s, 3H), 3.90 (s, 3H), 2.69 (s, 3H).
N-(4-Fluorobenzyl)-2-hydroxy-3-methoxy-5-(N-methylsulfamoyl)benzamide (18q)
To the solution of 17q (402mg, 1.05mmol) in DMF (20mL) was added LiCl (269mg, 6.31mmol). The above mixture was stirred at 110°C for 8h and excess DMF was removed under reduced pressure. The residue was diluted with DCM (10mL) and the organic phase was washed with brine (10mL), dried over anhydrous Na2SO4, filtered, concentrated under vacuum, the residue was chromatographed using PE/EA=1:1 to afford 18q (183mg, 65% yield) as yellow oil. 1H NMR (300MHz, CDCl3): δ 7.66-7.62 (m, 1H), 7.36-7.32 (m, 3H), 7.04-6.98 (m, 2H), 4.59 (d, J =5.4Hz, 2H), 3.94 (s, 3H), 2.60 (s, 3H).
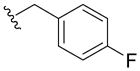
N-(4-Fluorobenzyl)-2,3-dihydroxy-5-(N-methylsulfamoyl)benzamide (5q)
To the solution of 18q (55mg, 0.15mmol) in DCM (2mL) was added 4N BBr3 (38uL). The above mixture was stirred at rt for 4h and treated with MeOH (0.5mL), concentrated under vacuum, the residue was chromatographied using PE/EA=1:1 to give 5q (12mg, 24% yield) as a red solid. 1H NMR (300MHz, CDCl3): δ 7.45 (s, 1H), 7.38-7.33 (m, 3H), 7.05 (t, J =8.4Hz, 2H), 4.61 (s, 2H), 2.62 (s, 3H). 13C NMR (100MHz, CDCl3): δ 168.4, 162.0 (d, 1JCF=244.8Hz), 149.0, 133.4, 129.4 (d, 3JCF=8.2Hz), 127.9, 118.5, 115.3 (d, 2JCF=21.6Hz), 114.5, 111.9, 42.6, 28.7. EI-MS: m/z 354 (M)+, 356 (M+2)+. HRMS (EI): calcd for C15H15FN2O5S (M)+ 354.0686, found 354.0682. Purity: system 1, 95.6% (method A, tR = 14.73 min); system 2, 98.9% (method F, tR = 12.95 min).
5-(N-tert-Butylsulfamoyl)-2,3-dimethoxybenzoic acid (16r)
Compound 16r was prepared from 15 and 2-methylpropan-2-amine according to the same procedure described for 16q. 16r was a white solid (379mg, 35% yield). 1H NMR (300MHz, CD3OD): δ 7.75 (d, J =2.1Hz, 1H), 7.58 (d, J =2.1Hz, 1H), 3.93 (s, 3H), 3.91 (s, 3H), 1.19 (s, 9H).
5-(N-tert-Butylsulfamoyl)-N-(4-fluorobenzyl)-2,3-dimethoxybenzamide (17r)
Compound 17r was prepared from 16r and (4-fluorophenyl)methanamine according to the same procedure described for 17q. 17r was a white solid (250mg, 50% yield). 1H NMR (300MHz, CDCl3): δ 8.25 (d, J =2.1Hz, 1H), 8.20-8.17 (m, 1H), 7.55 (d, J =2.1Hz, 1H), 7.35-7.30 (m, 2H), 7.04 (t, J =8.7Hz, 2H), 4.63 (d, J =5.4Hz, 2H), 3.94 (s, 3H), 3.88 (s, 3H), 1.26 (s, 9H).
5-(N-tert-Butylsulfamoyl)-N-(4-fluorobenzyl)-2-hydroxy-3-methoxybenzamide (18r)
Compound 18r was prepared from 17r according to the same procedure described for 18q. 18r was purified as a white solid (95mg, 88% yield). 1H NMR (300MHz, CDCl3): δ 7.78 (d, J =1.5Hz, 1H), 7.36 (d, J =1.5Hz, 1H), 7.32 (q, 2H), 7.02 (t, J =8.7Hz, 2H), 4.58 (d, J =5.7Hz, 2H), 3.93 (s, 3H), 1.21 (s, 9H).
5-(N-tert-Butylsulfamoyl)-N-(4-fluorobenzyl)-2,3-dihydroxybenzamide (5r)
Compound 5r was prepared from 18r according to the same procedure described for 5q. 5r was purified as a white solid (22mg, 30% yield). 1H NMR (300MHz, CDCl3): δ 7.77 (d, 1H, J =1.5Hz), 7.37-7.30 (m, 3H), 7.05-7.00 (m, 2H), 4.58 (d, J =5.7Hz, 2H), 1.21 (s, 9H). 13C NMR (100MHz, CDCl3): δ 169.4, 163.9 (d, 1JCF=242.5Hz), 153.9, 150.5, 136.2, 135.5, 131.1 (d, 3 JCF=7.9Hz), 120.3, 118.2, 117.9, 116.9 (d, 2JCF=19.4Hz), 44.1, 43.9. EI-MS: m/z 396 (M)+. HRMS (EI): calcd for C18H21FN2O5S (M)+ 396.1155, found 396.1143. Purity: system 1, 99.4% (method A, tR = 12.46 min); system 2, 99.1% (method F, tR = 12.67 min).
2,3-Dihydroxy-5-(piperidin-1-ylsulfonyl)benzoic acid (17s)
Compound 17s was prepared from 16p and BBr3 according to the same procedure described for 18p. 17s was purified as a white solid (195mg, 68% yield). 1H NMR (300MHz, CD3OD): δ 7.73 (d, J =2.1Hz, 1H), 7.28 (d, J =1.8Hz, 1H), 2.96 (t, J =5.7Hz, 4H), 1.67-1.59 (m, 4H), 1.48-1.41 (m, 2H).
N-(Furan-2-ylmethyl)-2,3-dihydroxy-5-(piperidin-1-ylsulfonyl)benzamide (5s)
Compound 5s was prepared from 17s and furan-2-ylmethanamine according to the same procedure described for 5a. 5s was purified as a white soild (14mg, 33% yield). 1H NMR (300MHz, CD3OD): δ 7.42 (d, J =1.2Hz, 1H), 7.36 (d, J =1.5Hz, 1H), 6.95 (brs, 1H), 6.39-6.36 (m, 2H), 5.98 (s, 1H), 4.64 (d, J =5.4Hz, 2H), 2.98 (t, J =5.4Hz, 4H), 1.66-1.62 (m, 4H), 1.47-1.41 (m, 2H). 13C NMR (100 MHz, CD3OD) δ: 170.3, 153.2, 148.6, 143.9, 131.4, 127.7, 120.2, 118.0, 117.2, 111.9, 109.0, 48.7, 37.7, 26.9, 25.0. ESI-MS: m/z 381.1 [M+H]+. HRMS (ESI): calcd for C17H10N2O6Na (M+Na)+ 403.0940, found 403.0989. Purity: system 1, 95.5% (method E, tR = 13.00 min); system 2, 98.4% (method G, tR = 7.76 min).
2,3-Dihydroxy-5-(piperidin-1-ylsulfonyl)-N-(4-(trifluoromethyl)benzyl) benzamide (5t)
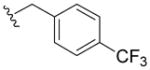
Compound 5t was prepared from 17s and (4-(trifluoromethyl)phenyl) methanamine according to the same procedure described for 5a. 5t was purified as an off white solid (51mg, 32% yield). 1H NMR (300MHz, CD3OD): δ 7.77 (d, J =1.8Hz, 1H), 7.63 (d, J =4.5Hz, 2H), 7.54 (d, J =4.8Hz, 2H), 7.25 (d, J =1.8Hz, 1H), 4.67 (d, J =6.0Hz, 2H), 2.97-2.93 (m, 4H), 1.83-1.79 (m, 4H), 1.73-1.70 (m, 2H). 13C NMR (100 MHz, CD3OD): δ 170.7, 154.8, 148.7, 144.9, 129.6, 127.8, 127.0, 126.9, 121.9, 120.1, 119.8, 118.1, 117.1, 114.3, 48.7, 44.3, 26.9, 25.0. EI-MS: m/z 458 (M)+. HRMS (EI): calcd for C20H21SN2O5F3 (M)+ 458.1123, found 458.1118. Purity: system 1, 99.6% (method C, tR = 17.35 min); system 2, 99.8% (method G, tR = 9.61 min).
N-(Cyclohexylmethyl)-2,3-dihydroxy-5-(piperidin-1-ylsulfonyl)benzamide (5u)
Compound 5u was prepared from 17s and cyclohexylmethanamine according to the same procedure described for 5a. 5u was purified as an off white solid (20mg, 21% yield). 1H NMR (300MHz, CDCl3): δ 8.07 (d, J =1.8Hz, 1H), 7.48 (d, J =1.8Hz, 1H), 3.04 (t, J = 5.1Hz, 4H), 1.84-1.41 (m, 11H), 1.29-1.14 (m, 4H), 1.07-0.96 (m, 4H).
N-(Thiophen-2-ylmethyl)-2,3-dihydroxy-5-(piperidin-1-ylsulfonyl)benzamide (5v)
Compound 5v was prepared from 17s and thiophen-2-ylmethanamine according to the same procedure described for 5a. 5v was purified as an off white solid (17mg, 23% yield). 1HNMR (300MHz, CDCl3): δ 8.12 (d, J =1.8Hz, 1H), 7.54 (d, J =1.8Hz, 1H), 7.34-7.25 (m, 3H), 4.31 (t, J =5.4Hz, 2H), 2.97-2.93 (m, 4H), 1.83-1.79 (m, 4H), 1.73-1.70 (m, 2H)..
2,3-Dimethoxy-5-nitrobenzoic acid (19)
To a stirred solution of 2,3-dimethoxybenzoic acid (1.96g, 10mmol) in conc. H2SO4 (10ml) was added the solution of HNO3 (0.43ml) in concentrated H2SO4 (4.5ml) dropwise at 0°C. After stirring at 0°C for 1h, the mixture was poured into ice water and diluted with DCM. The organic layer was washed with brine, then concentrated under vacuum. The residue was chromatographed using DCM/MeOH=10:1 to give 19 as a white solid (634 mg, 25%). 1H NMR (300MHz, CDCl3): δ8.26 (d, J =2.7Hz, 1H), 7.89 (d, J =2.7Hz, 1H), 4.01 (s, 3H), 3.99 (s, 3H), 3.95 (s, 3H).
N-(4-Fluorobenzyl)-2,3-dimethoxy-5-nitrobenzamide (20)
Compound 20 was prepared from 19 and (4-fluorophenyl)methanamine according to the same procedure described for 5a. 20 was a white solid (45%). 1H NMR (300MHz, CDCl3): δ 8.66 (d, J =2.7Hz, 1H), 8.09 (br, 1H), 7.88 (d, J =2.7Hz, 1H), 7.31–7.36 (m, 2H), 7.05 (d, J =8.7Hz, 2H), 4.64 (d, J =5.7Hz, 2H), 3.99 (s, 3H), 3.94 (s, 3H).
5-Amino-N-(4-fluorobenzyl)-2,3-dimethoxybenzamide (21)
To the solution of compound 20 (498 mg, 1.49 mmol) and concentrated HCl (0.62 mL) in 10 mL of THF, was added SnCl2 (1414 mg, 7.44 mmol). The reaction mixture was stirred for 8h at room temperature, then diluted with DCM (20 mL). Usual work-up followed by purification on column chromatography (DCM/MeOH =10:1) afforded 21 as a yellow solid (272 mg, yield 60%). 1H NMR (300MHz, CDCl3): δ7.29–7.34 (m, 2H), 6.99–7.04 (m, 3H), 6.39 (d, J =2.4Hz, 1H), 4.61 (d, J =6.0Hz, 2H), 3.83 (s, 3H), 3.70 (s, 3H).
N-(4-Fluorobenzyl)-2,3-dimethoxy-5-(methylsulfonamido)benzamide (22w)
Compound 22w was prepared from 21 and methanesulfonyl chloride according to the same procedure described for 16p. 22w was a white solid in yield of 80%. 1H NMR (300MHz, CDCl3): δ 7.86 (d, J =2.7Hz, 1H), 7.32–7.37 (m, 3H), 7.00–7.06 (m, 2H), 4.74 (d, J =5.7Hz, 2H), 3.91 (s, 3H), 3.82 (s, 3H), 2.92 (s, 3H).
5-Acetamido-N-(4-fluorobenzyl)-2,3-dimethoxybenzamide (22x)
Compound 22x was prepared from 21 and acetyl chloride according to the same procedure described for 16p. 22x was a white solid in yield of 88%. 1H NMR (300MHz, CDCl3): δ 8.07 (d, J =2.7Hz, 1H), 7.99 (br, 1H), 7.38 (d, J =2.7Hz, 1H), 7.28–7.33 (m, 2H), 7.00–7.06 (m, 2H), 4.63 (d, J = 5.7Hz, 2H), 4.31 (t, J =6.6Hz, 1H), 3.91 (s, 3H), 3.79 (s, 3H), 2.16 (s, 3H).
N-(4-Fluorobenzyl)-2,3-dihydroxy-5-(methylsulfonamido)benzamide (5w)
Compound 5w was prepared from 22w and BBr3 according to the same procedure described for 18p. 5w was a yellow solid (42%). 1H NMR (300MHz, CD3OD): δ 7.34–7.39 (m, 2H), 7.14 (d, J = 2.7Hz, 1H), 7.01–7.07 (m, 2H), 6.93 (d, J =2.7Hz, 1H), 4.54 (s, 2H), 4.28 (t, J =6.6Hz, 1H), 2.90 (s, 3H). MS (EI): m/z 354 (M)+.
5-Acetamido-N-(4-fluorobenzyl)-2,3-dihydroxybenzamide (5x)
Compound 5x was prepared from 22x and BBr3 according to the same procedure described for 18p. 5x was a yellow solid (47%). 1H NMR (300MHz, CD3OD): δ 7.34–7.38 (m, 3H), 7.14 (s, 1H), 7.01–7.07 (m, 2H), 4.54 (s, 2H), 4.28 (t, J =6.6Hz, 1H), 2.07 (s, 3H). MS (EI): m/z 318 (M)+, 319 (M+1)+.
4-((Dimethylamino)methyl)-2,3-dihydroxybenzoic acid (23)
To a stirred solution of 2,3-dihydroxybenzoic acid (300mg, 1.95mmol), dimethylamine hydrochloride (159mg, 1.95mmol), Et3N (544μL, 3.9mmol) in EtOH (20mL) was added HCHO(aq) (146 μL, 1.95mmol, 40%) at 0°C. The above mixture was refluxed overnight. The white precipitate was filtered and washed 3 times with anhydrous EtOH. The residue was dried under vacuum to give 23 (186mg, 45% yield) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 7.16 (d, J = 7.8Hz, 1H), 6.47 (d, J =7.5Hz, 1H), 4.14 (s, 1H), 2.72 (s, 6H). EI-MS m/z: 211 (M)+.
4-((Dimethylamino)methyl)-N-(4-fluorobenzyl)-2,3-dihydroxybenzamide (6a)
Compound 6a was prepared from 23 and (4-fluorophenyl)methanamine according to the same procedure described for 5a. 6a was purified as a white solid. (91mg, 61% yield) 1H NMR (300MHz, CD3OD): δ 7.36 (dd, J =5.4Hz, 8.4Hz, 2H), 7.27 (d, J =7.8Hz, 1H), 7.04 (t, J =8.7Hz, 2H), 6.64 (d, J =8.4Hz, 1H), 4.56 (s, 2H), 4.03 (s, 2H), 2.63 (s, 6H).
4-((Dimethylamino)methyl)-N-(furan-2-ylmethyl)-2,3-dihydroxybenzamide (6b)
Compound 6b was prepared from 23 and furan-2-ylmethanamine according to the same procedure described for 5a. 6b was purified as a white solid. (95mg, 63% yield) 1H NMR (300MHz, CD3OD): δ 7.32 (d, J =1.8Hz, 1H), 7.14 (d, J =7.8Hz, 1H), 6.47 (d, J =7.5Hz, 1H), 6.33 (d, J =8.7Hz, 2H), 4.63(s, 2H), 4.09 (s, 2H), 2.65 (s, 6H).
2-Hydroxy-3-methoxy-5-(piperidin-1-ylsulfonyl)benzoic acid (17w)
Compound 17w was prepared from 16p and BBr3 according to the same procedure as 18p. 17w was purified as a white solid (21mg, 16% yield). 1H NMR (300MHz, CD3OD): δ 7.73 (d, J =2.1Hz, 1H), 7.28 (d, J =1.8Hz, 1H), 3.98 (s, 3H), 2.96 (t, J =5.4Hz, 4H), 1.65-1.63 (m, 4H), 1.46-1.40 (m, 2H).
N-(Furan-2-ylmethyl)-2-hydroxy-3-methoxy-5-(piperidin-1-ylsulfonyl)benzamide. (6c)
Compound 6c was prepared from 17w and furan-2-ylmethanamine according to the same procedure described for 5a. 6c was purified as a yellow solid (51mg, 32% yield). 1H NMR (300MHz, CDCl3): δ 7.54 (d, J =1.8Hz, 1H), 7.24 (d, J =2.1Hz, 1H), 7.09 (brs, 1H), 6.38-6.34 (m, 1H), 6.32 (q, 1H), 6.23 (d, J =3.6Hz, 1H), 4.64 (d, J =6.0Hz, 2H), 3.95 (s, 3H), 2.98 (t, J =5.4Hz, 4H), 1.68-1.62 (m, 4H), 1.47-1.41 (m, 2H). 13C NMR (100MHz, CD3OD): δ 168.5, 154.8, 150.5, 149.1, 142.1, 125.3, 118.9, 114.3, 112.3, 110.3, 108.0, 56.4, 46.8, 36.3, 25.0, 23.2. EI-MS: m/z 394 (M)+. HRMS (EI): calcd for C18H22SN2O6 (M)+ 394.1199, found 394.1198. Purity: system 1, 99.4% (method E, tR = 13.60 min); system 2, 98.5% (method G, tR = 9.27 min).
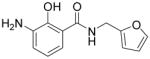
3-Amino-N-(furan-2-ylmethyl)-2-hydroxybenzamide (6d)
Compound 6d was prepared from 3-amino-2-hydroxybenzoic acid and furan-2-ylmethanamine according to the same procedure described for 5a. 6d (45mg, 30% yield) was purified as an off white solid. 1H NMR (300MHz, CDCl3): δ 12.38 (s, 1H), 7.37 (s, 1H), 6.81 (dd, J =1.5Hz, 7.2Hz, 1H), 6.74 (dd, J =1.5Hz, 7.8Hz, 1H), 6.66 (d, J =7.8Hz, 1H), 6.56 (brs, 1H), 6.36 (t, J =2.4Hz, 1H), 6.31 (d, J =3.0Hz, 1H), 4.62 (d, J =5.4Hz, 2H), 3.93 (brs, 2H). EI-MS: m/z 232 (M)+. HRMS (EI): calcd for C12H12N2O3 (M)+ 232.0848, found 232.0839. Anal. Calcd (found): C, 62.06 (61.89); H, 5.21 (4.93); N, 12.06 (12.37). Purity: system 1, 99.7% (method C, tR = 9.65 min); system 2, 97.0% (method G, tR = 7.77 min).
Homology modeling and molecular docking
Given the lack of a full length crystal structure of HIV-1 IN, a homology model was built based on the X-ray crystal structure of PFV intasome (PDB: 3OYA)27,30 a retro-lentivirus belonging to the same viral genus as HIV. For the generation of this model we used secondary structural alignment on the basis of 3D structures of PFV IN and HIV-1 IN.31 Prime v2.2 from Schrodinger, Inc was used for the modeling.32 As the nucleic acid residues in the binding sites (DC16 and DA17) are conserved we did not model DNA for HIV, instead we used PFV DNA and two Mg2+ ions in the binding site. The modeled protein was minimized using OPLS-2005 force filed within MacroModel.33 The structure of the modeled protein was validated by PROCHECK34 as 89.6 % residues fall within most favorable region and another 9.5% in additional allowed region. The RMSD value of the modeled protein with catalytic core domain crystal structure (PDB: 1BL3) was 3.18. HIV IN crystal structure (PDB: 3LPU) co-crystallized with a LEDGF/p75 inhibitor was used to dock compounds with LEDGF/p75 inhibitory activity. Compounds were docked into the binding site using Standard Precision method of Glide from Schrodinger, Inc28,29 with default values for all parameters. Prior to docking, compounds were minimized and all possible combinations of stereo isomers were generated by LigPrep from Schrodinger using OPLS-2005 force filed. Glide performed an exhaustive search of the positional, orientational, conformational space available to the docked ligand using a series of hierarchical filters and followed by energy optimization. Finally, the conformations were further refined via a Monte Carlo sampling that examines nearby torsional minima.29
Biological Materials, Chemicals and Enzymes
All compounds were dissolved in DMSO and the 10 mM stock solutions were stored at −20°C. Further dilutions were also performed in DMSO. The expression system used in purifying IN was a kind gift from Dr. Robert Craigie, Laboratory of Molecular Biology, NIDDK, NIH, Bethesda, MD. The oligonucleotides used in the HIV-1 IN catalytic activity assay were synthesized at the USC Norris Cancer Center microsequencing core facility. The γ [32P]-ATP was purchased from Perkin Elmer (Waltham, MA).
Preaparation of Oligonucleotide Substrate
The HIV-1 IN catalytic activity assay uses a 21′mer top strand: (5′-GTGTGGAAAATCTCTAGCAGT-3′), and a 21′mer bottom strand: (5′-ACTGCTAGAGATTTTCCACAC-3′). The top strand was labelled at the 5′end with γ [32P]-ATP by T4 polynucleotide kinase (Epicenter, Madison, WI). The mixture was then incubated at 95°C for 15 minutes to inactivate the kinase and the bottom strand was added in 1.5 molar excess. The strands were allowed to anneal by cooling the mixture slowly to room temperature. Any unincorporated material was subsequently removed by centrifuging the mixture through a Spin-25 mini-column (USA Scientific, Ocala, FL).
Integrase Catalytic Activity Assay
The extent of 3′-processing and strand transfer was analyzed by preincubating recombinant wild-type HIV-1 IN, at a final concentration of 200 nM, with the inhibitor in reaction buffer (50 mM NaCl, I mM HEPES, pH 7.5, 50 μM EDTA, 50 μM dithiothreitol, 10% glycerol (w/v), 7.5 mM MnCl2, 0.1 mg/mL bovine serum albumin, 10 mM 2-mercaptoethanol, 10% DMSO, and 25 mM MOPS, pH 7.2) at 30°C for 30 min. Then 20 nM of the 32P 5′end-labeled linear 21′mer substrate was added, and incubation was continued for an additional 1 h. Reactions were then quenched by the addition of an equal volume (16 μL) of loading dye (98% deionized formamide, 10 mM EDTA, 0.025% xylene cyanol, and 0.025% bromophenol blue). An aliquot (7 μL) was electrophoresed on a denaturing 20% polyacrylamide gel (0.09 M tris-borate pH 8.3, 2 mM EDTA, 20% acrylamide, 8 M urea).
Gels were dried, exposed in a PhosphorImager casstte, analyzed using a Typhoon 8610 Variable Mode Imager (Amersham Biosciences), and quantified using ImageQuant 5.2. The percent inhibition (%I) was calculated using the following equation: %I = 100 * [1 − (D − C)/(N− C)] where C, N, and D are the fractions of 21-mer substrate converted to 19-mer (3′-processing product) or strand transfer products for DNA alone, DNA plus IN, and IN plus drug, respectively. The IC50 values were determined by plotting the logarithm of drug concentration against percent inhibition of enzymatic activity to obtain the concentration that produced 50% inhibition.
LEDGF/p75–IN AlphaScreen assay
The AlphaScreen assay was performed according to the manufacturer’s protocol (Perkin–Elmer, Benelux). Reactions were performed in 25 μl final volume in 384-well Optiwell microtiter plates (Perkin-Elmer). The reaction buffer contained 25 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 0.01% (v/v) Tween-20 and 0.1% (w/v) bovine serum albumin. His6-tagged IN (300 nM final concentration) was incubated with the inhibitory compound for 30 min at 4°C. The compounds were added in varying concentrations spanning a wide range from 0.1 μM up to 100 μM. Afterwards, 100 nM Flag-LEDGF/p75 was added and incubation was prolonged for an additional hour at 4°C. Subsequently, 5 μl of Ni-chelate-coated acceptor beads and 5 μl anti-Flag donor beads were added to a final concentration of 20 μg/ml of both beads. Proteins and beads were incubated for 1 h at 30°C in order to allow association to occur. Exposure of the reaction to direct light was omitted as much as possible and the emission of light from the acceptor beads was measured in the EnVision plate reader (Perkin-Elmer) and analyzed using the EnVision manager software.
Cytotoxicity assays
(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), was used to assess cytotoxicity. H630 colon cancer cells were seeded in 96-well microtiter plates and allowed to attach overnight. Cells were subsequently treated with continuous exposure to corresponding drugs for 72 h. An MTT solution (final concentration of 0.5 mg/mL) was added to each well and cells were incubated for 4 h at 37°C. After removal of the supernatant, DMSO was added and the absorbance was read at 570 nm. The IC50 was then determined for each drug from a plot of log (drug concentration) versus percentage of cells killed.
Acknowledgments
The work in Y-Q L laboratory was supported by Science and Technology Commission of Shanghai Municipality (08JC1422200), National Natural Science Foundation of China (No. 81072527) and National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (Number: 2009ZX09301-001, 2009ZX09103-067). The work in NN laboratory was supported by funds from an NIH/NIAID (R21AI081610) grant.
References
- 1.Brown PO. Integration. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1997. p. 161. [PubMed] [Google Scholar]
- 2.Cowan JA. The Biological Chemistry of Magnesium. VCH; 1995. pp. 1–23. [Google Scholar]
- 3.Neamati N, Lin Z, Karki RG, Orr A, Cowansage K, Strumberg D, Pais GC, Voigt JH, Nicklaus MC, Winslow HE, Zhao H, Turpin JA, Yi J, Skalka AM, Burke TR, Jr, Pommier Y. J Med Chem. 2002;45:5661. doi: 10.1021/jm0201417. [DOI] [PubMed] [Google Scholar]
- 4.Johns BA, Svolto AC. Expert Opin Ther Patents. 2008;18:1225. [Google Scholar]
- 5.Dayam R, Al-Mawsawi LQ, Neamati N. Drugs R D. 2007;8:155. doi: 10.2165/00126839-200708030-00003. [DOI] [PubMed] [Google Scholar]
- 6.Evering TH, Markowitz M. Expert Opin Investig Drugs. 2008;17:413. doi: 10.1517/13543784.17.3.413. [DOI] [PubMed] [Google Scholar]
- 7.Nair V, Chi G. Rev Med Virol. 2007;17:277. doi: 10.1002/rmv.539. [DOI] [PubMed] [Google Scholar]
- 8.Ramkumar K, Serrao E, Odde S, Neamati N. Med Res Rev. 2010;30:890. doi: 10.1002/med.20194. [DOI] [PubMed] [Google Scholar]
- 9.Zhao H, Neamati N, Sunder S, Hong H, Wang S, Milne GW, Pommier Y, Burke TR., Jr J Med Chem. 1997;40:937. doi: 10.1021/jm960755+. [DOI] [PubMed] [Google Scholar]; (b) Neamati N, Hong H, Owen JM, Sunder S, Winslow HE, Christensen JL, Zhao H, Burke TR, Jr, Milne GW, Pommier Y. J Med Chem. 1998;41:3202. doi: 10.1021/jm9801760. [DOI] [PubMed] [Google Scholar]
- 10.Pommer Y, Marchand C, Neamati N. Antiviral Res. 2000;47:139. doi: 10.1016/s0166-3542(00)00112-1. [DOI] [PubMed] [Google Scholar]
- 11.Al-Mawasawi LQ, Dayam R, Their L, Witvrouw M, Debyser Z, Neamati N. Bioorg Med Chem Lett. 2007;17:6472. doi: 10.1016/j.bmcl.2007.09.102. [DOI] [PubMed] [Google Scholar]
- 12.Zhao XZ, Semenova EA, Vu BC, Maddali K, Marchand C, Hughes SH, Pommier Y, Burke TR., Jr J Med Chem. 2008;51:251. doi: 10.1021/jm070715d. [DOI] [PubMed] [Google Scholar]
- 13.Hazuda D, Iwamoto M, Wenning L. Annu Rev Pharmacol Toxicol. 2009;49:377. doi: 10.1146/annurev.pharmtox.011008.145553. [DOI] [PubMed] [Google Scholar]
- 14.Stanwell C, Ye B, Yuspa SH, Burke TR., Jr Biochem Pharmacol. 1996;52:475. doi: 10.1016/0006-2952(96)00250-x. [DOI] [PubMed] [Google Scholar]
- 15.Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-Simandle K, Ilina T, Clark AD, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold E. ACS Chem Biol. 2006;1:702. doi: 10.1021/cb600303y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovacic P. J Recept Signal Transduct. 2008;28:141. doi: 10.1080/10799890802084077. [DOI] [PubMed] [Google Scholar]
- 17.Al-Mawsawi LQ, Neamati N. ChemMedChem. 2011;6:228. doi: 10.1002/cmdc.201000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plewe MB, Butler SL, Dress KR, Hu Q, Johnson TW, Kuehler JE, Kuki A, Lam H, Liu W, Nowlin D, Peng Q, Rahavendran SV, Tanis SP, Tran KT, Wang H, Yang A, Zhang J. J Med Chem. 2009;52:7211. doi: 10.1021/jm900862n. [DOI] [PubMed] [Google Scholar]
- 19.Ciuff A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. Nat Med. 2005;11:1287. doi: 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- 20.Hombrouck A, De Rijck J, Hendrix J, Vandekerckhove L, Voet A, De Maeyer M, Witvrouw M, Engelborghs Y, Christ F, Gijsbers R, Debyser Z. PLoS Pathog. 2007;3:e47. doi: 10.1371/journal.ppat.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Rivera JA, Bueno MT, Morales E, Kugelman JR, Rodriguez DF, Llano M. J Virol. 2010;84:740. doi: 10.1128/JVI.01043-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christ F, Voet A, Marchand A, Nicole S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z. Nat Chem Biol. 2010;6:442. doi: 10.1038/nchembio.370. [DOI] [PubMed] [Google Scholar]
- 23.De Luca L, Barreca ML, Ferro S, Christ F, Iraci N, Gitto R, Monforte MM, Debyser Z, Chimirr A. ChemMedChem. 2009;4:1311. doi: 10.1002/cmdc.200900070. [DOI] [PubMed] [Google Scholar]
- 24.De Luca L, Ferro S, Gitto R, Barreca ML, Agnello S, Christ F, Debyser Z, Chimirri A. Bioorg Med Chem. 2010;18:7515. doi: 10.1016/j.bmc.2010.08.051. [DOI] [PubMed] [Google Scholar]
- 25.Du L, Zhao Y, Chen J, Yang L, Zheng Y, Tang Y, Shen X, Jiang H. Biochem Biophys Res Commun. 2008;375:139. doi: 10.1016/j.bbrc.2008.07.139. [DOI] [PubMed] [Google Scholar]
- 26.Cherepanov P, Ambrosio AL, Rahman S, Ellenberger T, Engelman A. Proc Natl Acad Sci USA. 2005;102:17308. doi: 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Nat. 2010;464:232. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. J Med Chem. 2004;47:1750. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 29.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 30.Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. Proc Natl Acad Sci USA. 2010;107:20057–20062. doi: 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krishnan L, Li X, Naraharisetty HL, Hare S, Cherepanov P, Engelman A. Proc Natl Acad Sci USA. 2010;107:15910. doi: 10.1073/pnas.1002346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prime, version 2.2. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 33.MacroModel, version 9.8. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 34.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Cryst. 1993;26:283. [Google Scholar]