

Abstract
Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most frequent cause of autosomal-dominant Parkinson’s disease (PD). The second known autosomal-dominant PD gene (SNCA) encodes α-synuclein, which is deposited in Lewy bodies, the neuropathological hallmark of PD. LRRK2 contains a kinase domain with homology to mitogen-activated protein kinase kinase kinases (MAPKKKs) and its activity has been suggested to be a key factor in LRRK2-associated PD. Here we investigated the role of LRRK2 in signal transduction pathways to identify putative PD-relevant downstream targets. Over-expression of wild-type [wt]LRRK2 in human embryonic kidney HEK293 cells selectively activated the extracellular signal-regulated kinase (ERK) module. PD-associated mutants G2019S and R1441C, but not kinase-dead LRRK2, induced ERK phosphorylation to the same extent as [wt]LRRK2, indicating that this effect is kinase-dependent. However, ERK activation by mutant R1441C and G2019S was significantly slower than that for [wt]LRRK2, despite similar levels of expression. Furthermore, induction of the ERK module by LRRK2 was associated to a small but significant induction of SNCA, which was suppressed by treatment with the selective MAPK/ERK kinase inhibitor U0126. This pathway linking the two dominant PD genes LRRK2 and SNCA may offer an interesting target for drug therapy in both familial and sporadic disease.
Keywords: LRRK2, Mitogen-activated protein kinases, ERK, α-Synuclein, Parkinson’s disease
1. Introduction
Parkinson’s disease (PD) is a chronic, progressive neurodegenerative disease clinically defined by the symptoms tremor, rigor and bradykinesia [1], and pathologically by a selective degeneration of dopaminergic neurons in the substantia nigra, together with the presence of Lewy bodies (LBs) and Lewy neurites [2]. To date, 13 disease-associated loci and 5 PD-causative genes have been identified, providing valuable insights into the molecular mechanisms of PD [3]. Whereas the genes PARKIN, PINK1 and DJ-1 cause autosomal-recessive early-onset PD, α-synuclein (SNCA) and leucine-rich repeat kinase 2 (LRRK2) are associated to autosomal-dominant PD.
SNCA [4] codes for a small presynaptic phosphoprotein with an intrinsic propensity to aggregate [2] that has been demonstrated to have a role in both inherited and idiopathic PD as major constituent of LBs [5]. The three described SNCA pathogenic mutations A53T, A30P and E46K increase SNCA fibril formation in vitro [6,7] in a seeding-dependent manner beyond a critical threshold [8]. This could be reached in patients for [wt]SNCA for example by up-regulation of SNCA expression due to genomic multiplications [9] or polymorphisms in promoter region [10,11].
The gene LRRK2 [12,13] encodes a large multi-domain (see Fig. 1A) ROCO protein [14] characterized by the presence of a Ras-of-Complex (Roc) domain with high similarity to small GTPases and a C-terminal-of-Roc (COR) domain involved in dimerization [15,16]. Additionally, LRRK2 has a second catalytic domain, a serine/threonine kinase with sequence homology to mitogen-activated protein kinase kinase kinases (MAPKKKs) [17]. These key signal transduction molecules are involved in a vast number of cellular functions such as proliferation, differentiation and survival and have been shown to play a crucial role in regulating neural functions [18,19].
Fig. 1.

Expression and distribution of LRRK2. (A) A LRRK2-specific rabbit polyclonal antibody was raised against the region 800–1000 amino acids (MID) between the ANK and LRR domains of human LRRK2. Location of the mutations used in the study is shown in red. R1441C in the COR domain and G2019S in the kinase domain are PD-associated mutations, whereas K1906N is a synthetic mutant with an impaired kinase activity. (B) Extracts of HEK293 cells transfected for 48 h with [wt]LRRK2 were run on a 6% polyacrylamide gel, immunoblotted with the antibody against LRRK2-MID (lower panel) and reprobed with an antibody against the HA tag (upper panel). LRRK2 protein is detected at approximately 280 kDa by both HA-specific and LRRK2-specific antibodies. The LRRK2 antibody detected also endogenous LRRK2 in extracts from mock-transfected cells. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
More than 50 LRRK2 variants have been identified in PD patients. To date R1441C/G, Y1699C, G2019S and I2020T have been proven to be pathogenic. Mutation G2019S has been regarded as the most common cause of dominant inherited as well as sporadic PD [20] and has been shown to significantly increase both auto- and MBP-phosphorylation in vitro compared to [wt]LRRK2 [21]. This effect together with the kinase-dependent toxicity of some of the mutants [22,23] suggests a mechanism of toxic gain-of-function, probably related to deregulation of LRRK2 kinase activity.
Increasing evidence indicates that the major pathology associated to mutations in LRRK2 and particularly to G2019S [13] is SNCA aggregation, pointing towards a causative link between LRRK2 mutations and SNCA pathology. However, it is unclear if LRRK2 directly binds to and phosphorylates SNCA [21,24].
MAPKKKs are enzymes often acting as initiators of signaling cascades that ultimately lead to transcriptional regulation [25]. Thus, we hypothesized a potential regulation of SNCA through a MAPK signaling cascade initiated by LRRK2. We systematically analyzed the activation state of the three MAPK modules (extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK), p38MAPK) after LRRK2 over-expression in HEK293 cells. Our results indicate that LRRK2 is a functional serine/threonine kinase that selectively activates the ERK pathway, but not the p38MAPK and JNK pathways, in a kinase-dependent manner. Interestingly, LRRK2 mutations R1441C and G2019S caused a significant delay of ERK activation. Activation of the ERK pathway by LRRK2 stimulated endogenous SNCA transcription, resulting in increased SNCA expression. Our results indicate a functional link between the two autosomal-dominant PD genes LRRK2 and SNCA through the activation of a specific kinase cascade that might be relevant in the development of new therapies for the treatment of both familial and sporadic PD.
2. Materials and methods
2.1. Vectors and constructs
The 7.58 kb LRRK2 gene was amplified with a high fidelity PCR System (Roche) by using four complementary fragments from human brain (BD Biosciences Clontech) and cerebellum cDNA libraries (Dr. K. Kaupmann, Novartis). Full-length cDNA was generated by subcloning the fragments in the mammalian expression vector pCI (Promega) using the three unique internal restriction sites AfeI, NdeI and ClaI. The C-terminal HA tag, as well as the missense mutations R1441C, K1906N and G2019S were inserted using the QuikChange™ site-directed mutagenesis kit (Stratagene) using wild-type plasmid pCI LRRK2 as template for the PCR reactions. The entire CMV promoter and coding sequences were verified by automated sequencing.
2.2. Cell culture and inhibitors
To generate LRRK2-inducible cells, 293-FT inducible Flp-In cells (Invitrogen) were used. Integration was achieved by transfection with WT, G2019S or R1441C LRRK2 in combination with the fip recombinase expressed from the pOG44 plasmid. 293-FT cells were selected based on Zeocin (100 μg/ml)/hygromycin (200 μg/ml) resistance. HEK293E cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum (PAA Laboratories), LRRK2-inducible cells additionally with penicillin/streptomycin, 2× non-essential amino acids (Gibco), 100 μg/ml hygromycin B and 15 μg/ml blasticidin.
Confluent cells were seeded in 6-well plates and transiently transfected with 1 μg of DNA per well using the lipofection reagent FuGene6 (Roche). Cells were lysed for 30 min at 4 °C in 100 μl lysis buffer (50 mM Tris–HCl pH=7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) with the protease inhibitor cocktail Complete Mini (Roche) and phosphatase inhibitors (NaF and Na3VO4). Protein concentration was determined by BCA assay (Pierce). LRRK2 expression in stable cell lines was induced by treatment with 1 μg/ml tetracycline.
To block the ERK–MAPK pathway, the specific phospho-MEK1/2 inhibitor U0126 (Cell Signaling) was used for 12 h prior to harvesting in a concentration of 10 μM.
2.3. Western blotting and antibodies
25 μg of cell lysate was loaded in 7.5% polyacrylamide gels and transferred onto PVDF membranes (Immobilon). Membrane was blocked for 1 h with 5% powdered milk/TBS-T (50 mM Tris pH=7.4, 150 mM NaCl, 0.1% Tween-20). Membranes were incubated overnight at 4 °C with primary antibodies diluted in 1% bovine serum albumin in TBS-T, followed by 3 washing steps for 10 min each one. Incubation with secondary antibodies diluted in 2% powdered milk/TBS-T was done for 1 h at room temperature.
All anti-MAPKs antibodies including ERK, p38MAPK and JNK, total and phosphorylated forms (Cell Signalling) were used at a concentration 1:2000. Primary antibodies anti-MID LRRK2 and anti-α-tubulin (Sigma-Aldrich) and secondary antibodies anti-mouse-HRP and anti-rabbit-HRP (Dako) were used at a concentration 1:15,000. The chemiluminescent substrate HRP Immobilon Western (Millipore) was used to visualize protein bands.
2.4. Quantitative real-time PCR (qRT-PCR)
Total mRNA was extracted with the RNeasy Kit (Qiagen), followed by reverse transcription with the Transcriptor First Strand cDNA Kit (Roche). The sequence between the exons 2 and 5 was amplified by qRT-PCR and the specific product quantified using the LightCycler 480 System (Roche). Detection of the product was done with highly specific fluorescent hybridization primers. The crossing-point (Cp) values were calculated by the second derivate method. A low copy number constitutive gene, the porphobilinogen deaminase (h-PBGD), was used as external standard and quantified with the h-PBGD Housekeeping Gene Set (Roche). Relative transcript levels were calculated as the ratio SNCA/PBGD, normalized to the expression level in the cells transfected with the empty vector.
2.5. ELISA assays
Quantification of endogenous SNCA protein was done using the Immunoassay Kit for human α-synuclein (Biosource), following manufacturer’s instructions. Cells were lysed for 30 min in specific lysis buffer (10 mM Tris–HCl pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% deoxycholate, and 1 mM PMSF).
2.6. Statistical analyses
All figures shown are representative of at least three independent experiments. Western blot data were quantified by ImageJ software. Data were analyzed as a change of percent pelleted from vehicle control conditions. Results are presented as mean±SEM. Statistical analyses were performed using Student’s t-test and ANOVA followed by Tukey’s post hoc testing.
3. Results
3.1. Expression and cellular distribution of LRRK2
LRRK2 expression was assessed by immunoblot 48 h after transfection of HEK293 cells with [wt]LRRK2/HA. Our rabbit polyclonal antibody directed against the middle (MID) region located between the ankyrin-like repeat and the leucine-rich repeat (LRR) domains of human LRRK2 (see Fig. 1A) [16] detected a strong band at approximately 280 kDa in transfected cells that was confirmed by reprobing the membrane with the antibody against the HA tag (Fig. 1B). We also detected a faint band in mock-transfected cells likely corresponding to endogenous LRRK2 (Fig. 1B). Western blot show no degradation products of LRRK2.
3.2. LRRK2 selectively activates the ERK pathway in a kinase-dependent manner
Because the kinase domain of LRRK2 shares homology to MAPKKK and these enzymes usually stimulate at least one of the three classical MAPK cascades, ERK, p38MAPK and JNK, we investigated putative modifications in the basal phosphorylation state of these pathways. HEK293E cells were transiently transfected for 48 h with [wt]LRRK2, mutant [G2019S]LRRK2 and the kinase-dead mutant [K1906N]LRRK2. Cell lysates were analyzed by immunoblot with antibodies recognizing only the activated dually phosphorylated form of the specific MAPK from each pathway. Over-expression of LRRK2 led to increased ERK1/2 phosphorylation, whereas no significant change was observed in the basal levels of phospho-p38MAPK and phospho-JNK (Fig. 2). Total protein levels of ERK1/2, p38MAPK and JNK remained unaltered. Moreover, ERK stimulation was dependent on LRRK2 kinase activity, since transfection with the kinase-dead LRRK2 mutant resulted in a low phospho-ERK1/2 signal. Although transfection with [K1906N] LRRK2 slightly increased phospho-ERK1/2 levels in HEK293 cells, they were stable over time. The most probable explanation for this effect is that this mutant is not completely catalytically dead, but may retain some residual kinase activity (Fig. 2). The mutant G2019S did not further enhance ERK1/2 phosphorylation in comparison to [wt]LRRK2 (Fig. 2).
Fig. 2.
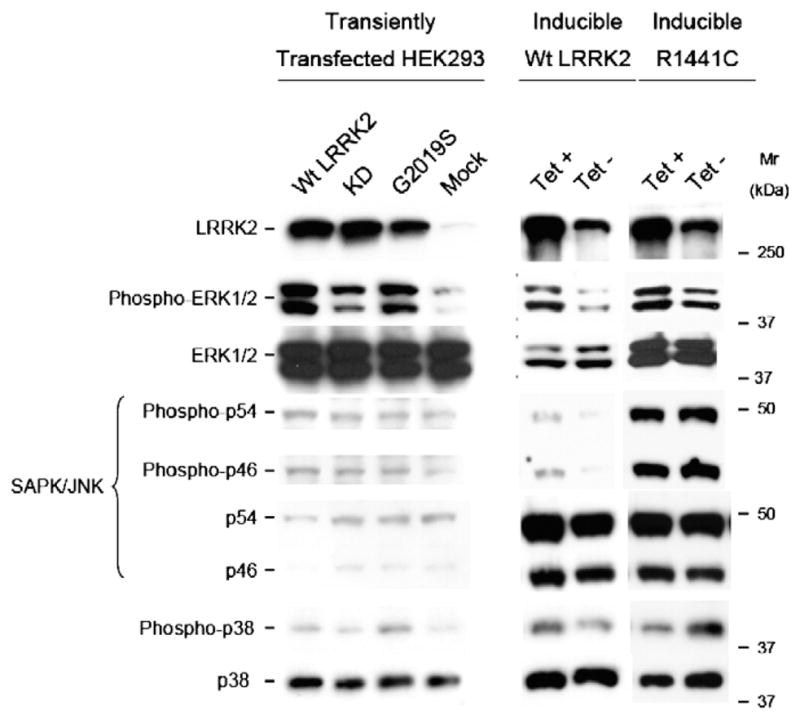
Activation of ERK1/2 in response to LRRK2 expression. 30 μg of protein lysate from LRRK2-inducible or HEK293 transiently transfected with wild-type, G2019S and kinase-dead LRRK2 cells was loaded on 7.5% polyacrylamide gels and subjected to immunoblot analysis using specific antibodies against the activated dually phosphorylated forms of ERK1/2, JNK and p38MAPK. Membranes were stripped and reprobed with antibodies against the total MAPK protein. Similar LRRK2 expression levels were assessed with the specific LRRK2 antibody. LRRK2 expression in inducible wild-type and R1441C cell lines was achieved by 48 h treatment with 1 μg/ml tetracycline. Immunoblots are representative from 3 independent experiments.
Stimulation of the ERK pathway by LRRK2 was further validated in two LRRK2-inducible Tet-On HEK293 cell lines expressing wild-type LRRK2 and the PD-associated R1441C mutant. As for transient transfection, tetracycline-induced expression of LRRK2 in cells led to augmented phospho-ERK1/2 levels compared to basal levels in non-treated cells. Although mild changes in the levels of phospho-p38MAPK and phospho-JNK were observed between induced and non-induced cells, they were not as clear and consistent as for ERK phosphorylation. No significant differences were observed in the total protein levels, apart from inducible R1441C cells that showed increased levels of total and phospho-JNK independently of LRRK2 expression (Fig. 2). Expression levels of LRRK2 in non-induced conditions were similar to those in untransfected HEK293E cells, whereas induction increased LRRK2 expression to the same extent as transient transfection (data not shown).
3.3. PD-associated LRRK2 mutations delay ERK activation
The duration and type of stimulus that activate the ERK cascade can lead to different activation profiles that might mediate different functions [26]. To investigate the kinetics of ERK activation by LRRK2 we treated [wt]LRRK2-inducible cell line with 1 μg/ml tetracycline for 0 to 48 h. Cell lysates were subjected to immunoblot and probed against phospho-ERK1/2. Phosphorylation of ERK1/2 peaked at 12 h after induction and remained elevated over time (Fig. 3). Since LRRK2 mutations did not alter intensity of ERK activation, we studied whether they influenced ERK temporal activation profile. R1441C inducible cells were treated with tetracycline for 0, 12, 24 and 48 h, and the cell lysates immunoblotted and probed against phospho-ERK1/2 and analyzed by band densitometry. Compared to [wt]LRRK2, mutant R1441C exhibited a significant delay of ERK1/2 phosphorylation (Fig. 3). Levels of active phospho-ERK1/2 after 12 h induction are about 60% higher in [wt]LRRK2-inducible cells than in R1441C mutants that only 48 h after induction were able to significantly phosphorylate ERK1/2 (relative intensity [RI]; wt_12h 1.611±0.254, n=6, vs wt_0h 0.996±0.171, n=5; p<0.05; wt_12h 1.611±0.254, n=6, vs R1441C_12h 1.011±0.084, n=7; p<0.05). Basal ERK1/2 phosphorylation levels were comparable in both cell lines.
Fig. 3.

Time course of ERK1/2 activation in inducible cell lines. [wt]LRRK2 and [R1441C] LRRK2 inducible HEK293 cells were treated with tetracycline for the indicated times and subjected to immunoblot analysis with antibodies against phospho- and total ERK1/2. Relative densitometry intensities for phospho-ERK1/2 immunoreactivity showed that over-expression of the PD-causative mutation R1441C led to a delayed activation of ERK1/2 compared to [wt]LRRK2. Each value is averaged from 4 independent experiments.
To determine whether the kinase mutant G2019S also led to a slower activation of ERK1/2 we transiently transfected G2019S LRRK2 into HEK293 cells. As in R1441C cell lines, phosphorylation of ERK1/2 by G2019S LRRK2 was also delayed to 48 h (Fig. 4A). Band densitometry analysis of phospho-ERK1/2 showed that, compared to wild-type LRRK2, the effect of mutant G2019S was even more robust than that of R1441C. Wild-type LRRK2 could significantly phosphorylate ERK1/2 already at 12 h (relative intensity [RI]; wt_12h 4.417± 0.876, n=4, vs Mock_12h 1.000±0.379, n=4; p<0.01), getting a maximum peak at 24 h with a 4-fold difference between wild-type and G2019S mutant (relative intensity [RI]; wt_24h 7.324±0.763, n=4, vs G2019S_24h 2.208±0.383, n=3; p<0.01). In contrast, transfection of HEK293 cells with the mutant G2019S only showed a significant increase in ERK1/2 phosphorylation after 48 h (relative intensity [RI]; G2019S 5.252±0.671, n=3, vs Mock 1.088±0.551, n=3; p<0.05), suggesting an impaired ability of this mutant to elicit a fast activation of the ERK cascade (Fig. 4B). Cells transfected with the kinase-dead version of LRRK2 showed levels of phospho-ERK1/2 similar to mock-transfected cells at all the different time points (Fig. 4B).
Fig. 4.

Delayed activation of ERK1/2 by LRRK2 mutant G2019S. (A) HEK293 cells were transfected with wild-type and G2019S mutant LRRK2 for 12, 24 and 48 h and subjected to immunoblot analysis. The samples were probed for activated phospho-ERK1/2 and total ERK1/2. (B) Relative band intensities for phospho-ERK1/2 immunoreactivity in wild-type, G2019S and kinase-dead LRRK2 at the indicated time points after transfection. Each value is averaged from 4 independent experiments. Mock = cells transfected with the empty vector pCI/HA.
3.4. PD-linked mutations delay phosphorylation of MEK2, upstream activator of ERK
We next sought to determine whether LRRK2 was also able to induce phosphorylation of the upstream MAPK/ERK kinases (MEK). HEK293 cells were transiently transfected with [wt]LRRK2 for 12, 24 and 48 h, immunoblotted and probed against phospho-MEK1/2 and phospho-ERK1/2. The HEK293 cells used here expressed predominantly MEK2. MEK1 protein in untransfected cells was not detectable (Fig. 5A). Mild phosphorylation of endogenous MEK2 was observed 12 h after wild-type, G2019S and kinase-dead mutant LRRK2 transfection, but with no significant difference compared to mock-transfected cells. At this time point, the relatively low MEK2 phosphorylation state compared to ERK1/2 might be due to different antibody sensitivities. Like ERK1/2, MEK2 phosphorylation after [wt] LRRK2 transfection reached a maximum peak at 24 h and remained elevated at 48 h (Fig. 5). Importantly, MEK2 phosphorylation via [G2019S]LRRK2 was delayed, exactly like ERK1/2 phosphorylation (Fig. 5). Transfection with MEK1 was used as a positive control for activation, showing that LRRK2 activation of the ERK pathway was comparable in intensity to that of the specific ERK1/2 activator MEK1 (Fig. 5A).
Fig. 5.

Delayed activation of the specific ERK1/2 upstream kinases MEK1/2. (A) HEK293 cells were transfected for 12, 24 and 48 h with the indicated LRRK2/HA constructs or with MEK1, non-treated or treated with the pharmacological inhibitor of the MEK/ERK kinase pathway U0126. Cell lysates were immunoblotted and probed for phospho- and total-MEK1/2 and for phospho- and total ERK1/2. (B) Relative band densitometry for phospho-MEK1/2 in transiently transfected HEK293 cells. Image is representative from 3 independent blots. Mock = cells transfected with the empty vector pCI/HA.
Treatment with the selective MEK1/2 inhibitor U0126 at the concentration of 10 μM for 12 h prior to harvesting efficiently prevented phosphorylation of both MEK and ERK (Fig. 5A) and was therefore used in subsequent experiments. Band densitometry analysis of at least 3 independent experiments allowed us to quantify MEK2 phosphorylation in cells transfected with the indicated LRRK2 constructs (Fig. 5B), showing again a 4-fold increase in phospho-MEK2 24 h after transfection with [wt]LRRK2 compared to basal MEK2 levels or to cells transfected with the mutant G2019S and the kinase-dead constructs (Fig. 5B) (relative intensity [RI]; wt_24h 14.915±3.388, n=3, vs G2019S_24h 4.677±1.689, n=3; p<0.05; wt_24h vs Mock_24h 1.00±0.600, n=4; p<0.01). The activation of MEK2 elicited by the mutant G2019S was delayed to 48 h (Fig. 5B), consistent with the activation observed in the downstream MAPK ERK1/2.
3.5. LRRK2 over-expression stimulates SNCA transcription in a kinase-dependent manner
To test the hypothesis of a putative transcriptional regulation of SNCA by LRRK2 we quantified endogenous levels of SNCA mRNA by real-time RT-PCR. Total mRNA was extracted 48 h after transfection of HEK293 cells with [wt]LRRK2, G2019S or the kinase-dead mutant K1906N. Over-expression of [wt]LRRK2 significantly increased the levels of endogenous SNCA mRNA about 2-fold compared to mock-transfected cells (relative quantity [RQ]; wt 1.756±0.136, n=23, vs Mock 1.000±0.2849, n=12; p<0.01) (Fig. 6). The kinase-dead mutant K1906N was not able to stimulate SNCA transcription over the basal levels, indicating that SNCA induction was dependent on LRRK2 kinase activity (Fig. 6). Transfection with the mutant G2019S up-regulated SNCA to approximately the same extent as [wt]LRRK2, but no further induction was reached. These results are in agreement with our observations of the activation in the ERK module.
Fig. 6.
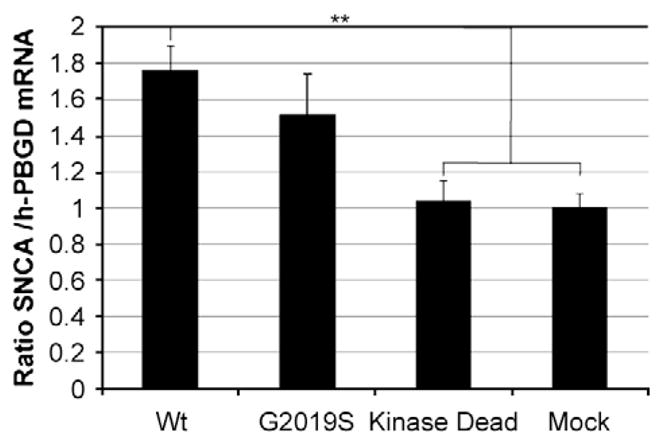
Up-regulation of SNCA by LRRK2. Endogenous mRNA levels of SNCA in HEK293 cells 48 h after transfection with the indicated LRRK2/HA constructs were determined by real-time qRT-PCR. Total RNA was purified on spin columns from cell lysates and quantified in the LightCycler with specific hybridization probes for SNCA. Results are given as the ratio between SNCA and the housekeeping gene h-PBGD.
Up-regulation of SNCA by LRRK2 was also consistently observed at the protein level by enzyme-linked immunosorbent assay (ELISA). Student’s t-test found that relative SNCA protein levels in cells transfected with [wt]LRRK2 are mildly but significantly increased compared to mock-transfected cells (relative quantity [RQ]; wt 109.127 ± 3.151, n = 11, vs Mock 100.000 ± 1.994, n = 10; p=0.0273), whereas ANOVA analysis only shows a trend towards increased SNCA levels (Table 1). Neither the mutant G2019S nor the kinase-dead LRRK2 constructs led to any significant induction of SNCA at the protein level. No significant differences in total protein were detected.
Table 1.
Protein up-regulation of SNCA by LRRK2. Expression of SNCA protein after 48 h transfection with wild-type, G2019S and kinase-dead LRRK2 constructs and with the empty vector (mock) was determined by a specific ELISA. Results are given as the ratio between SNCA and total protein.
| Construct | Average % | SD | SEM | n | p-value |
|---|---|---|---|---|---|
| wt | 109.1273 | 10.4509 | 3.151052 | 11 | 0.0273 |
| G2019S | 101.2743 | 17.5976 | 6.2217 | 8 | 0.8334 |
| K1906N(KD) | 105.9705 | 23.1845 | 8.1970 | 8 | 0.4443 |
| Mock | 100.0000 | 6.3068 | 1.9943 | 10 |
3.6. ERK activation mediates LRRK2-induced transcriptional up-regulation of SNCA
We showed that over-expressed LRRK2 is involved in transcriptional regulation of SNCA and, in parallel, was also able to stimulate the ERK pathway in HEK293 cells. It has been previously described that SNCA mRNA and protein levels are induced in PC12 cells after prolonged treatment with growth factors, being this signaling depending, at least in part, on the activation of the ERK pathway [27]. Therefore we studied whether ERK signaling was also mediating transcriptional regulation of SNCA by LRRK2. Total mRNA was extracted from HEK293 cells transfected with [wt]LRRK2 and treated or not with the inhibitor U0126 for 12 h prior harvesting.
Endogenous SNCA-specific mRNA was quantified by real-time RT-PCR. SNCA mRNA levels at 24 h were about 40% higher in cells transfected with [wt]LRRK2 than in mock-transfected cells, in agreement to observed MEK/ERK activation (ratio SNCA/housekeeping gene; wt_24h 1.430±0.193, n=4, vs Mock_12h 1.000±0.048, n=4; p<0.05) (Fig. 7). Furthermore, LRRK2-induced up-regulation of SNCA could be efficiently repressed by treatment with the pharmacological inhibitor of the ERK pathway U0126 (ratio SNCA/housekeeping gene; wt_24h 1.430±0.193, n=4, vs wt+U0126_24h 0.890±0.1208, n=3; p<0.05; wt_48h 1.760±0.136, n=3, vs wt+ U0126_48h 0.770±0.115, n=3; p<0.05) (Fig. 7), suggesting that in our system SNCA transcriptional regulation elicited by LRRK2 is also mediated by activation of the ERK/MAPK pathway. Treatment with the inhibitor U0126 alone did not induce any change in SNCA mRNA expression in mock-transfected cells (Fig. 7).
Fig. 7.

LRRK2-mediated induction of SNCA is repressed by the phospho-MEK1/2 inhibitor U0126. HEK293 cells transiently transfected with different LRRK2 constructs were treated with 10 μg/ml U0126 or vehicle (DMSO) for 12 h prior to harvesting. Total mRNA was extracted and SNCA-specific mRNA quantified by real-time RT-PCR to measure the induction levels of SNCA. Similar results were achieved in 3 independent experiments.
4. Discussion
SNCA deposition in LBs is both the neuropathological hallmark of PD, as well as the main pathological output of LRRK2-associated PD. However, no study up to date has been able to provide sufficient experimental evidence of how LRRK2 could affect this pathological process. The present study reveals a novel pathway that integrates the two dominant PD-causative genes LRRK2 and SNCA and might be of relevance for understanding the pathogenesis of PD. Here we show for the first time that LRRK2 is able to up-regulate SNCA transcription in HEK293 cells in a kinase-dependent manner via specific activation of the MAPK/ERK cascade. Furthermore, expression of the pathogenic LRRK2 mutants R1441C and G2019S led to a delayed activation and propagation of the signal. LRRK2 is a common cause of dominant parkinsonism, whereas SNCA, regarded as a rare cause of disease, plays a key role in both familial and sporadic disease as major component of LBs [5]. Hence, deciphering the pathway linking both genes would be highly valuable to understand the pathogenesis of PD and to identify new potential therapeutic targets.
LRRK2 has been recently reported to co-localize with early stages of aggregating SNCA in lower brainstem of PD and dementia with Lewy bodies (DLB) patients [28], suggesting that LRRK2 dysfunction might contribute to the early formation of LBs. The mechanisms involved in this process are, nevertheless, completely unknown. According to our results, these mechanisms might imply transcriptional up-regulation of SNCA by LRRK2, since over-expressed wild-type and G2019S LRRK2 stimulate by almost 2-fold endogenous SNCA transcription in transiently transfected HEK293 cells. These results are consistent with a previous report showing co-regulation of LRRK2 and SNCA mRNAs in rodent striatum [29], indicating the existence in vivo of common regulatory mechanisms for both genes.
Transcriptional stimulation of SNCA by LRRK2 was also translated into a mild but significant increase in SNCA protein expression. The inability of LRRK2 kinase-dead to elicit SNCA induction in our cell system indicates that this effect is kinase-dependent. Genetic regulation of SNCA expression levels is linked to the development of neuropathology, thus, induction of SNCA by LRRK2 points towards SNCA up-regulation as a potential pathogenic mechanism in patients with LRRK2-associated PD. For example, multiplications of the SNCA locus have been shown to cause familial PD [9] and SNCA over-expression has been suggested to contribute to the disease [10,11]. Thus, mutations affecting the kinase activity of LRRK2 could regulate SNCA protein levels, acting synergistically on the genetic variability of SNCA itself and modulating disease course or pathology.
Tracking down signaling from LRRK2 to SNCA might be crucial in order to understand LRRK2-associated pathogenesis and to further characterize this pathway. Since the kinase domain of LRRK2 shares structural homology with MAPKKKs, in this study we investigated the activation of any of the classical MAPK cascades ERK, p38MAPK or JNK by LRRK2. These pathways play a critical role in cell death and survival and have been repeatedly reported to be involved in different neurodegenerative diseases, including PD [30]. Here, we demonstrated that LRRK2 is a functional MAPKKK that initiates the ERK signaling cascade leading to the phosphorylation of both ERK1/2 and its upstream kinase MEK. Despite a potential cross-talk between LRRK2 and the ERK pathway as has been previously suggested in neuronal models [31,32], a precise characterization of the effect of LRRK2 on the cascade is still lacking. Surprisingly, in our study the mutant G2019S did not cause any further activation of ERK in comparison to wild-type LRRK2 as expected from an over-active kinase, but in agreement with our observation of similar SNCA induction levels by [wt]LRRK2 and [G2019S]LRRK2 expression. Many evidence in the literature show that, besides enzymatic activation, there are additional factors required for successful transmission of a specific signal in a MAPK cascade [33] such as interaction with scaffold proteins [34], cross-talk among MAPK pathways that can act in parallel or duration and intensity of the activation. While our findings of LRRK2 selectively activating the ERK module in transfected HEK293 cells are in accord with most studies also using neuronal cell lines [31,32,35], LRRK2 is capable of phosphorylating other module’s MAPKKs in vitro [36]. The JNK pathway was found the most activated MAPK module in lymphoblasts derived from [G2019S]LRRK2 PD patients [37]. Thus, cell-type specific differences in LRRK2 scaffolding and/or MAPK signaling crosstalks appear to fine-tune signal transduction by LRRK2 kinase.
Correlation between the duration of ERK activity and its downstream effects has been observed in several systems, including PC12 cells. In this model stimulation of the ERK pathway with different growth factors was shown to lead to different activation kinetics and downstream outputs. Whereas transient activation of the ERK cascade upon EGF stimulation promotes cell proliferation, sustained activation caused by stimulation with NGF has been shown to promote differentiation and neurite formation [38]. In addition, LRRK2 has been suggested to participate in neuritogenesis [32,39]. In this study we showed that the LRRK2 PD-associated mutations R1441C and specially G2019S delay ERK activation compared to the wild-type. Thus, delayed and/or shorter activation of ERK caused by mutations in LRRK2 may interfere in the process of neuronal differentiation, leading to altered neurite morphology. Indeed, treatment of differentiated SH-SY5Y cells with the MEK inhibitor U0126 has been reported to reduce LRRK2-neuritic shortening caused by expression of the LRRK2 mutant G2019S [32].
The ERK pathway has been previously reported to participate in SNCA transcriptional regulation in a PC12 cell model via a regulatory region lying within intron 1 of the SNCA gene [27], but no other PD-causative gene has ever been shown to be involved. We showed that LRRK2 effects on the ERK pathway and on SNCA regulation are related events. Activation of the ERK pathway mediates the induction of SNCA by LRRK2, since treatment with the specific inhibitor of the ERK pathway U0126 completely suppresses the transcriptional up-regulation of SNCA elicited by LRRK2.
5. Conclusion
Our results indicate a potential pathway linking LRRK2 and SNCA and contribute to clarify the means by which LRRK2 can specifically modify SNCA biology via activation of the ERK/MAPK cascade that ultimately leads to SNCA transcriptional up-regulation. Although future neuronal models are required to validate our findings, the literature discussed here provides good evidence that activation of the ERK/MAPK pathway and ERK-mediated regulation of SNCA also take place in neurons. This novel pathway opens a new window for the development of future therapeutic treatments for PD.
Acknowledgments
This work was supported by the Medical Faculty of the University of Tübingen (fortüne grant F.1313045), the Hertie Foundation, and the Helmholtz Alliance for Mental Health in an Aging Society.
Abbreviations
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun N-terminal kinase
- LB
Lewy body
- LRRK2
leucine-rich repeat kinase 2
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- PD
Parkinson’s disease
- SNCA
α-synuclein
Contributor Information
Iria Carballo-Carbajal, Email: icarballo@ir.vhebron.net.
Susanne Weber-Endress, Email: mail@susanne-weber.net.
Giorgio Rovelli, Email: giorgio.rovelli@novartis.com.
Diane Chan, Email: dchan.bu@gmail.com.
Benjamin Wolozin, Email: bwolozin@bu.edu.
Christian L. Klein, Email: christian.klein@encryption-web.de.
Nadja Patenge, Email: nadja.patenge@med.uni-rostock.de.
Thomas Gasser, Email: thomas.gasser@uni-tuebingen.de.
References
- 1.Dauer W, Przedborski S. Neuron. 2003;39(6):889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 2.Goedert M. Nat Rev Neurosci. 2001;2(7):492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 3.Biskup S, Gerlach M, Kupsch A, Reichmann H, Riederer P, Vieregge P, Wullner U, Gasser T. J Neurol. 2008;255(Suppl 5):8–17. doi: 10.1007/s00415-008-5005-2. [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassia-dou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 5.Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 6.Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. J Biol Chem. 2005;280(9):7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- 7.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, Citron M. J Biol Chem. 1999;274(14):9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 8.Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. J Biol Chem. 1999;274(28):19509–19512. doi: 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]
- 9.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 10.Chiba-Falek O, Nussbaum RL. Hum Mol Genet. 2001;10(26):3101–3109. doi: 10.1093/hmg/10.26.3101. [DOI] [PubMed] [Google Scholar]
- 11.Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Krüger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven C. JAMA. 2006;296(6):661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- 12.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood AB, Singleton AB. Neuron. 2004;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 13.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Neuron. 2004;44(4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Bosgraaf L, Van Haastert PJ. Biochim Biophys Acta. 2003;1643(1–3):5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Greggio E, Zambrano I, Kaganovich A, Beilina A, Taymans JM, Daniels V, Lewis P, Jain S, Ding J, Syed A, Thomas KJ, Baekelandt V, Cookson MR. J Biol Chem. 2008;283(24):16906–16914. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein CL, Rovelli G, Springer W, Schall C, Gasser T, Kahle PJ. J Neurochem. 2009;111(3):703–715. doi: 10.1111/j.1471-4159.2009.06358.x. [DOI] [PubMed] [Google Scholar]
- 17.Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. Trends Neurosci. 2006;29(5):286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Miloso M, Scuteri A, Foudah D, Tredici G. Curr Med Chem. 2008;15(6):538–548. doi: 10.2174/092986708783769731. [DOI] [PubMed] [Google Scholar]
- 19.Sweatt JD. Curr Opin Neurobiol. 2004;14(3):311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, Lynch T, Bhatia KP, Gasser T, Lees AJ, Wood NW. Lancet Neurol. 2008;7(7):583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Proc Natl Acad Sci USA. 2005;102(46):16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Neurobiol Dis. 2006;23(2):329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Nat Neurosci. 2006;9(10):1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 24.Qing H, Wong W, McGeer EG, McGeer PL. Biochem Biophys Res Commun. 2009;387(1):149–152. doi: 10.1016/j.bbrc.2009.06.142. [DOI] [PubMed] [Google Scholar]
- 25.Yang SH, Sharrocks AD, Whitmarsh AJ. Gene. 2003;320:3–21. doi: 10.1016/s0378-1119(03)00816-3. [DOI] [PubMed] [Google Scholar]
- 26.Pouyssegur J, Volmat V, Lenormand P. Biochem Pharmacol. 2002;64(5–6):755–763. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- 27.Clough RL, Stefanis L. FASEB J. 2007;21(2):596–607. doi: 10.1096/fj.06-7111com. [DOI] [PubMed] [Google Scholar]
- 28.Alegre-Abarrategui J, Ansorge O, Esiri M, Wade-Martins R. Neuropathol Appl Neurobiol. 2008;34(3):272–283. doi: 10.1111/j.1365-2990.2007.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westerlund M, Ran C, Borgkvist A, Sterky FH, Lindqvist E, Lundstromer K, Pernold K, Brene S, Kallunki P, Fisone G, Olson L, Galter D. Mol Cell Neurosci. 2008;39(4):586–591. doi: 10.1016/j.mcn.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 30.Silva RM, Kuan CY, Rakic P, Burke RE. Mov Disord. 2005;20(6):653–664. doi: 10.1002/mds.20390. [DOI] [PubMed] [Google Scholar]
- 31.Liou AK, Leak RK, Li L, Zigmond MJ. Neurobiol Dis. 2008;32(1):116–124. doi: 10.1016/j.nbd.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plowey ED, Cherra SJ, III, Liu YJ, Chu CT. J Neurochem. 2008;105(3):1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaul YD, Seger R. Biochim Biophys Acta. 2007;1773(8):1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Schaeffer HJ, Weber MJ. Mol Cell Biol. 1999;19(4):2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heo HY, Park JM, Kim CH, Han BS, Kim KS, Seol W. Exp Cell Res. 2009 doi: 10.1016/j.yexcr.2009.09.014. Electronic publication ahead of print. [DOI] [PubMed] [Google Scholar]
- 36.Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O’Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. Hum Mol Genet. 2006;15(2):223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- 37.White LR, Toft M, Kvam SN, Farrer MJ, Aasly JO. J Neurosci Res. 2007;85(6):1288–1294. doi: 10.1002/jnr.21240. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen TT, Scimeca JC, Filloux C, Peraldi P, Carpentier JL, Van Obberghen E. J Biol Chem. 1993;268(13):9803–9810. [PubMed] [Google Scholar]
- 39.MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. Neuron. 2006;52(4):587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]