

Abstract
TGX-221 is a highly potent phosphoinositide 3-kinases β (PI3Kβ) inhibitor that holds great promise as a novel chemotherapeutic agent to treat prostate cancer. However, poor solubility and lack of targetability limit its therapeutic applications. The objective of this present study is to develop a peptide-drug conjugate to specifically deliver TGX-221 to HER2 over-expressing prostate cancer cells. Four TGX-221 derivatives with added hydroxyl groups were synthesized for peptide conjugation. Among them, TGX-D1 exhibited a similar bioactivity to TGX-221, and it was selected for conjugation with a peptide promoiety containing a HER2-targeting ligand and a prostate specific antigen (PSA) substrate linkage. From this selection, the peptide-drug conjugate was proven to be gradually cleaved by PSA to release TGX-D1. Cellular uptake of the peptide-drug conjugate was significantly higher in prostate cancer cells compared to the parent drug. Moreover, both the peptide-drug conjugate and its cleaved products demonstrated comparable activities as the parent drug TGX-D1. Our results suggest that this peptide-drug conjugate may provide a promising chemotherapy for prostate cancer patients.
Keywords: TGX-221, prostate cancer, prodrug, peptide ligand, HER2, PSA
INTRODUCTION
Phosphoinositide 3-kinases (PI3Ks) are lipid enzymes that phosphorylate the 3′-OH of the inositol ring of phosphoinositides. It has been established that PI3Ks play a pivotal role in cell growth, proliferation, and survival,1 and PI3Ks have been divided into three classes (I, II, and III) according to their different structural features and in vitro lipid substrate specificities.2, 3 Interestingly, class I PI3Ks can be further divided into two subclasses (class IA p110α/β/δ and class IB p110γ).1 Recent emerging evidence indicates that PI3K isoforms (p110α/β/δ) possess an oncogenic potential through gain-of-function mutations or gene amplifications.4 Their major PI3K downstream signaling effector, protein kinase B (PKB, also known as Akt), has been found to be over-activated in human cancers, such as prostate cancer.5–7 However, a negative regulator of the PI3K signaling pathway, phosphatase and tensin homolog (PTEN), has long been reported to be inactive in most human cancers, including prostate cancer.8–10 Conditional knockout of PTEN in mouse prostate epithelium resulted in intraepithelial neoplasia.11, 12 PTEN-null cancers represent a significant portion (30~50%) of all prostate cancer cases,13 and they are always associated with an unfavorable prognosis.14 In PTEN-null prostate cancers, PI3Kβ (PI3K-p110β), rather than PI3Kα, has been proven to contribute to tumoregenesis and androgen-independent progression.15 Recently, for the first time, we have demonstrated that PI3Kβ is essential for the androgen-stimulated AR (androgen receptor) transactivation and cell proliferation.16 We also observed high expression of this isoform in malignant prostate tissues compared to nonmalignant compartments, and the expression level was significantly correlated with prostate cancer progression.16 The chemical inhibition or genetic knockout of PI3Kβ is believed to prevent AR-dependent gene expression and tumor growth. Given all of the evidence described above, PI3Kβ is considered an attractive target for the development of novel anti-cancer agents for prostate cancer, which is the most common malignance in American men.
TGX-221 is a novel, potent, isoform-specific, and cell-permeable small molecule inhibitor of PI3Kβ. The inhibition effect of TGX-221 on PI3Kβ is about 1000-fold over PI3Kα and PI3Kγ, and 20-fold over PI3Kδ.17 TGX-221 has been successfully used in animal models to suppress the PI3Kβ activity in vivo.18 However, the therapeutic potential of TGX-221 in cancer therapy is halted due to its poor solubility and the wide distribution of PI3Kβ in vivo. Organic solvent is generally required for intravenous administration of TGX-221, which may cause significant toxicities.18 Additionally, widely distributed PI3Kβ plays important roles in cell metabolism, growth and development.19 For example, knockdown of PI3Kβ leads to early embryonic lethality,20 glucose intolerance15 and platelet disaggregation.21 As a result, an improved solubility and the targeted delivery of TGX-221 to tumor cells are two essential prerequisites for its successful application in prostate cancer therapy.
Peptide-drug conjugation is a promising strategy for the targeted delivery of chemotherapy drugs. Unlike an antibody-drug conjugate, a peptide-drug conjugate is small in size, which results in excellent cell permeability and a high drug-loading capability. In our approach, a HER2 (Human Epidermal growth factor Receptor 2)-specific peptide was conjugated to TGX-D1, our TGX-221 derivative, and the drug was specifically delivered to the prostate cancer cells. HER2, a well-known membrane receptor in breast cancer, is also overexpressed in many prostate cancers.22 It has been revealed that 25% of untreated primary prostate tumors, 59% of localized tumors after hormone treatment, and 78% of castrate metastatic tumors overexpress the HER2 protein.23–25 Although the monoclonal anti-HER2 antibody has not shown a significant therapeutic effect in prostate cancer,26 the overexpressed HER2 on prostate cancers does provide a promising molecular target for targeted drug delivery systems. To further increase the tumor specificity, a PSA (prostate specific antigen) cleavable peptide (SSKYQ) was incorporated as a linker between the anti-HER2 peptide and TGX-D1. PSA is a prostate tissue-specific serine protease that is often over-expressed in prostate cancer cells, but not in other normal cells.27 In this study, the peptide conjugated TGX-D1 was specifically delivered to prostate cancer cells, and then cleaved by PSA to release the parent drug, which led to high cellular uptake of TGX-D1 by tumor cells.
EXPERIMENTAL SECTION
Materials
All starting reagents listed below were obtained from commercial sources and used without further purification. 2-amino-3-bromo-5-methylpyridine was purchased from Oakwood Products, Inc. (West Columbia, SC). Piperidine, Malonyl dichloride, methanesulfonyl chloride, aniline, 4-aminophenethyl alcohol, 2-aminophenethyl alcohol, 3-aminobenzyl alcohol and butyl vinyl ether were obtained from Sigma-Aldrich (St Louis, MO). Morpholine was purchased from Ricca chemical company (Arlington, Texas). Triisopropylsilane (TIPS), Trifluroacetic acd (TFA), Triethylamine, N, N-diisopropylethylamine (DIPEA) 4-dimethyl-aminopyridine (DMAP) and dichloro 1,1′-bis(diphenylphosphino)ferrocene palladium (PdCl2(dppf)) were purchased from Acros Organics (Morris Plains, NJ). 2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) was ordered from Genscript Inc. (Piscataway, NJ). Thiazolyl Blue was obtained from RPI Corp. (Prospect, IL). All the amino acids used in solid phase peptide synthesis were obtained from Anaspec Inc. (Fremont, CA). All the organic solvents for synthesis and HPLC were ordered from Fisher Scientific Inc. and used as received.
Synthesis of TGX-221, TGX-D2, TGX-D3 and TGX-D4
Compound 2, 3, and 4 were synthesized from commercially available 2-amino-3-bromo-5-methylpyridine (1) following Jackson’s report.28 For the synthesis of TGX-221 and its derivatives, compound 4 (1 mmol) was suspended in 20 ml of dry toluene. After adding 3 g of molecular sieves (4Å) and 3 mmol of aniline or its derivatives, the reaction was refluxed for 4 hours under nitrogen. The molecular sieves were removed, and the filtrate was concentrated under vacuum. The residue was re-dissolved in methanol, followed by adding 20mg of NaBH4 at 0°C. The reaction was continuously stirred for another hour at room temperature. After quenching the reaction with 10ml of saturated NH4Cl aqueous solution, the product was extracted with CHCl3. The organic phase was then washed with brine, dried and concentrated under vacuum. The final product was purified by silica gel chromatography (CHCl3/methanol, 50/1) to give a yield of 50~80%.
TGX-221
1H NMR (400 MHz, CDCl3) δ8.62 (s, 1H), 7.61(s, 1H), 7.13(m, 2H), 6.68(t, 1H), 6.45(d, 2H), 5.63(s, 1H), 5.15(q, 1H), 3.80(m, 4H), 3.65(m, 5H), 2.25(s, 3H), 1.54(d, 3H). ESI-MS calcd. for C21H24N4O2 364.4; found, 365.4.
TGX-D2
1H NMR (400 MHz, CDCl3) δ8.63(s, 1H), 7.57(s, 1H), 7.08(d, 1H), 7.00(t, 1H), 6.68(t, 1H), 6.25(d, 1H), 5.65(s, 1H), 5.18(m, 1H), 3.80(m, 6H), 3.65(m, 6H), 2.63(s, 1H), 1.6(d, 3H). ESI-MS calcd. for C23H28N4O3 408.5; found 409.3.
TGX-D3
1H NMR (400 MHz, CDCl3) δ8.65 (s, 1H), δ7.57 (s, 1H), δ7.09 (t, 1H), δ6.68 (d, 1H), δ6.55 (s, 1H), δ6.34 (d, 1H), δ5.66 (s, 1H), δ5.12 (m, 1H), δ3.93 (m, 1H), δ3.79 (m, 4H), δ6.67 (m, 4H), δ3.60 (m, 2H), δ3.27 (s, 3H), δ1.50 (d, 3H). ESI-MS calcd. for C22H26N4O3 394.5; found, 395.3.
TGX-D4
1H NMR (400 MHz, CDCl3) δ8.65(s, 1H), 7.60(s,1H), 6.94(d, 2H), 6.41(d, 2H), 5.63(s, 1H), 5.09(q, 1H), 3.80(m, 6H), 3.65(m, 4H), 2.71(t, 2H), 2.30(s, 3H), 1.58(d, 3H). ESI-MS calcd. for C23H28N4O3 408.5; found, 409.4.
Synthesis of compound 5
Methyl bromoacetate (5 mmol) and N,N-diisopropylethylamine (5 mmol) were added into a solution of TGX-221 (364 mg, 1 mmol) in CH3CN (50 ml), and the reaction mixture was stirred at 85°C for 24 hours. After cooling to room temperature, the reaction mixture was concentrated under vacuum. The remaining brown residue was dissolved with CHCl3 and then washed with water, brine and dried over Na2SO4. After concentration, the residue was purified by silica gel chromatography (chloroform/methanol, 100/1, yield ≈ 60%). 1H NMR (400 MHz, CDCl3) δ8.65(s, 1H), 7.58(s, 1H), 7.10(t, 2H), 6.70(t, 1H), 6.46(d, 2H), 5.64(s, 1H), 5.13(q, 1H), 4.20(s, 3H), 3.80(m, 4H), 3.65(m, 6H), 2.25(s, 3H), 1.57(d, 3H). ESI-MS calcd. for C24H28N4O4 436.5; found 437.3.
Synthesis of TGX-D1
LiCl (5 mmol) and NaBH4 (5mmol) were added to a solution of compound 5 (1 mmol) in THF (10 ml). After stirring at room temperature for 10 minutes, 20 ml of anhydrous ethanol was added to the reaction mixture. After twelve hours, an additional 5 mmol of LiCl and NaBH4 were added to the reaction mixture and stirred for another 12 hours. The reaction was quenched with saturated NH4Cl aqueous solution and then extracted with CHCl3. The organic layer was washed with water, brine, and dried over Na2SO4. After concentration, the residue was purified by silica gel chromatography (chloroform/methanol, 50/1, yield ≈ 50%). 1H NMR (400 MHz, CDCl3) δ8.64 (s, 1H), δ7.43 (s, 1H), δ7.20 (t, 2H), δ6.86 (d, 2H), δ6.76 (t, 1H), δ5.60 (s, 1H), δ3.81 (t, 1H), δ3.60 (m, 5H), δ3.51 (m, 2H), δ3.42 (t, 2H), δ3.37 (m, 2H), δ2.33 (s, 3H), δ1.63 (d, 2H). ESI-MS calcd. for C23H28N4O3 408.5; found 409.5.
Synthesis of NH2-L-TGX
Fmoc-Leu-OH (100 mg) and EDCI (50 mg) were added to TGX-D1 (1mmol) in dichloromethane (DCM), and the reaction was initiated by adding DMAP and stirring at room temperature overnight. After that time, water was added to the reaction, and the organic phase was separated, washed, and dried with Na2SO4. The intermediate was purified by silica gel chromatography (CHCl3/methanol, 100/1). To remove the Fmoc protecting groups, the intermediate was dissolved in 1.8 ml DCM, followed by adding 200 μl of piperidine. The reaction mixture was stirred at room temperature for 30 minutes. Finally, the solvent was evaporated under vacuum, and the residue was purified with silica gel chromatography (CHCl3/ethanol, 50/2.5). ESI-MS calcd. for C29H39N5O4 521.6; found 522.4.
Synthesis of NH2-SL-TGX
HATU (10 mg) and Boc-Ser(OtBu)-OH (20 mg) were dissolved in 5 ml of dried DMF under nitrogen, followed by adding 10 μl of DIPEA. After 10 minutes, NH2-L-TGX was added to initiate the coupling reaction. After 12 hours, the organic solvent was evaporated and the residue was purified by silica gel chromatography. The purified intermediate was then dissolved in the mixture of TFA/DCM/TIPS (v/v/v, 50/45/5). The mixture was stirred for 3 hours, followed by concentration and purification with HPLC (Abs. 268nm). ESI-MS calcd. for C32H44N6O6 608.7; found 609.6.
Synthesis of the peptide-drug conjugate
Undeprotected peptide synthesis
All undeprotected peptide intermediates were synthesized by an Fmoc solid-phase peptide synthesis method. The resin used for peptide synthesis was 2-chlorotrityl resin. HATU was used as the coupling reagent and DIPEA served as the base. The undeprotected peptides were cleaved from the resin by acetic acid/trifluoroethanol/DCM (v/v/v, 10/20/70) at room temperature after 2 hours. After removing the resin by filtration, the reaction was concentrated under vacuum at room temperature. The crude products were purified by small silica gel chromatography (CHCl3/Methanol, 50/2.5~50/10).
Coupling and deprotection
The un-deprotected peptides (0.02 mmol) and purified NH2-L-TGX-D1 (0.01 mmol) were dissolved in 3ml of dried DMF. Two equivalents of HATU (0.02 mmol) and four equivalents of DIPEA (0.04 mmol) were added to the solution. After stirring at room temperature overnight, the reaction was quenched by a saturated NH4Cl aqueous solution. The solvent was then removed, and the brown residue was reconstituted in 20ml of CHCl3, followed by washing with water. After concentration under vacuum, the crude product was purified by silica gel chromatography (CHCl3/methanol, 50/2.5~50/10). The residual protecting groups in the peptide drug conjugate were removed by acidic cleavage. Briefly, the purified compound was dissolved in a mixture of DCM and TFA (1ml/1ml) and stirred for 2 hours at room temperature. To increase the yield, 50 μl of scavenging agent such as triisopropylsilane (TIPS) was added into the reaction. The reaction mixture was then concentrated, and the final product was purified by reverse phase HPLC. The molecular weight was confirmed by LC/MS. Ac-SSKYQSL-TGX: ESI-MS calcd. for C60H85N13O16 1244.4; found 1245.2. Ac-KCCYSLGGGSSKYQSL-TGX-D1 (KCC-TGX): ESI-MS calcd. for C96H141N23O27S2 2113.4; found 705.5 (M+3)3+; 1056.4 (M+2)2+.
Cytotoxicity assay
LNCaP cells were seeded in 96-well plates at a density of 1×104 cells/well (DU145 cells were seeded at a density of 3×103 cells/well due to its fast growth rate). After 24 hours, TGX-221 and its derivatives (TGX-D1, TGX-D2, TGX-D3 and TGX-D4) were added, and the cells were incubated at 37°C for 72 hours. The relative cell numbers were determined using a MTT assay. IC50 was calculated by fitting a concentration-response curve using Graphpad Prism 5 (GraphPad Software, Inc.).
Androgen induced proliferation assay
LNCaP cells were seeded in 96-well plates 12 hours before the assay. The medium was changed to serum-free medium containing 1nM of R1881 (a synthetic androgen), followed by adding TGX-221 and its derivatives. After incubation for 72 hours, the proliferation rate was examined by the Alamar Blue-based proliferation assay (Promega Corp., WI).
Androgen induced gene expression assay
The luciferase reporter plasmid controlled by a synthetic androgen response element (ARE-LUC) has been described previously.16 LNCaP cells were seeded in 6-well plates and transfected with 1.0μg of ARE-LUC using Lipofectamine on the following day. After 24 hours, the medium was changed to RPMI-1640 containing 2% FBS and R1881 (1.0nM). TGX-221 and TGX-D1 were added to the cells and incubated for 24 hours. After washing with PBS, the cells were lysed, and the total protein concentration of the cell lysate was measured by a BCA protein assay (Pierce Biotech., IL). Luciferase activity of each sample was determined and normalized to the protein concentration.
In vitro release of TGX-D1 from the peptide-drug conjugate
Drug release from NH2-SL-TGX
Stock solution of NH2-SL-TGX (10 mM in DMSO, 0.5% formic acid) was diluted to a final concentration of 100 μM with buffer (50 mM Tri-Cl, 100 mM NaCl, pH 7.4) and then incubated at 37°C. At selected time intervals, a 50-μl aliquot was removed and mixed with 50 μl of 0.1% TFA to quench the reaction. The samples were stored at −30°C for HPLC analysis.
Drug release from Ac-SSKYQSL-TGX
Ac-SSKYQSL-TGX was mixed with freshly prepared PSA solution (10Rg/ml) in buffer (50 mM Tri-Cl, 100mM NaCl, pH 7.4) to give a final concentration of 100 μM. The mixture was incubated at 37°C, and 50-μl aliquot were quenched with an equal volume of 0.1% TFA in methanol at selected time intervals. After incubation at room temperature for 10 minutes, the samples were centrifuged at 12,000g for 10 minutes, and the supernatant was collected for HPLC analysis.
The samples were assayed by a Waters HPLC system (717 plus autosampler, 600 controller pump, 486 tunable absorbance detector) on a C-18 reverse phase column (4.6×250mm, 5Rm). The mobile phase was methanol/water (65/35, v/v) in 0.1% formic acid, and the flow rate was set at 1.0 ml/min. Samples were tested as 30-μl injections, and the drug concentration was monitored by UV detector at 268nm.
Immunostaining
LNCaP cells cultured on 96-well plates were fixed in ice-cold acetone/methanol (1/1, v/v) for 10 minutes, followed by blocking with 1% BSA for 1 hour. The cells were then incubated with the biotinylated peptides for 1 hour in TBST buffer (0.1% Tween-20 in 0.1M Tri-Cl, pH 7.4). After washing to remove the unbound peptides, streptavidin-FITC (Pierce, Rockford, IL) was added, and the cells were examined with a Leica DMI3000 fluorescence microscope (Leica Microsystems GmbH, Wetzlar, Germany).
Aqueous solubility assay
The aqueous solubilities of TGX-221, TGX-D1 and KCC-TGX were assayed by the thermodynamic method. Briefly, 1 mg of TGX-221, TGX-D1, or KCC-TGX was suspended in 100 μl of water, and the mixture was shaken at room temperature for 3 days. The un-dissolved drug crystals were removed by centrifugation at 12,000g for 10 minutes. The supernatant was diluted, and the drug concentration was quantified by HPLC.
Stability assay of KCC-TGX
The stability of KCC-TGX was analyzed in PBS, cell culture medium and mouse serum. KCC-TGX was diluted to 5 μM in PBS (pH 7.5), cell culture medium (10% FBS in RMPI 1640) or 50% mouse serum. Next, the solutions were incubated at 37°C for their indicated time interval. A 20-μl aliquot was mixed with 60μl of acetonitrile containing 0.1% formic acid to precipitate the proteins. The samples were centrifuged at 12,000g for 10 minutes, and 50μl of the supernatant was collected for HPLC quantification.
Cellular uptake study
LNCaP cells were plated in 96-well plates at a density of 2×104 cells/well and incubated for 12 hours before the study. The cells were washed once with PBS and incubated with freshly prepared drug solutions in RPMI-1640 medium at 37°C. One hour after the incubation, the drug was removed and the cells were washed three times with cold PBS. The cells were then harvested and kept at −80°C for the LC-MS/MS assay. The total protein concentration of the cell lysate was quantified by a BCA protein assay kit (Pierce, Rockford, IL), and the cellular uptake was normalized to the total amount of protein for each sample.
In vitro competitive receptor binding assay
To inhibit the uptake of KCC-TGX by the anti-HER2 ligand Ac-KCCYSL, LNCaP cells were pre-incubated with the indicated concentrations of Ac-KCCYSL at 37°C for 1 hour. KCC-TGX was then added and incubated with the cells for another 30 minutes. The cells were finally washed and lysed as described above. Cellular uptake of KCC-TGX was analyzed by LC-MS/MS.
LC-MS/MS analysis of TGX-221 derivatives and the peptide-drug conjugate
Drug molecules were extracted from the cells using a liquid-liquid extraction technique as reported.29 TGX-221 was used as the internal standard during the extraction and analysis. Ethyl acetate (AcOEt) was utilized to extract the drug from the aqueous layer. All samples were hydrolyzed to release the parent drug TGX-D1 for LC-MS/MS analysis. Briefly, the cells were lysed by three cycles of a freeze-thaw procedure, and 100 μl of the clear cell suspension was incubated with 34 μl of 4N NaOH for 1 hour. Phosphate buffer (68 μl, 2N) was then added to neutralize the pH. After adding the internal standard TGX-221, each sample was vortexed with 200 μl of AcOEt for 5 minutes. The organic layer was separated from the aqueous layer by centrifugation, and 150 μl of the organic layer were transferred to a tube and dried under vacuum. The residue was reconstituted with 50 μl of acetonitrile for LC-MS/MS analysis.
LC-MS/MS analysis was performed on a QTrap® 3200 LC/MS/MS mass spectrometer equipped with a Shimadzu Prominence HPLC system. Detection was operated in the multiple-reaction monitoring (MRM) mode. The precursor and product ions were 409.1/272.1 for TGX-D1, and 365.1/272.1 for TGX-221. The turbo ion spray setting and collision gas pressure were optimized (IS Voltage of 5500V; temperature of 300 °C; nebulizer gas of 40 psi; curtain gas of 40 psi). Ion source parameters employed a declustering potential (DP) of 31 V; collision energy (CE) of 16 V; entrance potential (EP) of 5 V; and collision cell exit potential (CXP) of 4V. The detection limit of TGX-D1was 10 nM.
Statistical Analysis
Data were expressed as the mean ± standard deviation (SD). Difference between any two groups was determined by ANOVA. P<0.05 was considered statistically significant.
RESULTS
Structure of the peptide-drug conjugate
The chemical structure of the peptide-drug conjugate (KCC-TGX) is depicted in Figure 1A. This peptide-drug conjugate consists of five parts: i) the parent drug TGX-D1; ii) a self-cyclizing linker (SL); iii) the PSA cleavable peptide (SSKYQ); iv) a peptide spacer (GGG); and iv) a HER2-targeting peptide (KCCYSL). The peptide-drug conjugate could be activated by PSA cleavage to release the parent drug TGX-D1.
Figure 1.

(A) Structure of the peptide-drug conjugate KCC-TGX. KCC-TGX contains a HER2-targeting peptide, a GGG spacer, a PSA cleavable substrate, a self-cyclizing linker and the parent drug TGX-D1. (B) The activation mechanism of KCC-TGX. KCC-TGX is expected to be cleaved at the Gln-Ser site by PSA to release NH2-SL-TGX. The free amino group of Ser (S) in NH2-SL-TGX attacks the ester carbonyl group and releases the parent drug TGX-D1. The dipeptide Ser-Leu self-cyclizes to form diketopiperazines (DKP).
Figure 1B illustrates the release of TGX-D1 from KCC-TGX upon binding to HER2 on prostate cancer cells. Briefly, KCC-TGX was specifically delivered to the prostate cancer cells, and cleaved by PSA to form NH2-SL-TGX which was further cleaved by self-cyclization to release the active parent drug TGX-D1. PSA’s enzymatic activity only exists in the microenvironment of the prostate cancer cells, but not in systemic circulation due to the presence of PSA inhibitors (α1-antichymotrypsin and α2-macroglobulin) in the blood.30 Therefore, the PSA-cleavable linker was expected to remain stable in systemic circulation.
Synthesis of the TGX-221 derivatives
Because TGX-221 does not contain any functional groups for conjugation with peptides, we synthesized four TGX-221 derivatives (TGX-D1, TGX-D2, TGX-D3, and TGX-D4) with additional hydroxyl groups, as illustrated in Figure 2. Compounds 2, 3, and 4 were synthesized from compound 1, as reported.28 TGX-221 was synthesized according to the same report with a modification. In line with the reported procedure,18 the Schiff base was generated successfully after refluxing in toluene. However, the reported conditions (NaBH4 in toluene) could not reduce the Schiff base to the amine, even after increasing the temperature to 100°C. This result is probably due to the poor solubility of NaBH4 in toluene. After changing the solvent from toluene to methanol, a complete conversion was achieved in 30 minutes at room temperature. TGX-D2, TGX-D3 and TGX-D4 were also prepared by the same route. Compound 5 was synthesized from TGX-221 by substituting the secondary aniline with a methyl acetate. to do this conversion, the ester group of compound 5 was converted to a primary alcohol (TGX-D1) by LiCl/NaBH4 with a yield of 40~60%.
Figure 2.

Synthesis of TGX-221 and its derivatives. Reagents and conditions: (a) malonyl dichloride, DCM, rt; (b) CH3SO2Cl, Et3N, morpholine; (c) I. PdCl2(dppf), DIPEA, butyl vinyl ether; II. 1N HCl, rt; (d) I. 4Å MS, toluene, reflux; II. NaBH4, methanol; (e) methyl bromoacetate, DIPEA, CH3CN; (f) LiCl, NaBH4, THF/EtOH.
Bioactivity screening of the TGX-221 derivatives
We next examined the bioactivities of these TGX-221 derivatives in two prostate cancer cell lines, LNCaP and DU145. LNCaP represents a PTEN-null prostate cancer cell line that is sensitive to TGX-221 treatment, while DU145 is PTEN-wild and unresponsive to TGX-221. As shown in Figure 3A and B, TGX-221 exhibits the highest activity in LNCaP cells with an IC50 of 1.07 μM. Among the four TGX-221 derivatives, TGX-D1 showed the best activity (IC50=2.67 μM) in LNCaP cells. Both TGX-221 and TGX-D1 showed a negligible activity in DU145 cells (IC50>100 μM). TGX-D2, TGX-D3 and TGX-D4 also demonstrated activities in the LNCaP cells with IC50 of 6.9 μM, 8.8 μM, and 17.5 μM, respectively. DU145 cells did not respond to these derivatives with the exception of TGX-D3, which had an IC50 of 52.9 μM.
Figure 3.

Bioactivity screening of TGX-221 and its derivatives. TGX-D1 showed comparable effects as TGX-221 in cytotoxicity, the inhibition of androgen-induced cell proliferation and gene expression. (A) Structures of the TGX-221 derivatives and their IC50 values in LNCaP (PTEN-null) and DU145 (PTEN-wild) cells. (B) Cytotoxicities of TGX-221 and TGX-D1 are examined on LNCaP cells by MTT assay. (C) Both TGX-221 and TGX-D1 inhibit androgen-induced cell proliferation. LNCaP cells were treated with TGX-221 and TGX-D1 in R1881-containing serum-free mediumfor 3 days before the Alarm Blue assay. (D) Both TGX-221 and TGX-D1 inhibit androgen-induced gene expression. The ARE-LUC reporter was transfected into LNCaP cells and the cells were stimulated with R1881 (1.0 nM) in the presence or absence of TGX-221 or TGX-D1 for 24 hours before being subjected to the luciferase assay. The results are represented as the mean ± SD (n=3).
PI3Kβ has been reported to be essential for androgen-stimulated AR transactivation and cell proliferation in prostate cancer cells.16 Androgen-stimulated cell proliferation and gene expression are dramatically reduced by silencing the PI3Kβ gene with siRNA or inhibiting its activity with the PI3Kβ-selective inhibitor TGX-221.16 TGX-D1 demonstrated comparable effects as TGX-221 on the inhibition of androgen-induced cell proliferation (Figure 3C) and gene expression (Figure 3D), suggesting that TGX-D1 has a similar potency to TGX-221 to specifically inhibit PI3Kβ activity. As a result, TGX-D1 was selected for conjugation with the peptide promoiety.
Synthesis of the peptide-drug conjugate
Synthesis of NH2-SL-TGX is depicted in Figure 4A. Fmoc-Leu-OH was conjugated to the hydroxyl group of TGX-D1 via an ester bond. After deprotection, Boc-Ser(tBu)-OH was coupled to the amino group of the NH2-L-TGX. The dipeptide-drug conjugate (NH2-SL-TGX) was obtained by deprotecting the peptide and purifying it by HPLC using acidic conditions. Neutral and basic environments were avoided for NH2-SL-TGX because of its instability.
Figure 4.

Synthesis of the peptide-drug conjugate. (A) Synthesis of NH2-SL-TGX. Reaction conditions: (a) Fmoc-Leu-OH, EDCI, DMAP (cat), DCM; (b) 20% piperidine in DCM; (c) Boc-Ser(OtBu)-OH, EDCI, DMAP (cat), DCM; (d) 50% TFA in DCM. (B) Synthesis of KCC-TGX. Reaction conditions: (a) AcOH/trifluroethanol/DCM (10/20/70), rt; (b) HATU, DIPEA, NMP; (c) TFA/DCM/TIPS (47.5/47.5/5), rt.
For the synthesis of KCC-TGX, undeprotected peptide KCCYSLGGGSSKYQS including the PSA substrate, a GGG spacer, and the HER2-specific peptide was synthesized independently, and then coupled to NH2-L-TGX (Figure 4B). The undeprotected peptide KCCYSLGGGSSKYQS was constructed on 2-Cl-Trt-resin using solid-phase synthesis. The N-terminal of the peptide was protected with an acetyl group to enhance the stability of the peptide-drug conjugate. Coupling reagent HATU was used to increase the yield and suppress the racemization. The undeprotected peptide-drug conjugate (KCC-TGX) was isolated by silica gel column, followed by de-protection and purification by HPLC. The peptide-drug conjugate exhibited good solubility, and no organic solvent was required in its drug release and bioactivity studies.
Drug release from the peptide-drug conjugate
Release of the parent drug from the peptide-drug conjugate is essential to elicit the therapeutic effect in prostate cancer cells. According to our hypothesis, KCC-TGX (Ac-KCCYSLGGGSSKYQSL-TGX) would be delivered to prostate cancer cells and cleaved on the cell surface by PSA. Additionally, the released NH2-SL-TGX would undergo a self-cyclization reaction to release the parent drug TGX-D1 in physiological pH (Figure 5A). To test this hypothesis, we monitored the release of TGX-D1 from NH2-SL-TGX in vitro by HPLC. The data presented in Figure 5B and 5C illustrated that TGX-D1 dissociated readily from the dipeptide-drug conjugate NH2-SL-TGX in PBS buffer (pH 7.4) at 37°C. The release profile followed zero-order kinetics with a half-life of 8 hours.
Figure 5.

In vitro drug release from the intermediate NH2-SL-TGX. NH2-SL-TGX underwent self-cyclization to release the parent drug TGX-D1. The dipeptide Ser-Leu self-cyclizes to form DKP in a neutral pH. (A) Illustration of the self-cyclization reaction. (B) HPLC chromatogram monitored at 268nm. (C) Release profile of TGX-D1 from NH2-SL-TGX. The results are represented as the mean ± SD (n=3).
We further examined the hypothesis using Ac-SSKYQSL-TGX that only contained the PSA cleavable peptide sequence SSKYQ.31 PSA enzymes expressed in prostate cancers recognize SSKYQ and cleave it between the residues Gln (Q) and Ser (S) to form the dipeptide-drug conjugate (NH2-SL-TGX) which undergoes a self-cyclization reaction to release the parent drug TGX-D1 in a physiological pH (Figure 6A). Our selection of the dipeptide SL as the linker between the PSA substrate and TGX-D1 was not only for the self-cyclizaiton reaction, it was also because the most efficient PSA cleavage occurs between Gln (Q) and Ser (S) that are followed by Leu (L).32 To monitor the PSA cleavage and drug release, the peptide-drug conjugate Ac-SSKYQSL-TGX was incubated with PSA at 37°C and then assayed by HPLC. As Figure 6B and 6C depict, Ac-SSKYQSL-TGX was gradually cleaved by PSA to release the intermediate NH2-SL-TGX. The generation of NH2-SL-TGX by PSA cleavage was faster than the self-cyclization rate at the beginning, leading to an increase of NH2-SL-TGX. However, a gradual and steady release of the parent drug TGX-D1 was also observed, suggesting that the peptide-drug conjugate could readily release the active TGX-D1 in prostate cancer cells.
Figure 6.

PSA-mediated drug release from the peptide-drug conjugate (Ac-SSKYQSL-TGX). Ac-SSKYQSL-TGX was cleaved by PSA to release the intermediate NH2-SL-TGX which underwent self-cyclization to release the parent drug TGX-D1. (A) Illustration of PSA activation of the peptide-drug conjugate. (B) HPLC chromatogram monitored at 268nm. (C) Release profile of TGX-D1 from Ac-SSKYQSL-TGX. The results are represented as the mean ± SD (n=3).
Cytotoxicity of the peptide-drug conjugates
We next examined whether the released parent drug TGX-D1 was still active to induce cytotoxicity in the prostate cancer cells. As demonstrated in Figure 7, NH2-SL-TGX, Ac-SSKYQSL-TGX and KCC-TGX induced similar cytotoxicities in the LNCaP cells. The IC50 values for NH2-SL-TGX, Ac-SSKYQSL-TGX and KCC-TGX obtained were 5.54μM, 4.20μM and 5.27μM, respectively, which are close to the IC50 (2.67 μM) of the parent drug TGX-D1. These results clearly indicated the efficient release of the parent drug TGX-D1 from the peptide-drug conjugate.
Figure 7.

Cytotoxicity of NH2-SL-TGX (A), Ac-SSKYQSL-TGX (B) and KCC-TGX (C) in LNCaP cells. LNCaP cells were incubated with the peptide-drug conjugates for 72 hours, and cytotoxicity was measured by a MTT assay. The results are represented as the mean ± SD (n=3).
High affinity of the HER2-targeting peptide with LNCaP cells
It has been demonstrated that HER2 is highly expressed in a significant proportion of prostate cancer cells including LNCaP cells.33, 34 KCCYSL is a HER2-targeting peptide that was generated against HER2-positive breast cancer cells using phage display technology.35 KCCYSL is relatively small, and its affinity to the HER2 receptor is well defined.35, 36 To verify its affinity to LNCaP cells, C-terminus biotinylated KCCYSL was incubated with LNCaP cells and then stained with streptavidin-FITC. KCCYSL exhibited high-affinity binding on the LNCaP cells at a low concentration (10 μM) in comparison to the control peptide DPRATPGSK (Figure 8). No significant fluorescence was observed for this control peptide even when the concentration was increased to 100 μM (data not shown). These results further support the rationale of using the HER2-targeting peptide for targeted drug delivery to prostate cancer cells.
Figure 8.

Immunostaining of LNCaP cells with biotinylated anti-HER2 peptide. After blocking with 1% BSA in PBS, the fixed LNCaP cells were incubated with the biotinylated peptide for 1 hour in TBST buffer. After washing with TBS, the cells were incubated with streptavidin-FITC for 20 minutes and the nucleus was stained with DIPA for cell tracking. The cells were visualized using a Leica DMI3000 microscope at a 200x magnification.
Cellular uptake of KCC-TGX
We finally investigated whether the HER2-targeting peptide enhanced the delivery of the peptide-drug conjugate, KCC-TGX, to prostate cancer cells. Cellular uptake of TGX-D1 and its peptide conjugates were first evaluated on LNCaP cells at a concentration of 0.5μM after 1 hour of incubation (Figure 9A). Both TGX-D1 and Ac-SSKYQSL-TGX showed low uptake in LNCaP cells, while NH2-SL-TGX showed a high uptake. By contrast, KCC-TGX, whichcontains the HER2 targeting peptide, exhibited the highest uptake, indicating that the HER2-specific peptide (KCCYSL) dramatically increased the cellular uptake of KCC-TGX. The time-course of drug uptake in LNCaP cells was also studied (Figure 9B). While KCC-TGX showed the highest uptake extent, the uptake rate was comparable to that of TGX-D1 and NH2-SL-TGX, suggesting that coupling of the anti-HER2 peptide increases the drug affinity to prostate cancer cells without compromising its uptake rate.
Figure 9.

Cellular uptake of the peptide-drug conjugates in LNCaP cells. (A) Cellular uptake in LNCaP cells at a concentration of 0.5 μM. (B) The time-course of cellular uptake in LNCaP cells. (C) Dipeptide Ser-Leu concentration-dependent inhibition of the uptake of NH2-SL-TGX. (D). The HER2- specific ligand AcKCCYSL inhibits the uptake of KCC-TGX in LNCaP cells. The results are represented as the mean ± SD (n=3). (* P < 0.05; ** P < 0.01)
It is interesting to note that NH2-SL-TGX demonstrated a higher cellular uptake in comparison to Ac-SSKYQSL-TGX and TGX-D1. The possible explanation might be that NH2-SL-TGX can be taken up through peptide transporters that are expressed on many cancer cells.37 In line with this hypothesis, Ac-SSKYQSL-TGX did not show increased uptake either because only di- and tri-peptide can be actively transported by peptide transporters.38 However, to test this hypothesis, LNCaP cells were incubated with 0.5μM of NH2-SL-TGX in the presence of dipeptide Ser-Leu (0, 3.16, 10, and 100 μM) at 37°C for 1 hour. As Figure 9C revealed, the uptake of NH2-SL-TGX showed a concentration-dependent decrease after co-incubation with the dipeptide Ser-Leu, which competed for the same transporters on the prostate cancer cells. The peptide transporters were almost completely saturated by Ser-Leu at 10 μM, and the uptake of NH2-SL-TGX was accordingly inhibited by 75%.
To demonstrate whether the uptake of KCC-TGX by LNCaP cells is mediated by the anti-HER2 peptide ligand, we pre-incubated the cells with the anti-HER2 peptide and then applied KCC-TGX to the cells. As Figure 9D revealed, the uptake of Ac-KCCYSL was significantly inhibited by the pre-incubation with the HER2-specific ligand, which confirmed the specific binding of KCC-TGX to LNCaP cells via the recognition of the anti-HER2 peptide.
Aqueous solubility and stability of KCC-TGX
The aqueous solubility of KCC-TGX was assayed by the thermodynamic method at room temperature. The aqueous solubility of TGX-221 is approximately 147.5 μg/ml which is considered to be water-insoluble according to the United States Pharmacopeia (USP) (Table 1). Surprisingly, the solubility of TGX-D1 is only 33.5 μg/ml which is much lower than that of TGX-221. However, after conjugating to the peptide promoiety, its solubility increased to more than 9.895 mg/ml, which is 60-fold higher than that of TGX-D1 in terms of molarity. The substantial increase in aqueous solubility is probably due to the hydrophilic peptide promeity Ac-KCCYSLGGGSSKYQSL-OH in KCC-TGX.
Table 1.
Solubilities of TGX-221, TGX-D1 and KCC-TGX
| Compound | Aqueous solubility
|
|
|---|---|---|
| μg/ml | μM | |
| TGX-221 | 147.5±17.6 | 405.2±48.2 |
| TGX-D1 | 33.5±6.6 | 82.1±16.2 |
| KCC-TGX | ≥9899.1±465.4 | ≥4684.9±465.4 |
The stability of KCC-TGX was measured in three different solutions. KCC-TGX was found to be relatively stable in PBS and cell culture medium at 37°C. After 4 hours of incubation, 7% and 12% of KCC-TGX were found to be degraded in PBS and cell culture medium, respectively (Figure 10). However, KCC-TGX was quickly degraded in mouse serum with a half-life of 15 minutes, and nearly 83% of the KCC-TGX molecules were degraded in mouse serum after 60 minutes. The quick degradation of KCC-TGX is probably due to the labile ester linker was used between the peptide and TGX-D1. However, it should be noted that the hydrolysis rate of the ester linker is species dependent, and the stability is much higher in human plasma than that in mouse serum.39 Therefore, the degradation of KCC-TGX in human plasma is expected to be much slower.
Figure 10.
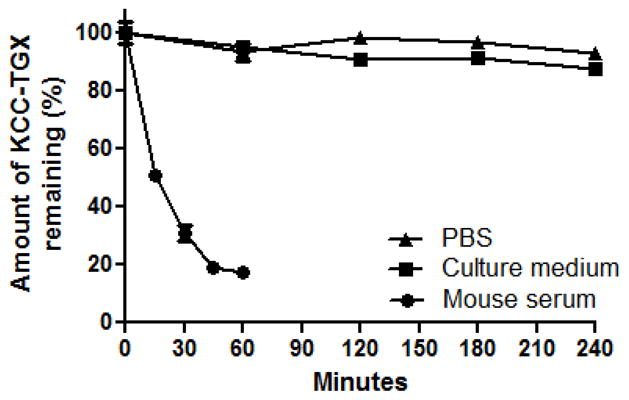
The stability of KCC-TGX in PBS, cell culture medium and mouse serum. Results were represented as mean ± SD (n=3).
DISCUSSION
In this study, we conjugated a peptide promoiety that contains a HER2-specific peptide and a PSA-cleavable substrate to TGX-221, a highly potent PI3Kβ inhibitor that shows a great promise for prostate cancer therapy. Despite the essential role of PI3Kβ in prostate cancer development, the therapeutic application of TGX-221 is limited due to its poor solubility and lack of specificity to prostate cancer cells. Therefore, the aim of this study is to overcome these two problems by attaching a peptide promoiety to TGX-221. Targeted delivery of this peptide-conjugate drug was achieved by considering two mechanisms. First, the HER2-specific peptide binds to HER2 on the surface of prostate cancer cells. Second, PSA, which exists in the microenvironment of prostate cancer cells cleaves the peptide and releases the active parent drug.
Because TGX-221 does not contain a functional group for peptide conjugation, we first synthesized four TGX-221 derivatives (Figure 2) by attaching a -OH group at different sites. Among them, TGX-D1 was selected for peptide conjugation due to its similar activity compared to TGX-221 (Figure 3)
HER2 has been reported to be overexpressed in many cancers such as breast cancer and prostate cancer. Although the role of its overexpression in prostate cancer is not fully understood, the HER2 receptor has been successfully exploited as a molecular target for targeted drug delivery to prostate cancer.33, 40 Anti-HER2 antibody is the first type of targeting moiety used for an antibody-drug conjugate. However, its large size compromises its cellular uptake, and the antibody-drug conjugate is unable to reach the tumor interior readily. Furthermore, one antibody molecule can only be conjugated with 1–3 drug molecules, which leads to a low drug loading (<1%). As a result, only highly potent drugs (IC50=10−9–10−11M) can be used for this antibody-drug approach.41 By contrast, a peptide ligand is a more appropriate targeting moiety due to its excellent cell permeability, small molecular weight, ease of production, and flexibility in chemical conjugation.42 The size of the peptide-drug conjugate is small enough to efficiently penetrate tissues, and drug loading can be as high as 20~30%. In this study, a 16-mer peptide (KCCYSLGGGSSKYQSL) was conjugated to TGX-D1 to form the peptide-drug conjugate (MW 2113.4), in which a drug loading of 19.4% was achieved. This 16-mer peptide promoiety is composed of a HER2-targeting peptide, a GGG spacer, a PSA substrate peptide and a self-cycling dipeptide (Figure 1A).
PSA-activated peptide-drug conjugates have been well studied in recent years.32, 43 For example, L-377202, a peptide-doxorubicin prodrug hydrolyzed by PSA, has already entered clinical trials.44 However, none of these peptide-drug conjugates include tumor-specific ligands, which makes accumulation in the cancer cells. Here, we reported the first application of a HER2-specific ligand to deliver the PSA-activated TGX-221 prodrug to prostate cancer cells. Uptake of the peptide-drug conjugate KCC-TGX in prostate cancer cells is approximately 10-fold that of the peptide-drug conjugate (Ac-SSKYQSL-TGX) that only contains the PSA cleavage linker (Figure 9A). In addition, the peptide-mediated cellular uptake is fast, and equilibrium is quickly reached in less than 10 minutes, demonstrating a rapid uptake of the prodrug in prostate cancer cells (Figure 9B). This rapid uptake is critical to minimize the loss of the peptide-drug conjugate caused by kidney excretion after systemic administration. Compared to the quick uptake, PSA cleavage of the peptide-drug conjugate is slow in in vitro studies (Figure 6). However, a much more efficient cleavage in vivo is expected because the prostate cancer interstitial fluid contains a substantial level of enzymatically active PSA (50~500 μg/ml), which is 5~50 times higher than we used in the in vitro cleavage study.45 As demonstrated in Garsky’s study, the half-life of a similar PSA substrate peptide was reduced to 10~30 minutes when 340 μg/ml of PSA was used.32 In comparison, the self-cyclization of NH2-SL-TGX might be the rate-limiting step in the release of the parent drug in vivo.
One interesting observation was that the dipeptide-drug conjugate NH2-SL-TGX exhibited a higher cellular uptake than TGX-D1 and Ac-SSKYQSL-TGX (Figure 9A). In other words, the peptide transporters that are believed to be widely expressed in various cancers could take up NH2-SL-TGX readily.37 In line with this hypothesis, we have found that co-incubation with the dipeptide Ser-Leu suppressed the cellular uptake of NH2-SL-TGX (Figure 9C). Therefore, we reasonably believe that PSA enzymes cleave the KCC-TGX bound on the cell surface to release NH2-SL-TGX, which are then transported into the cells through the peptide transporters. However, KCC-TGX can also be directly taken up by HER2 receptor-mediated endocytosis.
In summary, a multi-component peptide-drug conjugate was synthesized for TGX-221. This peptide-drug exhibits high cellular uptake in LNCaP cells. PSA enzymes activate the peptide-drug conjugate by cleaving the PSA substrate to release NH2-SL-TGX, which undergoes a self-cyclization to release the parent drug TGX-D1. This peptide-drug conjugate exhibits a much higher cellular uptake in prostate cancer cells in comparison to the parent drug, indicating its tremendous potential as a targeted therapy for prostate cancer patients.
Acknowledgments
This project has been supported by awards from National Cancer Institute (NCI), NIH (1R21CA143683-01) and National Institute of Alcohol Abuse and Alcoholism (NIAAA), NIH (1R21AA017960-01A1) to Dr Kun Cheng, and also partially by a Department of Defense grant (W81XWH-09-1-0455) and KU Masonic Foundation to Dr Benyi Li. The authors also would like to express thanks for the financial support from a start-up package at the University of Missouri-Kansas City.
References
- 1.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7 (8):606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436 (1–2):127–50. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 3.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784(1):159–85. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Vogt PK, Bader AG, Kang S. Phosphoinositide 3-kinase: from viral oncoprotein to drug target. Virology. 2006;344 (1):131–8. doi: 10.1016/j.virol.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 5.Ayala G, Thompson T, Yang G, Frolov A, Li R, Scardino P, Ohori M, Wheeler T, Harper W. High levels of phosphorylated form of Akt-1 in prostate cancer and non-neoplastic prostate tissues are strong predictors of biochemical recurrence. Clin Cancer Res. 2004;10 (19):6572–8. doi: 10.1158/1078-0432.CCR-04-0477. [DOI] [PubMed] [Google Scholar]
- 6.Kreisberg JI, Malik SN, Prihoda TJ, Bedolla RG, Troyer DA, Kreisberg S, Ghosh PM. Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Res. 2004;64 (15):5232–6. doi: 10.1158/0008-5472.CAN-04-0272. [DOI] [PubMed] [Google Scholar]
- 7.Le Page C, Koumakpayi IH, Alam-Fahmy M, Mes-Masson AM, Saad F. Expression and localisation of Akt-1, Akt-2 and Akt-3 correlate with clinical outcome of prostate cancer patients. Br J Cancer. 2006;94 (12):1906–12. doi: 10.1038/sj.bjc.6603184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulholland DJ, Dedhar S, Wu H, Nelson CC. PTEN and GSK3beta: key regulators of progression to androgen-independent prostate cancer. Oncogene. 2006;25 (3):329–37. doi: 10.1038/sj.onc.1209020. [DOI] [PubMed] [Google Scholar]
- 9.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15 (4):356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275 (5308):1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 11.Lei Q, Jiao J, Xin L, Chang CJ, Wang S, Gao J, Gleave ME, Witte ON, Liu X, Wu H. NKX3.1 stabilizes p53, inhibits AKT activation, and blocks prostate cancer initiation caused by PTEN loss. Cancer Cell. 2006;9 (5):367–78. doi: 10.1016/j.ccr.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 12.Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, Wu H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci U S A. 2006;103 (5):1480–5. doi: 10.1073/pnas.0510652103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57 (22):4997–5000. [PubMed] [Google Scholar]
- 14.Yoshimoto M, Cunha IW, Coudry RA, Fonseca FP, Torres CH, Soares FA, Squire JA. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br J Cancer. 2007;97 (5):678–85. doi: 10.1038/sj.bjc.6603924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454 (7205):776–9. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Q, Youn H, Tang J, Tawfik O, Dennis K, Terranova PF, Du J, Raynal P, Thrasher JB, Li B. Phosphoinositide 3-OH kinase p85alpha and p110beta are essential for androgen receptor transactivation and tumor progression in prostate cancers. Oncogene. 2008;27 (33):4569–79. doi: 10.1038/onc.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong D, Yamori T. Phosphatidylinositol 3-kinase inhibitors: promising drug candidates for cancer therapy. Cancer Sci. 2008;99 (9):1734–40. doi: 10.1111/j.1349-7006.2008.00891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, Kenche V, Anderson KE, Dopheide SM, Yuan Y, Sturgeon SA, Prabaharan H, Thompson PE, Smith GD, Shepherd PR, Daniele N, Kulkarni S, Abbott B, Saylik D, Jones C, Lu L, Giuliano S, Hughan SC, Angus JA, Robertson AD, Salem HH. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med. 2005;11 (5):507–14. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- 19.Shaywitz AJ, Courtney KD, Patnaik A, Cantley LC. PI3K enters beta-testing. Cell Metab. 2008;8 (3):179–81. doi: 10.1016/j.cmet.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bi L, Okabe I, Bernard DJ, Nussbaum RL. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome. 2002;13 (3):169–72. doi: 10.1007/BF02684023. [DOI] [PubMed] [Google Scholar]
- 21.Canobbio I, Stefanini L, Cipolla L, Ciraolo E, Gruppi C, Balduini C, Hirsch E, Torti M. Genetic evidence for a predominant role of PI3Kbeta catalytic activity in ITAM- and integrin-mediated signaling in platelets. Blood. 2009;114 (10):2193–6. doi: 10.1182/blood-2009-03-208074. [DOI] [PubMed] [Google Scholar]
- 22.Scher HI. HER2 in prostate cancer--a viable target or innocent bystander? J Natl Cancer Inst. 2000;92 (23):1866–8. doi: 10.1093/jnci/92.23.1866. [DOI] [PubMed] [Google Scholar]
- 23.Signoretti S, Montironi R, Manola J, Altimari A, Tam C, Bubley G, Balk S, Thomas G, Kaplan I, Hlatky L, Hahnfeldt P, Kantoff P, Loda M. Her-2-neu expression and progression toward androgen independence in human prostate cancer. J Natl Cancer Inst. 2000;92 (23):1918–25. doi: 10.1093/jnci/92.23.1918. [DOI] [PubMed] [Google Scholar]
- 24.Oman IASH, Drobnjak M, Morris M, Fazzari M, Cordon-Cardo C. HER-2/neu membrane overexpression in prostate cancer (PC) 2000. p. 41. abstract 4572. [Google Scholar]
- 25.Morris MJRV, Kelly WK, Slovin SF, Kenneson KI, Osman I. A phase II trial of herceptin alone and with taxol for the treatment of prostate cancer. Proc ASCO. 2000:19. abstract 330. [Google Scholar]
- 26.Ziada A, Barqawi A, Glode LM, Varella-Garcia M, Crighton F, Majeski S, Rosenblum M, Kane M, Chen L, Crawford ED. The use of trastuzumab in the treatment of hormone refractory prostate cancer; phase II trial. Prostate. 2004;60 (4):332–7. doi: 10.1002/pros.20065. [DOI] [PubMed] [Google Scholar]
- 27.Watt KW, Lee PJ, M’Timkulu T, Chan WP, Loor R. Human prostate-specific antigen: structural and functional similarity with serine proteases. Proc Natl Acad Sci U S A. 1986;83 (10):3166–70. doi: 10.1073/pnas.83.10.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaun J. Inhibition Of Phosphoinositide 3-Kinase Beta. WO/2004/016607 International Patent. 2004
- 29.Agarwal S, Boddu SH, Jain R, Samanta S, Pal D, Mitra AK. Peptide prodrugs: improved oral absorption of lopinavir, a HIV protease inhibitor. Int J Pharm. 2008;359 (1–2):7–14. doi: 10.1016/j.ijpharm.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Denmeade SR, Nagy A, Gao J, Lilja H, Schally AV, Isaacs JT. Enzymatic activation of a doxorubicin-peptide prodrug by prostate-specific antigen. Cancer Res. 1998;58 (12):2537–40. [PubMed] [Google Scholar]
- 31.Kumar SK, Roy I, Anchoori RK, Fazli S, Maitra A, Beachy PA, Khan SR. Targeted inhibition of hedgehog signaling by cyclopamine prodrugs for advanced prostate cancer. Bioorg Med Chem. 2008;16 (6):2764–8. doi: 10.1016/j.bmc.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garsky VM, Lumma PK, Feng DM, Wai J, Ramjit HG, Sardana MK, Oliff A, Jones RE, DeFeo-Jones D, Freidinger RM. The synthesis of a prodrug of doxorubicin designed to provide reduced systemic toxicity and greater target efficacy. J Med Chem. 2001;44 (24):4216–24. doi: 10.1021/jm0101996. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Liu B, Schmidt M, Lu Y, Wels W, Fan Z. Antitumor effect of an HER2-specific antibody-toxin fusion protein on human prostate cancer cells. Prostate. 2001;47 (1):21–8. doi: 10.1002/pros.1043. [DOI] [PubMed] [Google Scholar]
- 34.Agus DB, Scher HI, Higgins B, Fox WD, Heller G, Fazzari M, Cordon-Cardo C, Golde DW. Response of prostate cancer to anti-Her-2/neu antibody in androgen-dependent and -independent human xenograft models. Cancer Res. 1999;59 (19):4761–4. [PubMed] [Google Scholar]
- 35.Karasseva NG, Glinsky VV, Chen NX, Komatireddy R, Quinn TP. Identification and characterization of peptides that bind human ErbB-2 selected from a bacteriophage display library. J Protein Chem. 2002;21 (4):287–96. doi: 10.1023/a:1019749504418. [DOI] [PubMed] [Google Scholar]
- 36.Kumar SR, Quinn TP, Deutscher SL. Evaluation of an 111In-radiolabeled peptide as a targeting and imaging agent for ErbB-2 receptor expressing breast carcinomas. Clin Cancer Res. 2007;13 (20):6070–9. doi: 10.1158/1078-0432.CCR-07-0160. [DOI] [PubMed] [Google Scholar]
- 37.Mitsuoka K, Miyoshi S, Kato Y, Murakami Y, Utsumi R, Kubo Y, Noda A, Nakamura Y, Nishimura S, Tsuji A. Cancer detection using a PET tracer, 11C-glycylsarcosine, targeted to H+/peptide transporter. J Nucl Med. 2008;49 (4):615–22. doi: 10.2967/jnumed.107.048231. [DOI] [PubMed] [Google Scholar]
- 38.Terada T, Sawada K, Irie M, Saito H, Hashimoto Y, Inui K. Structural requirements for determining the substrate affinity of peptide transporters PEPT1 and PEPT2. Pflugers Arch. 2000;440 (5):679–84. doi: 10.1007/s004240000339. [DOI] [PubMed] [Google Scholar]
- 39.Ayral-Kaloustian S, Gu J, Lucas J, Cinque M, Gaydos C, Zask A, Chaudhary I, Wang J, Di L, Young M, Ruppen M, Mansour TS, Gibbons JJ, Yu K. Hybrid inhibitors of phosphatidylinositol 3-kinase (PI3K) and the mammalian target of rapamycin (mTOR): design, synthesis, and superior antitumor activity of novel wortmannin-rapamycin conjugates. J Med Chem. 2010;53 (1):452–9. doi: 10.1021/jm901427g. [DOI] [PubMed] [Google Scholar]
- 40.Goldstein D, Gofrit O, Nyska A, Benita S. Anti-HER2 cationic immunoemulsion as a potential targeted drug delivery system for the treatment of prostate cancer. Cancer Res. 2007;67 (1):269–75. doi: 10.1158/0008-5472.CAN-06-2731. [DOI] [PubMed] [Google Scholar]
- 41.Hughes B. Antibody-drug conjugates for cancer: poised to deliver? Nat Rev Drug Discov. 2010;9 (9):665–7. doi: 10.1038/nrd3270. [DOI] [PubMed] [Google Scholar]
- 42.Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 2010;146 (3):264–75. doi: 10.1016/j.jconrel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar SK, Williams SA, Isaacs JT, Denmeade SR, Khan SR. Modulating paclitaxel bioavailability for targeting prostate cancer. Bioorg Med Chem. 2007;15 (14):4973–84. doi: 10.1016/j.bmc.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 44.DiPaola RS, Rinehart J, Nemunaitis J, Ebbinghaus S, Rubin E, Capanna T, Ciardella M, Doyle-Lindrud S, Goodwin S, Fontaine M, Adams N, Williams A, Schwartz M, Winchell G, Wickersham K, Deutsch P, Yao SL. Characterization of a novel prostate-specific antigen-activated peptide-doxorubicin conjugate in patients with prostate cancer. J Clin Oncol. 2002;20 (7):1874–9. doi: 10.1200/JCO.2002.07.001. [DOI] [PubMed] [Google Scholar]
- 45.Denmeade SR, Sokoll LJ, Chan DW, Khan SR, Isaacs JT. Concentration of enzymatically active prostate-specific antigen (PSA) in the extracellular fluid of primary human prostate cancers and human prostate cancer xenograft models. Prostate. 2001;48 (1):1–6. doi: 10.1002/pros.1075. [DOI] [PubMed] [Google Scholar]