

Abstract
Background
Nosocomial infections often lead to sepsis and multi-system organ failure in critically injured patients including burn and trauma patients. A better understanding of the bacterial response to the host immune system is essential to develop better antimicrobials against pathogens. Pseudomonas aeruginosa, combats host-initiated oxidant stress through expression of the transactivating factor, OxyR. Here we have tested the premise that OxyR regulates Pseudomonal cytotoxicity through secreted exotoxin production.
Methods
Wild-type P. aeruginosa (PAO1) and a deletion mutant lacking the oxyR gene (ΔoxyR) were grown for 18 hours in Luria broth and the supernatant containing the secreted products was removed using centrifugation. Secreted proteins were isolated using ammonium sulfate precipitation. ER-MP20+ myeloid progenitor cells were harvested from the bone marrow of C57Blk/6J mice. These cells were differentiated into dendritic cells and macrophages. Various concentrations (0–20 μg/100 μL) of the bacterial proteins were added to the medium and cells were allowed to differentiate for 7 days. Cellular viability was then assayed using a proliferation assay. These studies were repeated on two other macrophage cell lines, human U937 and murine P388D1.
Results
At a protein concentration of 5 μg/100 μL PAO1 supernatant protein, cellular proliferation was significantly reduced to 4.2±2.8% compared to untreated controls, while the ΔoxyR supernatant protein remained at 103.3±4.0% of untreated controls (P < 0.05). Similar significant results were seen in the U937, P388D1, and ER-MP20+ derived macrophage cells. Conclusions: Taken together, our data indicates that OxyR regulates the secretion of potent cytotoxic factors by P. aeruginosa.
Keywords: Sepsis, Bacterial Infection, Dendritic Cells, Macrophages, Bone Marrow Progenitor
Introduction
Nosocomial infections continue to be a major source of morbidity and mortality for patients in a hospital setting. Nowhere is this problem greater than in the intensive care units where the sickest and most immunocompromised patients are treated. Hospital acquired infections such as catheter line sepsis, ventilator associated pneumonia, and surgical wound site infections are steadily rising [1]. This is especially true in the burn and trauma patient population where nosocomial infections consistently outnumber those seen in the medical ICUs [2].
In addition to this problem, bacterial resistance to antibiotics continues to rise as well. Several strains of bacteria are becoming multi-drug resistant to antibiotics, and in many instances they are limiting our choices of systemic treatment to antibiotics and antimicrobials that possess a high degree of tissue toxicity. While antibiotic resistance is rising, the discovery of new antibiotic classes has failed to keep pace. In addition, novel treatments, like immunotherapy, have failed to produce meaningful treatment alternatives in large clinical trials [3, 4]. A potential reason for the ineffectiveness of many of the sepsis immunotherapies is perhaps they are monotherapies directed against a single component of the inflammatory cascade [5]. Because the immune response to inflammation and infection is a complex cascade of pro- and anti-inflammatory molecules that change with duration and severity of a critical injury, it is understandable that monotherapies like anti-TNF-α antibody and IL-1 receptor antagonists have not proven to be effective treatment modalities in sepsis.
As the incidence of sepsis is rising, much of the current research in the fields of critical care and sepsis has focused on host-response [6]. While this focus has generated much useful information, unfortunately it has provided only a partial picture of the pathophysiology of sepsis [7]. Similar to the responses of the host to the pathogen centered on eradicating the microbial insult, the bacterial pathogen also employs specific mechanisms that allow them to evade immune detection and ensure their survival [8]. Through processes such as adherence to host tissue, active evasion of the immune system cells, and direct damage to the host through exotoxin production, bacteria are able to initiate, disseminate and sustain infections. In order to combat bacterial infections, especially multi-drug resistant nosocomial infections, and to devise pathogen specific antimicrobial therapy, it is essential to understand the interrelationship between the responses of the host and the bacterial pathogen.
Pseudomonas aeruginosa is an aerobic, non-lactose fermenting Gram-negative bacterium that has developed several multi-drug resistant strains [9]. In fact, the number of resistant strains has risen 15% over the last five years and is projected to maintain this rate [1]. 9.9% of all ventilator associated pneumonias are caused by P. aeruginosa which is second only to Staphylococcus aureus [1]. While P. aeruginosa pneumonia or burn wound infection is a problem, the much larger problem is the resultant systemic spread of the bacterium causing sepsis and multi-system organ failure. Once critically ill patients are infected, it is extremely difficult to eradicate the P. aeruginosa infection. Mortality approaches 40% in this population [10].
The elimination of Pseudomonal infections is difficult due to the combination of antibiotic resistance and expression of multiple virulence factors [11, 12]. These virulence factors can attack the host and inactivate host immune cell processes. Especially potent are the exotoxins secreted by the bacteria. Proteases, elastases, exotoxin A, and the exoenzymes T, U, and S are all examples of highly virulent substances that directly attack host immune cells [13].
Initial host defense against an invading bacterial pathogen is mediated through bacterial phagocytosis and killing by neutrophils and macrophages. This killing predominantly involves generation of reactive oxygen metabolites such as superoxide, hydrogen peroxide, hydroperoxides, and nitric oxide [14]. Different pathogens handle oxidant stress in different ways depending on their relative susceptibility to a given oxidant stress. For example, Yersinia pestis, which is more susceptible to nitric oxide and peroxynitrate, upregulates specific enzymes that neutralize these oxidants but not those that combat hydrogen peroxide and superoxide [15]. Although P. aeruginosa is susceptible to nitrite derivatives [16], the immune cell mediated oxidant stress is negated through the transactivator OxyR that regulates the production of catalases and hydroperoxidases. OxyR is a transactivator that regulates the production of the catalase KatB and alkyl hydroperoxidases AhpB and AhpCF in P. aeruginosa [17]. Previous studies have shown that oxyR expression contributes to the virulence of P. aeruginosa [18, 19].
Since secreted toxins and proteolytic enzymes play a significant role in the pathogenicity of P. aeruginosa, and oxyR expression is an essential component of its virulence, we postulated that exotoxin mediated pathogenicity of P. aeruginosa may be regulated by oxyR expression. Our work has shown an inhibition of dendritic cell differentiation when mice were infected with wild-type P. aeruginosa on their burn wound but not with an isogenic oxyR mutant bacteria [20]. Since the innate immune response is vital to the primary response to bacterial pathogens, we chose to look at the effect of secreted Pseudomonal proteins on the preeminent antigen-presenting immune cells, dendritic cells (DC) and macrophages (MØ). Moreover, these cells are targets for inactivation by bacterial toxins.
Materials and Methods
Bacteria
Three different strains of P. aeruginosa bacteria were used in these experiments [17, 19]. 1) PAO1, a wild-type strain of P. aeruginosa; 2) ΔoxyR, a deletion mutant of PAO1 lacking the oxyR gene; and 3) ΔoxyR + poxyR (poxyR), the ΔoxyR mutant complemented with a plasmid expressing oxyR. Finally, we tested a PA14 strain of P. aeruginosa, a pathogenic strain similar to PAO1. Type III secretion in PA14 was blocked by the addition of a plasmid expressing type III dominant negative mutant [pscNK176G(T3DN)] (type III block).
Secreted Bacterial protein isolation
Bacteria were grown for 18 hours at 37 °C in 100 mL Luria broth. PAO1, ΔoxyR, and poxyR were grown in the presence of carbenicillin (20 μg/mL) while PA14 with type III block was grown with rifampin (20 μg/mL). Bacterial cultures were centrifuged at 3000 g to isolate the bacterial pellet from the supernatant, and the supernatant was then centrifuged again at 10,000 g. The resultant clear supernatant containing the secreted proteins was lyophilized and then re-suspended in 30 mL of sterile water. Bacterial proteins were precipitated with ammonium sulfate (0.568 g/mL) at 4 °C and pH 7.4. The precipitated proteins were isolated by centrifugation and then re-suspended in 3 mL of sterile water. The protein solution was placed into 6–8,000 kDa cut-off dialysis membranes, and the remaining ammonium sulfate was removed using large volume (3 × 4 L) dialysis for 3 d against 0.9% normal saline at 4 °C. The dialysate containing the secreted proteins was then sterilized through a 0.22 μm polysulfonate filter, which also removed endotoxin. Protein concentration was determined using a commercially available BCA protein assay kit (Pierce, Rockford, IL). For heat inactivation studies, the protein solution was heated at 85 °C for 10 min immediately prior to use.
Cell line preparation
The human myelomonocytic cell line U937 and a murine macrophage cell line P388D1 were sub-cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (10 μg/mL). On the day of the experiment, the cells (5 × 103) were suspended in Isocove’s medium (100 μL) supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (10 μg/mL) in 96 well plates. Phorbol myristate acetate (0.5 ng/mL) was added to the U937 cells to differentiate them into macrophages. Bacterial proteins (0–20 μg/100 μL) were added to the wells and incubated for 72 hr at 37 °C in 5% CO2 atmosphere.
Animals
C57Blk/6J male mice aged 6–8 weeks and weighing 24–30 grams were used for the experiments. Mice were acclimated in a climate controlled facility for one week prior to use with 12 h light-dark cycles and food and water ad libitum. The Loyola University Medical Center Animal Care and Use Committee approved all protocols used in this study.
Bone marrow cell isolation
Mice were euthanized by CO2 asphyxiation and bilateral femurs were removed. Total bone marrow cells were harvested by flushing each pair of femurs in 5 mL of McCoy’s medium supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (10 μg/mL). An aliquot from each sample was diluted in 3% acetic acid to lyse red blood cells, and the total nucleated bone marrow cells were then counted using a Neubauer hemocytometer.
Isolation of myeloid progenitor cells
Total nucleated bone marrow cells (2×107) were resuspended in phosphate buffered saline (PBS, 100 μL) containing EDTA (2 mM), 0.5% bovine serum albumin, penicillin (100 U/mL), and streptomycin (10 μg/mL). Cells were then labeled with rat anti-mouse ER-MP20+ monoclonal antibody (1:10 v/v) for 15 min at 4 °C. The cells were then washed and re-suspended in 100 μL of PBS with magnetic microbead conjugated goat anti-rat IgG (1:5 v/v) for 15 min at 4 °C. The cells were then passed through a MACS MS column (Miltenyi Biotec, Auburn, CA) while in the presence of a magnetic field. The MS column was then removed from the magnet and ER-M20+ bone marrow progenitors were eluted by flushing the column with 1 mL of PBS. Total ER-MP20+ cells were determined by counting an aliquot in a Neubauer hemocytometer.
Differentiation of ER-MP20+ progenitor cells
ER-MP20+ progenitor cells were differentiated into DC or MØ with appropriate growth factors. ER-MP20+ cells (5 × 104) were suspended in Isocove’s medium (100 μL) supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (10 μg/mL) in 96 well plates. Progenitors were differentiated into DC with rmGM-CSF (50 ng/mL) and rmIL-4 (10 ng/mL). Another set of ER-MP20+ progenitors were differentiated into MØ with rmM-CSF (50 ng/mL). Immediately, various concentrations of the bacterial proteins (0–20 μg/100 μL) were added to each well. Cells were incubated for 7 days at 37 °C in 5% CO2 atmosphere.
Proliferation assay
After day 3 (U937, P388D1) or day 7 (ERMP 20+ derived DC and MØ), cellular viability was determined using a commercially available tetrazolium/formazan proliferation assay (Promega, Madison, WI) and absorbance was read at 490 nm on a spectrophotometer (Molecular Devices, SpectraMax Plus 384).
Endotoxin measurement
To ensure there was no bacterial endotoxin contaminating the secreted bacterial protein samples, lipopolysaccharide (LPS) was assayed in the samples using a commercially available limulus amebocyte lysate test (Cambrex, Walkersville, MD).
Statistical analysis
Absorbance values were normalized among tests by dividing by control (untreated cells) values. Statistical analysis for more than two groups was accomplished using a one-way ANOVA followed by a post hoc Student-Newman-Keuls multiple comparison test. Statistical analysis between two groups was accomplished using a Students t-test. A p-value less than 0.05 was considered significant.
Results
Proliferation of cells exposed to secreted Pseudomonal protein
The cell surface marker, ER-MP20, has been shown to be expressed on monocyte committed myeloid progenitors which include monoblasts and promonocytes and these cells appear to lose the ER-MP20+ expression as they differentiate into either MØ or DC dependent on the specific growth factor cocktail [21–24]. Figure 1A is a representative sample of ER-MP20+ derived DC proliferation in response to the addition of supernatant proteins from the three different P. aeruginosa strains. Data shown in this figure represents a dose-response curve of the effect of secreted proteins isolated from either PAO1, ΔoxyR, or poxyR on cellular proliferation of ER-MP20+ progenitor derived DC. Wild-type PAO1 protein causes marked inhibition of proliferation once the concentration reaches 5 μg/100 μL. On the contrary, secreted proteins from ΔoxyR did not inhibit cellular proliferation. In fact, concentrations up to 160 μg/100 μL were tested with minimal decrease in proliferation (data not shown). Finally, when ΔoxyR was complemented in trans with poxyR, secreted protein was as potent as PAO1 in its anti-proliferative capacity.
Figure 1.
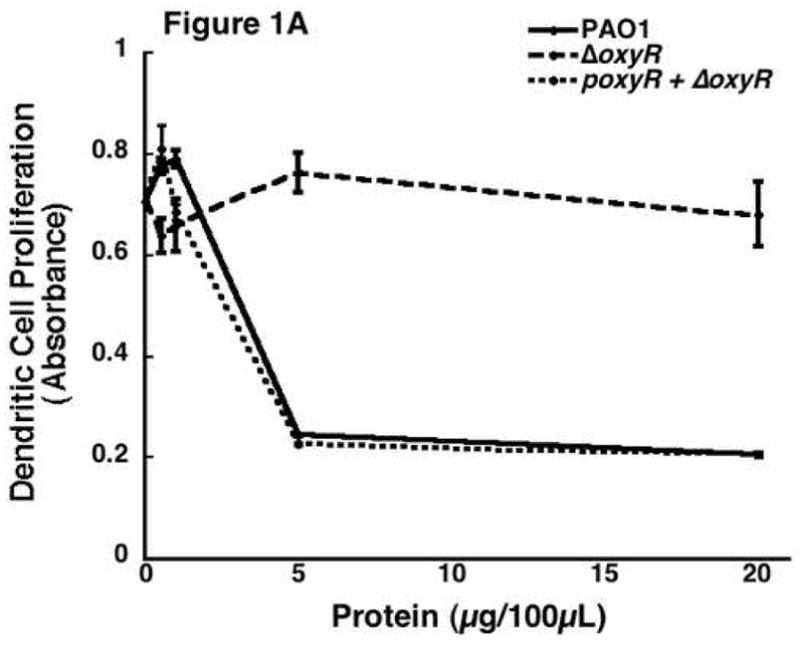
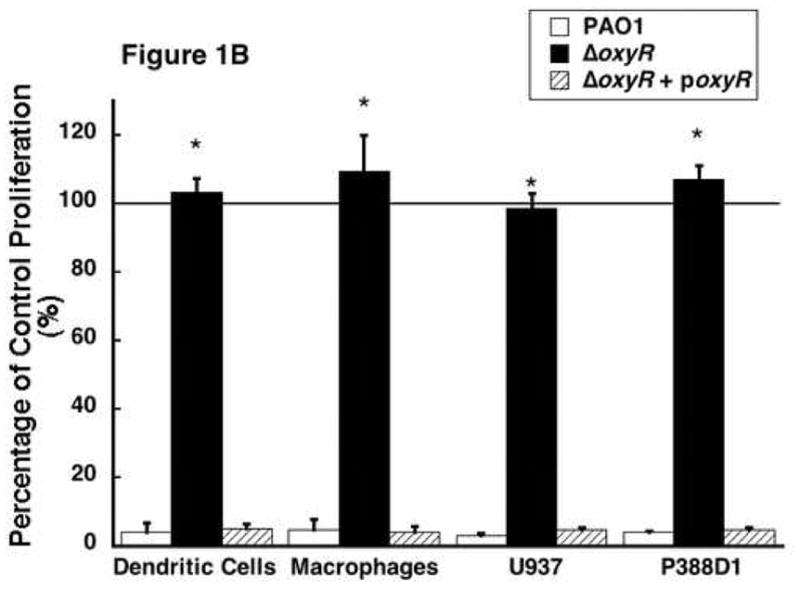
The effect of Pseudomonas aeruginosa supernatant protein on cell proliferation. ER-MP20+ progenitor derived DC and MØ as well as U937 and P388D1 MØ were allowed to incubate at 37 °C in Isocove’s solution with and without secreted bacterial proteins. Secreted proteins were isolated from the following bacterial strains: PAO1, a wild-type P. aeruginosa, ΔoxyR, a mutant P. aeruginosa missing the oxyR gene, and ΔoxyR + poxyR, an oxyR mutant complemented with an oxyR containing plasmid. Three to seven days later, cellular activity was determined using a cellular proliferation assay and measuring absorbance at 490 nm. A) A dose-response curve of the ER-MP20+ progenitor derived DC. Absorbance is plotted versus the concentration of protein added to the cells. PAO1 is represented by the solid line, ΔoxyR the dashed line, and ΔoxyR + poxyR the dotted line. Data represent the mean (N=3) ± the standard deviation. B) Graphical representation of the cellular proliferation assay for four different types of cells tested at 5 μg/100 μL protein concentration. Data is represented as a percentage of the absorbance of untreated control cells. The numbers are the mean (N=3) ± standard deviation. * p < 0.05 for PAO1 vs. ΔoxyR in all four groups.
The results seen in figure 1A are not specific for progenitor derived DC. Similar results were observed when ER-MP20+ derived MØ, human myelomonocytic cell line U937, and a murine macrophage cell line P388D1 were treated with secreted proteins from PAO1, ΔoxyR and poxyR (Figure 1B). In these experiments, the minimal concentration of secreted proteins that was found to be most effective as determined by the dose-response experiment (5 μg/100 μL) was employed. At this concentration, PAO1 protein reduces proliferation to 4.2±2.8% of untreated control cells in progenitor derived DC. ΔoxyR protein completely reversed the anti-proliferative response of PAO1 (103.3±4.0% of control), while the poxyR protein reduced proliferation to 5.1±1.3% of control values. These trends were replicated when the other three cell lines were studied: ER-MP20+ MØ (PAO1=4.7±3.1%, ΔoxyR=109.2±10.6%, poxyR=4.2±1.7%), U937 (PAO1=3.1±0.8%, ΔoxyR=98.5±4.3%, poxyR=4.9±0.7%), and P388D1 (PAO1=4.2±0.4%, ΔoxyR=106.9±4.2%, poxyR=4.9±0.6%). Therefore, P. aeruginosa secreted exotoxins limit host cell proliferation, and the transactivating gene, oxyR, partially regulates this.
In an effort to confirm the results that are seen are due to a protein in the supernatant, we denatured the supernatant proteins by heating to 85 °C for 10 minutes. These samples were tested on the proliferation of the same four cell lines and allowed to incubate. The results are shown in figure 2A. This is a dose-response curve showing the ER-MP20+ progenitor derived DC proliferation in response to varying concentrations of the PAO1 and ΔoxyR protein after heat inactivation. As seen in this representative curve, the PAO1 protein lost its ability to inhibit cell proliferation while the ΔoxyR protein did not change from the previous studies. Figure 2B further established that heat inactivation abrogated the ability of secreted proteins isolated from the different bacterial strains to influence cellular proliferation with all the four cell lines tested. There is no significant inhibition in any of the groups: ER-MP20+ DC (PAO1=94.6±7.1%, ΔoxyR=95.4±6.7%), ER-MP20+ MØ (PAO1=92.1±5.2%, ΔoxyR=91.4±6.2%), U937 MØ (PAO1=105.7±9.6%, ΔoxyR=90.1—7.9%), and P388D1 MØ (PAO1=109.1±7.9%, ΔoxyR=102.0±4.4%). In summary, heat inactivation of the bacterial supernatant proteins eliminated any inhibition in cellular proliferation. These results lend credence to the fact that the supernatant’s ability to decrease proliferation is due to a heat-labile protein.
Figure 2.
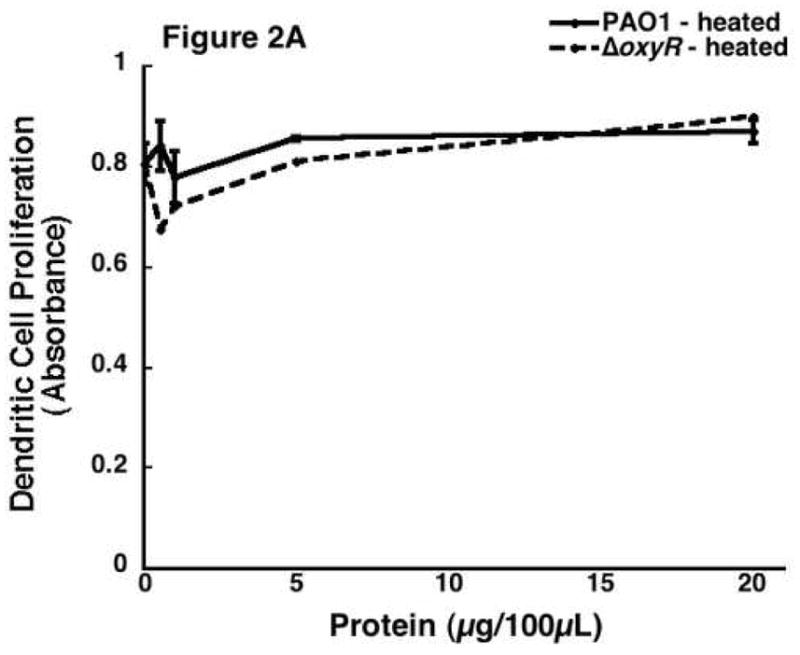
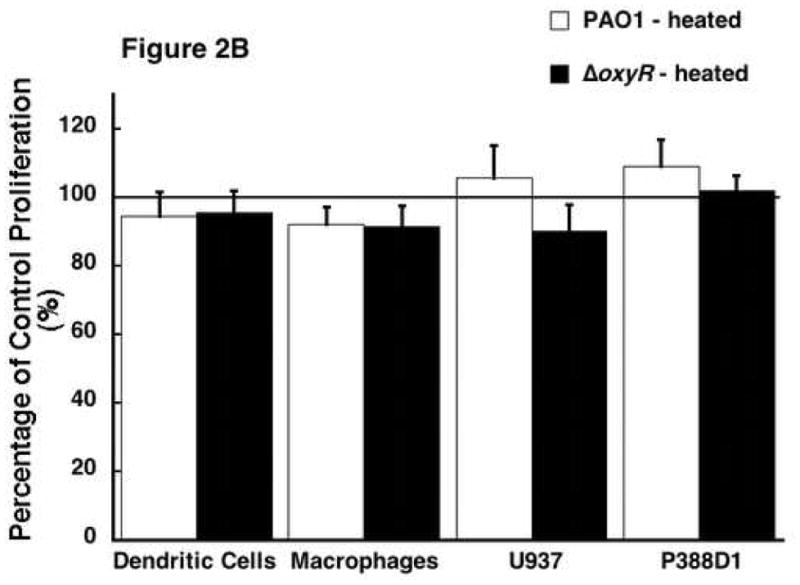
The effect of heat inactivated Pseudomonal supernatant protein on cellular proliferation. Bacterial protein was heated to 85 °C for 10 minutes then applied to all four cell lines. A) A dose response curve from a representative sample of ER-MP20+ progenitor derived DC. PAO1 (solid line) and ΔoxyR (dashed line) are shown as absorbance vs. concentration of bacterial protein. Data is represented as the mean (N=3) ± the standard deviation. B) Graphical representation of heat inactivated PAO1 protein (white bar) vs. ΔoxyR protein (black bar) for all four cell lines at 5 μg/100 μL. The numbers are the mean (N=3) ± standard deviation.
As previously mentioned, oxyR gene expression has been demonstrated to be primarily responsible for controlling the oxygen radical defense system of P. aeruginosa. In particular, OxyR has been shown to control the production of the secreted catalase KatB, the periplasmic AhpB, the cytoplasmic AhpCF [18]. While catalase in itself has never been shown to inhibit cellular activity, nor does a reliable mechanism exist for this process, it does represent the major end product of OxyR stimulation. Therefore, we added a physiological level of bovine liver catalase (100 U/mL, Roche, Nutley, NJ) to supernatant protein from ΔoxyR bacteria. This catalase previously has been shown to replicate the antioxidant effects of Pseudomonal catalase. [18] This combination was then added to the ER-MP20+ progenitor derived DC. Our results showed addition of catalase to the secreted proteins of ΔoxyR did not inhibit proliferation over a range of protein concentrations (figure 3A). ΔoxyR protein with catalase was then added to the remaining three cell lines and the results were all in accordance, that is there was no reduction in cellular proliferation. As seen in figure 3B, ER-MP20+ DC exposed to ΔoxyR and catalase displayed cellular activity at 107.0±17.3% of untreated cells. Similar numbers were observed for the rest of the groups: ER-MP20+ MØ 111.5±14.1%, U937 94.6±4.3%, and P388D1 111.3±11.0%.
Figure 3.
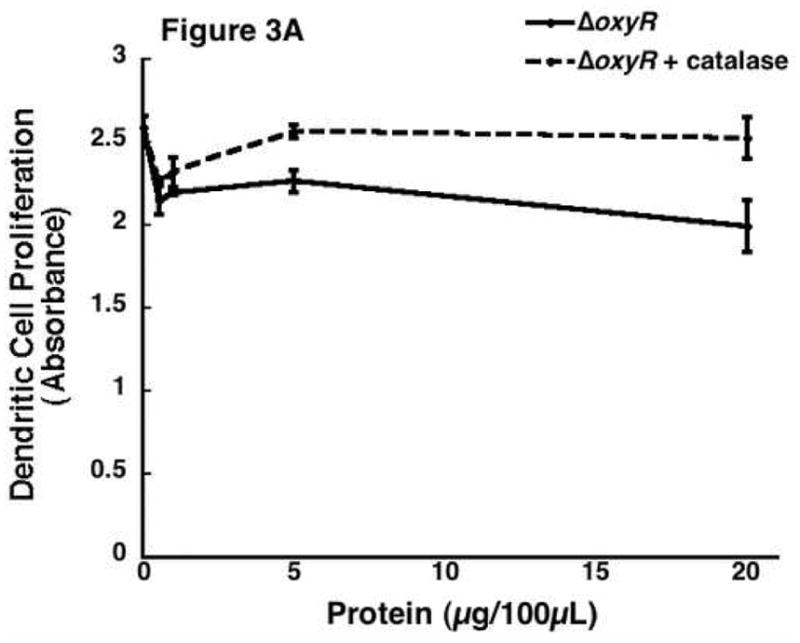
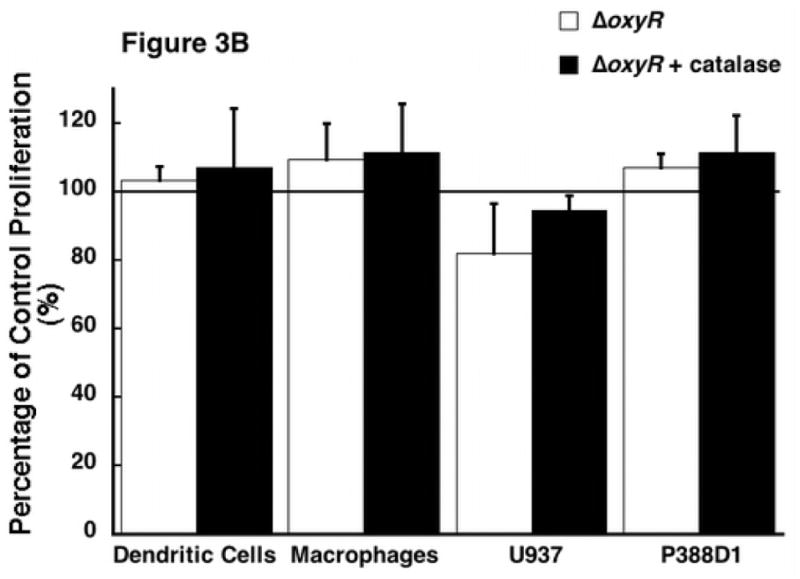
The effect of the addition of catalase to the ΔoxyR supernatant protein and its resultant effect on cellular proliferation. 100 U/mL of catalase was added to the supernatant protein from ΔoxyR and then added to all four cell lines. A) A dose-response curve from a representative sample of ER-MP20+ derived DC plotting ΔoxyR (solid line) and ΔoxyR + catalase (dashed line) as absorbance vs. protein concentration. Data is represented as the mean (N=3) ± the standard deviation. B) Graphical representation of ΔoxyR protein (white bar) vs. ΔoxyR protein + catalase (black bar) for all four cell lines at 5 μg/100 μL. The numbers are the mean (N=3) ± standard deviation.
There are multiple secretion systems in Gram-negative bacteria that are responsible for the export of proteins [25]. Many of the potent exotoxins produced by P. aeruginosa are regulated by the type III secretion system. The type III secretion system is designed to deliver virulent exotoxins such as ExoY, T, U, and S directly into host immune cells to inactivate them [26]. The type III secretion system involves a needle-like complex in the bacterial cell membrane that pierces host cells and allows direct injection into the host cytosol [27]. In order to determine if any of the type III secreted exotoxins are responsible for the observed anti-proliferative response in PAO1 and whether OxyR plays any role in the secretion of type III secreted exotoxins, we prepared and tested the supernatant secreted proteins from P. aeruginosa that had impaired type III secretion systems via repressor gene blockage. The premise is if type III secreted exotoxins were responsible for the anti-proliferative effect of secreted proteins isolated from PAO1, then type III block should reverse the loss of viability induced by PAO1. Figure 4A represents data from experiments where supernatant proteins from PAO1, ΔoxyR, and type III block were added to ER-MP20+ progenitor derived DC. Secreted protein preparation from type III block did cause a significant reduction in a manner similar to PAO1. Figure 4B shows the results when these proteins were applied to all four cell lines. As compared to untreated controls, all the cells exposed to type III block all had reduced activity. In other words, type III block behaved like PAO1. The numerical results are as follows: ER-MP20+ DC=2.9±0.7%, ER-MP20+ MØ=4.1±1.5%, U937=12.1±5.6%, and P388D1=7.6±3.7%. Therefore, the pathogenicity seen in P. aeruginosa in terms of reduced cellular proliferation is not due to exotoxins secreted from the type III secretion pathway.
Figure 4.
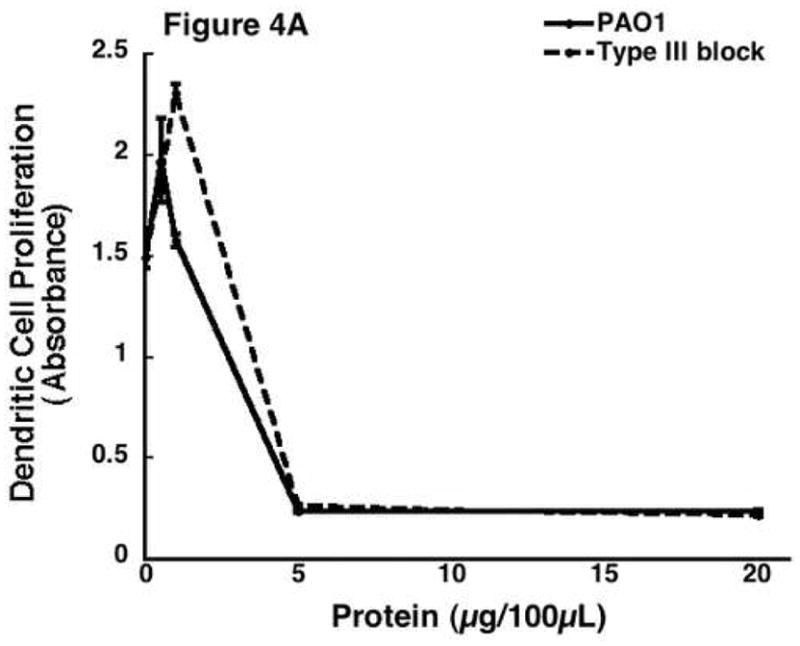
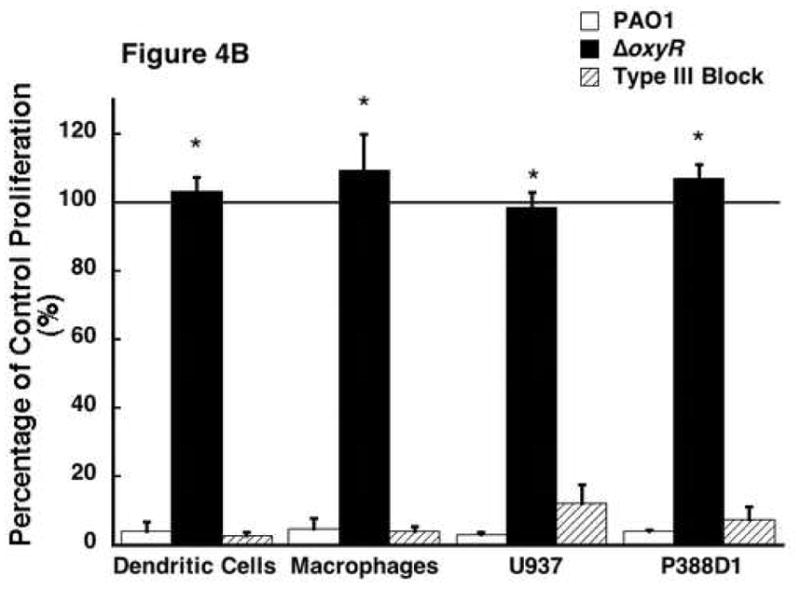
The effect of supernatant protein from secretion deficient Pseudomonas on cellular proliferation. Supernatant protein was isolated from P. aeruginosa blocked through a repressor gene (type III block). The protein was then added to all four cell lines as previously described. A) A dose response curve from a representative sample of ER-MP20+ derived DC plotting PAO1 (solid line) and type III block (dashed line) as absorbance vs. protein concentration. Data is represented as the mean (N=3) ± the standard deviation. B) Graphical representation of cellular proliferation for all four cell lines when treated with PAO1 (white bar), ΔoxyR (black bar), and type III block (dashed bar) supernatant protein at 5 μg/100 μL. The numbers are the mean (N=3) ± standard deviation. * p < 0.05 for PAO1 vs. ΔoxyR for all four cell lines.
Discussion
This present study contributes new evidence that the oxyR gene affects Pseudomonal pathogenicity through the production and secretion of exotoxins. More specifically, the presence of oxyR expression is required for P. aeruginosa’s ability to inhibit host cell macrophage and dendritic cell proliferation through secreted exotoxins. This study also builds on the premise that bacterial exotoxins rather than endotoxin is the major pathogenic determinant in the pathophysiology of sepsis, and therefore understanding the regulation of exotoxin production by P. aeruginosa in the tissue environment of sepsis and the host-response to the exotoxins are essential components in our understanding of sepsis.
The major function of OxyR has been well established to be the production of catalase in response to oxidant stressors applied to the bacteria. However, OxyR appears to control several other aspects of bacterial pathogenicity, for example DNA repair and resistance to aminoglycosides [17, 18]. OxyR is in fact necessary for the full expression of virulence of the organism. However, no attempts have yet been made to characterize what OxyR dependent virulence is comprised of and where its actions are exerted. This study attempts to elucidate this by narrowing OxyR’s effects to Pseudomonal secreted proteins. Wild-type P. aeruginosa, as of 2006, is known to secrete 86 different exotoxins [28]. While they serve many different functions for the bacteria, a significant portion is directly designed to interact and inhibit host immune cell functions. We have shown that exotoxins secreted in the cultured supernatant from P. aeruginosa are able to inhibit the proliferation of host dendritic cells and macrophages. This is not without precedent as many systems have already been described whereby P. aeruginosa exotoxins cause host cell apoptosis [29], necrosis [30], and inactivation of mitotic components [31]. However, none of these processes has as of yet been linked to the OxyR system. In fact, the OxyR regulon has historically been earmarked for only the stress response to reactive oxygen species.
While secreted exotoxins are no doubt vital to the pathogenesis of the bacterial organism, the cell membrane of P. aeruginosa also contains endotoxin or LPS, which confers a large amount of virulence to the organism. In an effort to remove all the LPS from our protein samples, protein was isolated by ammonium sulfate precipitation. This step should eliminate any non-protein components. In addition, the protein samples were passed through polysulfonate filters that should remove any remaining LPS. The limulus test to assay LPS was preformed on the isolated samples. The proteins contained on average 110.7±13.1 pg/mL of endotoxin. This level is approximately 1000–2000 times smaller than the LPS concentrations used to activate immune cells to make cytokines [32, 33]. Therefore, LPS did not play a role in our findings. However, we would not expect LPS to cause a reduction in cellular activity as LPS will actually prevent the apoptosis of host immune cells since its goal is activation rather than suppression of the immune system [34].
Since the most studied proteins under the control of OxyR are the catalases, we added catalase back to oxyR mutant protein samples to see if any inhibition of cell activity occurred. This hypothesis was unlikely because catalase has never been shown to possess any cytotoxic activity. The oxyR mutant bacterial supernatant actually contains the same amount of catalase as wild-type, however it is the deployment of catalase that is impaired [18]. The addition of the catalase to the mutant protein samples did not cause any significant inhibition of cellular activity. Therefore, OxyR must control the production of other pathogenic bacterial proteins in addition to catalase.
P. aeruginosa uses several mechanisms to secrete exotoxins out of the cell. The type III secretion system has been developed specifically to secrete exotoxins to inactivate or impair the host’s ability to phagocytose and kill the pathogens [35]. Although the type III secreted exotoxins usually are directly injected into cells, they are secreted into the extracellular environment and are biochemically active [36, 37]. Our results demonstrated that when the type III secretion system was blocked in P. aeruginosa, the secreted proteins isolated from these bacteria were as cytotoxic as the PAO1, which is in direct contrast to the results obtained with ΔoxyR. P. aeruginosa with the type III block did inhibit cellular viability which suggests that the cyto-protection endowed by OxyR is unlikely to be due any of the proteins secreted by the type III system.
This present study has shown that the oxyR gene of P. aeruginosa partly regulates bacterial pathogenicity through the production and/or secretion of bacterial exotoxins. This global transactivator is a powerful mechanism for the regulation of secreted bacterial exotoxins. Other transactivators in P. aeruginosa have been found to regulate pathogenesis through exotoxin production [38]. However, this is the first time that the transactivator OxyR has been identified as controlling secreted exotoxins that are cytotoxic toward host immune cells. In order to combat bacterial infections, new diagnostic tests and novel antimicrobials are needed. While the bacterial endotoxins no doubt are important to bacterial virulence, exotoxins too contribute a major portion of bacterial pathogenicity. Much has been learnt about the way bacterial exotoxins interact with the host immune system. Hopefully, further elucidation of these mechanisms will lead to the new diagnostics and therapeutics that can change the spectrum of how bacterial infections are treated.
Acknowledgments
This work was supported by grants from the Falk Foundation and the National Institutes of Health, RO1-GM42577 (RLG) and RO1-67836 (DJH).
Footnotes
Presented at the 2nd Annual Academic Surgical Congress (Association for Academic Surgery), Phoenix, AZ, February 2007.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.National Nosocomial Infections Surveillance (NNIS) System Report data summary from January 1992 through June 2004. Am J Infect Control. 2004;32:8. doi: 10.1016/S0196655304005425. [DOI] [PubMed] [Google Scholar]
- 2.Wibbenmeyer L, Danks R, Faucher L, Amelon M, Latenser B, Kealey GP, Herwaldt LA. Prospective analysis of nosocomial infection rates, antibiotic use, and patterns of resistance in a burn population. J Burn Care Res. 2006;27:2. doi: 10.1097/01.BCR.0000203359.32756.F7. [DOI] [PubMed] [Google Scholar]
- 3.Abraham E, Anzueto A, Gutierrez G, Tessler S, San Pedro G, Wunderink R, Dal Nogare A, Nasraway S, Berman S, Cooney R, Levy H, Baughman R, Rumbak M, Light RB, Poole L, Allred R, Constant J, Pennington J, Porter S. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet. 1998;351:9107. [PubMed] [Google Scholar]
- 4.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412. N Engl J Med. 2006;355:10. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 5.Abraham E. Why immunomodulatory therapies have not worked in sepsis. Intensive Care Med. 1999;25:6. doi: 10.1007/s001340050903. [DOI] [PubMed] [Google Scholar]
- 6.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:16. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 7.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:2. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 8.Wilson JW, Schurr MJ, LeBlanc CL, Ramamurthy R, Buchanan KL, Nickerson CA. Mechanisms of bacterial pathogenicity. Postgrad Med J. 2002;78:918. doi: 10.1136/pmj.78.918.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Obritsch MD, Fish DN, MacLaren R, Jung R. National surveillance of antimicrobial resistance in Pseudomonas aeruginosa isolates obtained from intensive care unit patients from 1993 to 2002. Antimicrob Agents Chemother. 2004;48:12. doi: 10.1128/AAC.48.12.4606-4610.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bregeon F, Papazian L, Visconti A, Gregoire R, Thirion X, Gouin F. Relationship of microbiologic diagnostic criteria to morbidity and mortality in patients with ventilator-associated pneumonia. JAMA. 1997;277:8. [PubMed] [Google Scholar]
- 11.Bassler BL. Small talk. Cell-to-cell communication in bacteria. Cell. 2002;109:4. doi: 10.1016/s0092-8674(02)00749-3. [DOI] [PubMed] [Google Scholar]
- 12.Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:5418. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 13.Lyczak JB, Cannon CL, Pier GB. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect. 2000;2:9. doi: 10.1016/s1286-4579(00)01259-4. [DOI] [PubMed] [Google Scholar]
- 14.Delves PJ, Roitt IM. The immune system. First of two parts. N Engl J Med. 2000;343:1. doi: 10.1056/NEJM200007063430107. [DOI] [PubMed] [Google Scholar]
- 15.Sebbane F, Lemaitre N, Sturdevant DE, Rebeil R, Virtaneva K, Porcella SF, Hinnebusch BJ. Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc Natl Acad Sci U S A. 2006;103:31. doi: 10.1073/pnas.0601182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoon SS, Coakley R, Lau GW, Lymar SV, Gaston B, Karabulut AC, Hennigan RF, Hwang SH, Buettner G, Schurr MJ, Mortensen JE, Burns JL, Speert D, Boucher RC, Hassett DJ. Anaerobic killing of mucoid Pseudomonas aeruginosa by acidified nitrite derivatives under cystic fibrosis airway conditions. J Clin Invest. 2006;116:2. doi: 10.1172/JCI24684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochsner UA, Vasil ML, Alsabbagh E, Parvatiyar K, Hassett DJ. Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J Bacteriol. 2000;182:16. doi: 10.1128/jb.182.16.4533-4544.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hassett DJ, Alsabbagh E, Parvatiyar K, Howell ML, Wilmott RW, Ochsner UA. A protease-resistant catalase, KatA, released upon cell lysis during stationary phase is essential for aerobic survival of a Pseudomonas aeruginosa oxyR mutant at low cell densities. J Bacteriol. 2000;182:16. doi: 10.1128/jb.182.16.4557-4563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lau GW, Britigan BE, Hassett DJ. Pseudomonas aeruginosa OxyR is required for full virulence in rodent and insect models of infection and for resistance to human neutrophils. Infect Immun. 2005;73:4. doi: 10.1128/IAI.73.4.2550-2553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melstrom KA, Muthu K, He LK, Smith J, Szilagyi A, Hassett D, Gamelli R, Shankar R. J Am Coll Surg. 3 Supplement. Vol. 203. 2006. OxyR gene expression of Pseudomonas aeruginosa regulates bone marrow dendritic cell differentiation and function after burn sepsis. [Google Scholar]
- 21.de Bruijn MF, Slieker WA, van der Loo JC, Voerman JS, van Ewijk W, Leenen PJ. Distinct mouse bone marrow macrophage precursors identified by differential expression of ER-MP12 and ER-MP20 antigens. Eur J Immunol. 1994;24:10. doi: 10.1002/eji.1830241003. [DOI] [PubMed] [Google Scholar]
- 22.Leenen PJ, de Bruijn MF, Voerman JS, Campbell PA, van Ewijk W. Markers of mouse macrophage development detected by monoclonal antibodies. J Immunol Methods. 1994;174:1–2. doi: 10.1016/0022-1759(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 23.Muthu K, Deng J, Romano F, He LK, Gamelli R, Shankar R, Jones SB. Thermal injury and sepsis modulates beta-adrenergic receptors and cAMP responses in monocyte-committed bone marrow cells. J Neuroimmunol. 2005;165:1–2. doi: 10.1016/j.jneuroim.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Santangelo S, Gamelli RL, Shankar R. Myeloid commitment shifts toward monocytopoiesis after thermal injury and sepsis. Ann Surg. 2001;233:1. doi: 10.1097/00000658-200101000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kostakioti M, Newman CL, Thanassi DG, Stathopoulos C. Mechanisms of protein export across the bacterial outer membrane. J Bacteriol. 2005;187:13. doi: 10.1128/JB.187.13.4306-4314.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vance RE, Rietsch A, Mekalanos JJ. Role of the type III secreted exoenzymes S, T, and Y in systemic spread of Pseudomonas aeruginosa PAO1 in vivo. Infect Immun. 2005;73:3. doi: 10.1128/IAI.73.3.1706-1713.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galan JE, Collmer A. Type III secretion machines: bacterial devices for protein delivery into host cells. Science. 1999;284:5418. doi: 10.1126/science.284.5418.1322. [DOI] [PubMed] [Google Scholar]
- 28.Winsor GL, Lo R, Sui SJ, Ung KS, Huang S, Cheng D, Ching WK, Hancock RE, Brinkman FS. Pseudomonas aeruginosa Genome Database and PseudoCAP: facilitating community-based, continually updated, genome annotation. Nucleic Acids Res. 2005;33(Database issue) doi: 10.1093/nar/gki047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Usher LR, Lawson RA, Geary I, Taylor CJ, Bingle CD, Taylor GW, Whyte MK. Induction of neutrophil apoptosis by the Pseudomonas aeruginosa exotoxin pyocyanin: a potential mechanism of persistent infection. J Immunol. 2002;168:4. doi: 10.4049/jimmunol.168.4.1861. [DOI] [PubMed] [Google Scholar]
- 30.Dacheux D, Toussaint B, Richard M, Brochier G, Croize J, Attree I. Pseudomonas aeruginosa cystic fibrosis isolates induce rapid, type III secretion-dependent, but ExoU-independent, oncosis of macrophages and polymorphonuclear neutrophils. Infect Immun. 2000;68:5. doi: 10.1128/iai.68.5.2916-2924.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shafikhani SH, Engel J. Pseudomonas aeruginosa type III-secreted toxin ExoT inhibits host-cell division by targeting cytokinesis at multiple steps. Proc Natl Acad Sci U S A. 2006;103:42. doi: 10.1073/pnas.0605949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng J, Muthu K, Gamelli R, Shankar R, Jones SB. Adrenergic modulation of splenic macrophage cytokine release in polymicrobial sepsis. Am J Physiol Cell Physiol. 2004;287:3. doi: 10.1152/ajpcell.00562.2003. [DOI] [PubMed] [Google Scholar]
- 33.Muthu K, Deng J, Gamelli R, Shankar R, Jones SB. Adrenergic modulation of cytokine release in bone marrow progenitor-derived macrophage following polymicrobial sepsis. J Neuroimmunol. 2005;158:1–2. doi: 10.1016/j.jneuroim.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80:8. [PubMed] [Google Scholar]
- 35.Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:2. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu H, Coburn J, Collier RJ. The eukaryotic host factor that activates exoenzyme S of Pseudomonas aeruginosa is a member of the 14-3-3 protein family. Proc Natl Acad Sci U S A. 1993;90:6. doi: 10.1073/pnas.90.6.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee VT, Pukatzki S, Sato H, Kikawada E, Kazimirova AA, Huang J, Li X, Arm JP, Frank DW, Lory S. PseudolipasinA is a Specific Inhibitor for PhospholipaseA2 Activity of Pseudomonas aeruginosa Cytotoxin ExoU. Infect Immun. 2006 doi: 10.1128/IAI.01184-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong KF, Jayawardena SR, Indulkar SD, Del Puerto A, Koh CL, Hoiby N, Mathee K. Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB beta-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob Agents Chemother. 2005;49:11. doi: 10.1128/AAC.49.11.4567-4575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]