

Abstract
XBP-1 is a CREB/ATF family transcription factor highly expressed in hepatocellular carcinomas. Here we report that XBP-1 is essential for liver growth. Mice lacking XBP-1 displayed hypoplastic fetal livers, whose reduced hematopoiesis resulted in death from anemia. Nevertheless, XBP-1-deficient hematopoietic progenitors had no cell-autonomous defect in differentiation. Rather, hepatocyte development itself was severely impaired by two measures: diminished growth rate and prominent apoptosis. Specific target genes of XBP-1 in the liver were identified as αFP, which may be a regulator of hepatocyte growth, and three acute phase protein family members. Therefore, XBP-1 is a transcription factor essential for hepatocyte growth.
Keywords: Transcription factors, liver development, acute phase protein, CREB/ATF family, apoptosis, hepatocyte.
Members of the CREB(CRE-binding protein)/ATF family of transcription factors form dimers and bind to cAMP response elements found in a large number of cellular promoters. The diversity of genes regulated by this large group of transcription factors is reflected in the essential functions of individual factors in fetal survival, neurological development, bone growth, and immune system activation (Reimold et al. 1996a; Rudolph et al. 1998; Maekawa et al. 1999). Recently, an important role in coordinating the timing of hepatocyte proliferation in the regenerating liver was demonstrated for the CREB/ATF family member CREM (CRE modulator; Servillo et al. 1998). In addition, ATF-3 was found to be induced in regenerating liver with the kinetics of an early response gene (Chen et al. 1996).
The functions of a further CREB/ATF family member, XBP-1 (X-box binding protein-1), have not been defined in detail. This transcription factor is expressed ubiquitously in adults but is found mainly in exocrine glands and bone precursors in the embryonic mouse (Liou et al 1990; Clauss et al. 1993). In vitro studies have demonstrated down-regulation of the XBP-1 gene by BSAP (B cell-specific activator protein), dimerization of XBP-1 protein with c-Fos, and a decrease in MHC class II gene expression when antisense XBP-1 sequences are introduced into Raji cells (Ono et al. 1991; Reimold et al. 1996b). Recently, the expression of XBP-1 was found to be increased dramatically in hepatocellular carcinomas (Kishimoto et al 1998), raising the possibility that XBP-1 may have a function in the liver, as well.
Results
Disruption of the XBP-1 gene
To define the actions of the XBP-1 protein in vivo, the XBP-1 gene was disrupted in embryonic stem (ES) cells by replacing a 0.8-kb fragment containing parts of exons 1 and 2, as well as the intervening intron with a neomycin resistance gene, resulting in a frameshift of the remaining amino acids (Fig. 1A). One of three ES clones that had undergone targeted disruption of the XBP-1 gene transmitted the disrupted allele to offspring and XBP-1+/− mice were intercrossed to generate XBP−/− mice, as demonstrated by Southern blot analysis of genomic DNA from embryos (Fig. 1B). Northern blot analysis of total cellular RNA made from +/+ and −/− embryos revealed an absence of a correctly sized XBP-1 transcript in −/− samples and the appearance of a fainter, higher molecular weight transcript (Fig. 1C), which also hybridized with a neomycin cDNA probe. Western blot analysis of extracts from XBP-1 +/+, +/−, and −/− fetal livers using a monoclonal antiserum specific for XBP-1 revealed an absence of immunoreactive XBP-1 protein in the −/− samples (Fig. 1D).
Figure 1.

Disruption of the XBP-1 gene. (A) Parts of XBP-1 exons 1 and 2 were deleted and replaced by the neo resistance gene, in the opposite orientation from XBP-1. (B) Southern blotting of genomic DNA from yolk sacs of XBP-1 +/+, +/−, and −/− embryos. (C) Northern blot of whole embryo RNA from XBP-1 +/+ and −/− embryos, probed with 32P-labeled XBP-1 coding region cDNA. (D) Western blot of nuclear cell extracts made from XBP-1 +/+ and −/− fetal livers and probed with XBP-1 monoclonal antibody. (E) Diminished survival of XBP-1−/− embryos seen in offspring of XBP-1+/− × XBP-1+/− matings genotyped by Southern blotting of genomic DNA. No live XBP-1−/− embryos were identified past E14.5. (ND) Not determined.
Embryonic lethality from liver hypoplasia in XBP-1−/− mice
Matings of heterozygous XBP-1 mice produced no −/− live births. Of >400 pups born, no homozygotes were obtained, suggesting that XBP-1 is necessary for survival. Genotyping of litters harvested from serial timed matings revealed embryonic lethality beginning at E12.5 (Fig. 1E). By E13.5, XBP-1−/− embryos could be recognized by their growth retardation, pale coloration, and hypoplastic livers. Inspection of −/− livers showed them to be markedly reduced in size compared to normal E13.5 livers (Fig. 2A), and total liver cell counts were 15% of +/+ livers at this age. Histologic analysis of −/− E14.5 livers revealed reduced cellularity and increased empty space, indicating less dense packing of cells in the −/− as compared to the control livers (Fig. 2B,C).
Figure 2.
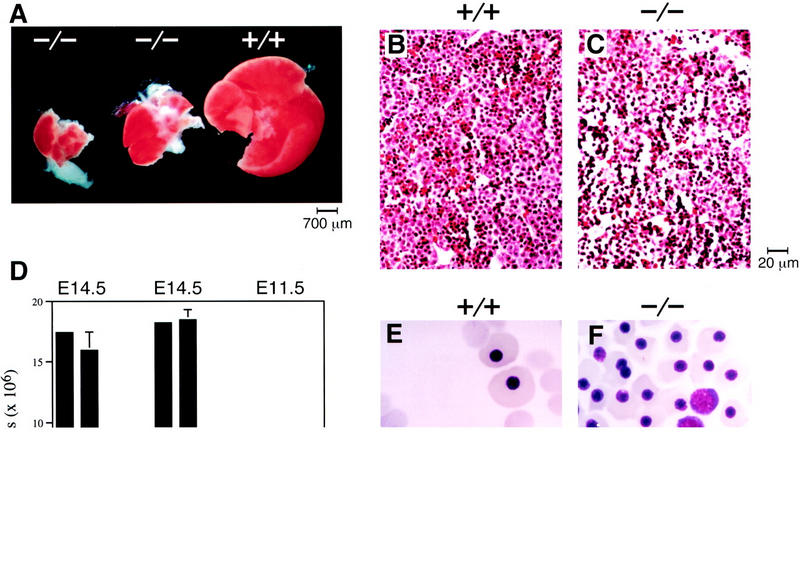
Liver hypoplasia and anemia in XBP-1-deficient embryos. (A) Hypoplasia of E13.5 XBP-1−/− embryonic livers (middle and left) compared to a XBP-1+/+ littermate control (right). (B,C) Sections of E14.5 +/+ and −/− livers, hematoxylin–eosin stain, 200× magnification. (D) Peripheral blood counts of +/+ and −/− E14.5 and E11.5 embryos. (E,F) Cytospin preparations of blood from E13.5 +/+ and −/− embryos. Nucleated red blood cells represent cells of yolk sac origin; nonnucleated cells derive from liver hematopoiesis.
Because the fetal liver becomes the main hematopoietic organ by E13.5, fetal blood counts were determined to assess whether severe liver hypoplasia correlated with abnormal red blood cell production. Anemia became evident in XBP-1−/− embryos after E11.5 as hematopoiesis switched from the yolk sac to the fetal liver. By E14.5, the total blood counts of surviving XBP-1−/− mice were 20% of the values found in normal littermates (Fig. 2D). Cytospin preparations of peripheral blood at E13.5 revealed that −/− erythroid cells were predominantly immature, nucleated cells of yolk sac origin, whereas +/+ erythroid cells were 80% liver-derived nonnucleated cells (Fig. 2E,F). Cytospin preparations of E13.5 liver cells demonstrated the presence of erythroid and myeloid lineage cells at all stages of maturity in −/− specimens, though in reduced numbers consistent with the hypoplastic livers (not shown).
To compare the potential of +/+ and −/− hematopoietic progenitor cells to develop into the erythroid and myeloid lineages, in vitro methylcellulose colony assays were performed using fetal AGM (aorto-gonad mesonephros), yolk sac, or liver as the source of progenitor cells (Muller et al. 1994; Wang et al. 1997). All three of these tissues gave rise to equivalent numbers of erythroid and myeloid colonies from +/+ and −/− samples (not shown), indicating that pluripotent hematopoietic stem cells are found in all three locations. Although −/− fetal livers are severely hypoplastic, some hematopoietic stem cells do migrate there and have the potential to give rise to all hematopoietic lineages, as also seen in cytospin preparations of fetal liver (not shown). Because the XBP-1−/− hematopoietic committed progenitor cells were able to proliferate and differentiate normally when tested in vitro, the anemia seen in these embryos cannot be attributed to a hematopoietic cell-autonomous defect. These findings were reinforced by an analysis of chimeric mice derived from XBP-1-deficient ES cells in the RAG-2-deficient complementation system. In these animals, the livers were almost exclusively of RAG-2-deficient cell origin, whereas reconstitution of hematopoietic elements such as B and T lymphocytes from XBP-1-deficient precursors occurred readily (A.M. Reimold, F. Alt, and L.H. Glimcher, in prep.). Therefore, although XBP-1-deficient ES cells do not contribute to normal liver development, they are not defective in the reconstitution of hematopoietic elements.
XBP-1 expression in the developing liver
The fetal liver begins its development as an outpouching of foregut endoderm (Gualdi et al. 1996). At E10.5, the liver bud was found to express high levels of XBP-1 mRNA by in situ hybridization (Fig. 3). As this represents a time point before significant population of the liver by hematopoietic cells, it demonstrates that XBP-1 is expressed in hepatic parenchymal cells. Northern blotting of RNA also showed high levels of XBP-1 mRNA in hepatic stromal cell lines and in HepG2 hepatocarcinoma cells (not shown). This evidence further supports a role for XBP-1 in liver growth rather than in hematopoietic cell division and differentiation.
Figure 3.
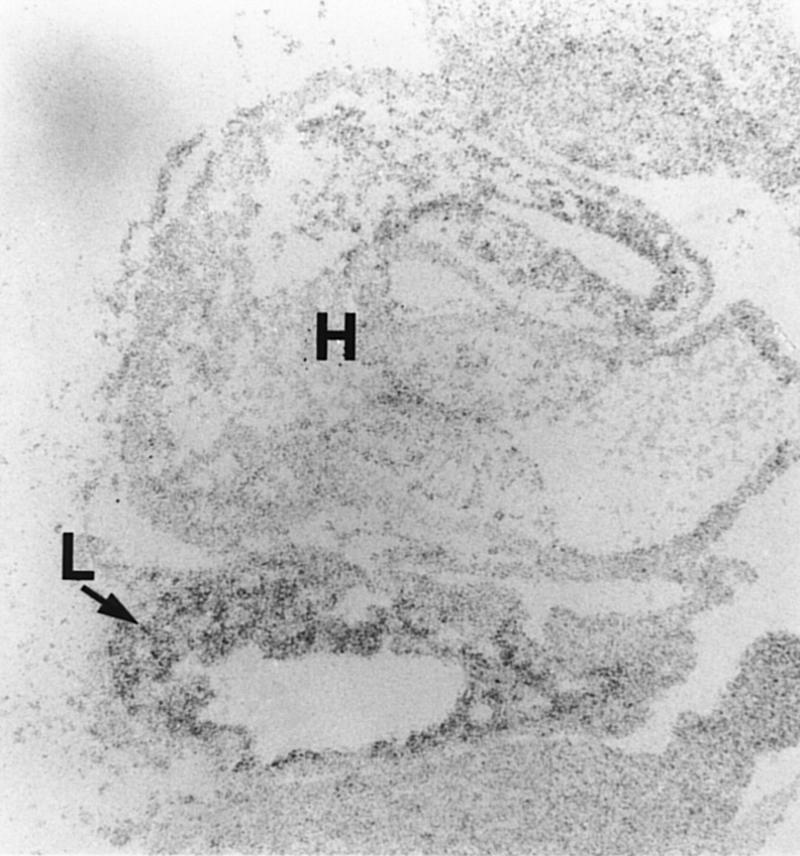
XBP-1 expression during liver growth. In situ hybridization of a wild-type E10.5 embryo showing XBP-1 signal in the developing liver. (L) Liver; (H) Heart.
Reduced growth rate and increased apoptosis in XBP-1-deficient livers
Two mechanisms that account for the severely hypoplastic livers in XBP-1−/− embryos were identified: reduced growth rate and increased apoptosis. To directly demonstrate reduced cell growth, mice at day 13.5 of pregnancy were injected with BrdU, and the fetuses were harvested and analyzed for BrdU incorporation. Wild-type fetal livers showed heavy nuclear staining in the entire liver, whereas XBP-1-deficient samples had less staining with significant areas remaining unstained, especially toward the center of the liver (Fig. 4A–D). The reduced BrdU incorporation in XBP-1−/− livers directly demonstrates a subnormal rate of growth in this organ.
Figure 4.
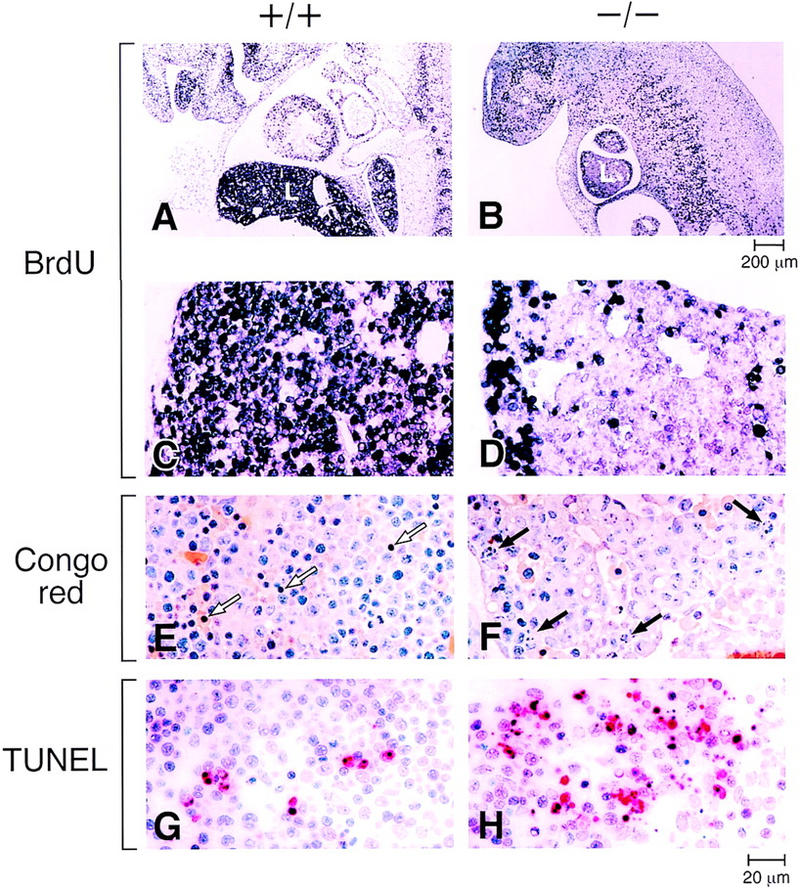
Reduced growth rate and increased apoptosis in XBP-1-deficient livers. (A,B) BrdU labeling of E13.5 XBP-1+/+ and XBP-1−/− embryos. (C,D) Higher power views. The −/− liver (L) is smaller in size and labels less intensely than the +/+ control. (E,F) Congo red staining of E13.5 livers. Arrows indicate morphologically apoptotic cells, which were mainly hematopoietic cells in +/+ samples and hepatocytes in −/− samples. (G,H) TUNEL staining (in red) of E13.5 +/+ and −/− livers.
Apoptosis was identified morphologically (Fig. 4E,F) and by TUNEL staining of E13.5 fetal livers (Fig. 4G,H), showing a markedly elevated rate of apoptotic hepatocytes in XBP-1−/− samples. In contrast, the apoptotic cells identified in +/+ livers were generally of the myeloid lineage (Fig. 4E). The use of chloroacetate esterase, which stains select myeloid lineage cells but not the liver parenchyma, was used to confirm that hepatocytes, but not hematopoietic cells, accounted for most of the apoptotic cells in −/− liver samples (not shown).
XBP-1 induction after partial hepatectomy
To further study the role of XBP-1 in liver growth and gene induction, partial hepatectomies were performed on normal adult mice. In this model the remnant liver reverts to a quasifetal phenotype and undergoes rapid cell division to re-establish the original weight of the organ within 10 days (Michalopoulos and DeFrances 1997). Among the first steps in this process is the activation of preformed transcription factors such as NF-κB or STAT3 (Taub 1996). These factors then induce transcription of immediate early genes, many of which encode transcription factors such as AP-1, NF-κB, and certain CREB/ATF family members such as CREB and CREM (Servillo et al. 1998; Taub 1996). Results of partial hepatectomies in mice now demonstrate that XBP-1 is an immediate early gene in this process, induced within 30 min of surgery but having a prolonged peak of induction past 16 hr (Fig. 5A). This time course is similar to the induction of C/EBPβ(CCAAT/enhancer binding proteinβ), which peaks slightly earlier at 14 hr post-hepatectomy. It is expected that XBP-1 acts as a homodimer or heterodimer to bind at CRE-like sites and, in turn, up-regulates the delayed-early genes involved in liver regeneration. The decreased liver cell proliferation seen in XBP-1−/− embryos indicates that XBP-1, like CREB and CREM, is a CRE-binding transcription factor involved in hepatic proliferation.
Figure 5.

XBP-1 induction in the acute phase response. (A) Gene induction after partial hepatectomy. Total cellular RNA collected in a time course after partial hepatectomy in +/+ mice was probed sequentially with the cDNAs for XBP-1, C/EBPβ, and control β2 microglobulin. (B) Northern blot of mRNA prepared from +/+ (E12.5 or E13.5) or −/− (E13.5) fetal livers, or from the embryo torsos with the livers removed (body). The blot was sequentially hybridized with probes for α1AT, αFP, transthyretin, apoA-1, vitamin D binding protein (VDBP), C-reactive protein (CRP), serum amyloid P (SAP), and γ-actin. (C) Transfections of an α1AT reporter construct or an α1AT reporter containing a mutation of the −1100-bp presumptive XBP-1 binding site (mut α1AT; the ACGT core changed to GTAG) (Clauss et al. 1996), along with an XBP-1 expression plasmid (XBP) or a frameshifted mutant XBP-1 plasmid (XBP-Δ) (Reimold et al. 1996b).
Identification of XBP-1 target genes in liver
To identify target genes for XBP-1 action in the liver, differential hybridization to microarray chips was compared in samples derived from E13.5 +/+ or −/− livers. The genes for α1-antitrypsin (α1AT) and α-fetoprotein (αFP) were found to be expressed at significantly reduced levels in −/− samples, as also shown in Northern blots of +/+ and −/− fetal liver mRNA (Fig. 5B). α1AT belongs to the family of acute phase proteins, whereas αFP is expressed strongly in dividing hepatocytes. Two other acute phase proteins, transthyretin and apolipoprotein A1 (apoA-1) were also found to have decreased levels of mRNA in −/− livers, whereas multiple other acute phase proteins and hepatocyte-expressed genes [vitamin D-binding protein, C-reactive protein, serum amyloid P, α1-acid glycoprotein, hepatocyte growth factor (HGF)] were expressed equally in +/+ and −/− samples (Fig. 5B; data not shown). This indicates that XBP-1-deficient hepatocytes have defects in the synthesis of specific gene products rather than a global reduction in transcription.
To demonstrate direct regulation of the α1AT promoter by XBP-1, transient transfections were performed using an XBP-1 expression plasmid and α1AT luciferase reporter constructs in the HepG2 cell line. Although this cell line contains endogenous XBP-1, overexpression of XBP-1 could transactivate the α1AT promoter by 3.5-fold (Fig. 5C). As controls for specificity of the XBP-1 effect, the use of a frameshifted XBP-1 expression construct or the mutation of an XBP-1 target site in the α1AT promoter eliminated all transactivation. Transactivation of an αFP reporter construct by the XBP-1 expression plasmid was also demonstrated in vitro (not shown). Therefore, XBP-1 has a specific transcriptional effect on these two hepatocyte-expressed genes.
Discussion
The phenotype of XBP-1-deficient embryos shows similarities to several mouse models in which gene disruptions result in abnormal liver development. Disruption of the homeobox gene Hlx resulted in initial liver specification but only minimal growth and represents an earlier, more severe defect than XBP-1 deficiency (Hentsch et al. 1996). Abnormal liver growth has been described in HGF/SF (hepatocyte growth factor/scatter factor)-deficient embryos with a phenotype similar to the XBP-1−/− embryos: loosened liver structure, enlarged sinusoidal spaces, and dissociation of parenchymal cells (Schmidt et al. 1995; Uehara et al. 1995). However, HGF/SF mRNA levels are normal in XBP-1−/− embryos and in vitro treatment of hepatocytes with HGF/SF did not lead to induction of XBP-1 (data not shown). Therefore, XBP-1 is unlikely to be an important element in the HGF/SF signaling pathway. MTF-1 deficiency resulted in a dissociated hepatic epithelial compartment and enlarged sinusoids, but unlike XBP-1−/− embryos, no significant decrease in liver size and no anemia (Gunes et al. 1998). Deficiency in c-Jun also led to hypoplasia and dissociation of liver cells and death by E15.5 (Hilberg et al. 1993; Johnson et al. 1993), but c-jun mRNA levels were normal in XBP-1−/− embryos. A small liver can be the result of abnormal hematopoietic cell proliferation, as described in c-myb-deficient embryos (Mucenski et al. 1991), or abnormal erythroid cell proliferation and differentiation, as seen in Rb−/− embryos (Jacks et al. 1992; Lee et al. 1992). In contrast, we found normal in vitro proliferative and differentiative function of XBP-1−/− hematopoietic progenitor cells. The homing of hematopoietic cells to the liver was the presumed defect in the absence of β1 integrin (Faessler and Meyer 1995; Hirsch et al. 1996), but XBP-1−/− livers showed normal hematopoietic potential on a per-cell basis. Finally, apoptosis has contributed to liver failure in Rb−/−, RelA−/− (Beg et al. 1995), and HGF/SF−/− embryos (Schmidt et al. 1995; Uehara et al. 1995), as was also seen in XBP-1−/− embryos. XBP-1 is not known to act directly upstream of these multiple factors that are important in normal hepatogenesis, but its potential to be a downstream target for these factors has not been evaluated in most cases. In addition, it is possible that XBP-1−/− hepatic parenchymal cells may be defective in their ability to support proliferation of hematopoietic progenitor cells. Analysis of mRNA has not identified altered levels of β1 integrin, HGF/SF, c-myb, c-kit ligand, AML-1, Rb, or c-jun in XBP-1−/− embryos (not shown), making these genes unlikely to be major targets of regulation by XBP-1 during liver development.
Instead, our data have identified four genes expressed in hepatocytes during liver growth as specific targets of XBP-1. The induction of acute phase proteins represents liver-specific gene activation, whether during embryonic development or in adults after injury from partial hepatectomy, inflammation, infection, toxins, or malignancy. The systemic role of some acute phase proteins has been defined in detail, such as the serum protease inhibition by α1AT or the antiatherogenic properties of apoA-1 (Andersson 1997; Gabay and Kushner 1999). A reduction in the levels of these acute phase proteins reflects a decrease in specific hepatocyte protein synthesis, but individually, deficiency of these genes is not known to result in the defects observed in XBP-1-deficient livers. However, the regulation of αFP by XBP-1 provides one possible explanation for the growth defect seen in XBP-1-deficient hepatocytes. αFP is highly expressed in fetal liver and yolk sac and represents one of the earliest phenotypic markers of the rapidly growing fetal liver as it develops from the foregut endoderm (Gualdi et al. 1996). αFP expression is down-regulated in adult liver but becomes strongly induced when hepatocytes are stimulated to resume growth, such as after partial hepatectomy or in hepatocellular carcinomas. Interestingly, the times when αFP levels are highest are also the times of strong XBP-1 expression in the liver. Although the precise role of αFP has not been established, it has been proposed that its major function is to promote cell growth in the liver, possibly by sequestering estrogen, which otherwise has antiproliferative effects (for review, see Chen et al. 1997). We have demonstrated that in the absence of XBP-1, αFP is expressed only at reduced levels and that liver growth is severely impaired. Our data establish that XBP-1 is involved in the growth and survival of hepatocytes and, through its regulation of acute phase protein genes, also in the expression of liver-specific genes. These studies have demonstrated that XBP-1 directly controls a subset of the liver's protein synthetic activity and that normal liver growth cannot be achieved in the absence of XBP-1 protein.
Materials and methods
Disruption of the XBP-1 gene
The targeting construct was generated by cloning the disrupted XBP-1 fragment into the vector pBS/TK, followed by linearization and introduction into D3 ES cells by electroporation. The cells were grown in neomycin and Gancyclovir to achieve positive/negative selection, and resistant clones were tested for homologous integration of the disrupted XBP-1 fragment by performing Southern blots of genomic DNA. Three clones had correctly targeted XBP-1 and were injected into blastocysts, transferred to pseudopregnant females, and produced chimeric offspring. One clone resulted in germ-line transmission of the disrupted allele.
Phenotype analysis
Methylcellulose colony assays were performed as described (Wang et al. 1997), with cultures using erythropoietin alone or erythropoietin, IL-1, IL-3, and G-CSF. For in situ hybridization, wild-type embryos were sectioned and probed for XBP-1 as described (Clauss et al. 1993). To perform BrdU labeling and TUNEL assays, pregnant mice were injected IV with 0.3 ml of 50 mg/ml BrdU (Sigma) in PBS. Embryos were harvested after 1 hr and fixed in Carnoy's fixative. After embedding in paraffin, detection of BrdU was performed following the instructions of the BrdU labeling kit (Roche). The TUNEL assay was performed using the Cell Death Kit (Roche). Partial hepatectomies removed the left and caudate lobes of the liver from wild-type adult mice. Remaining liver tissue was harvested in a time course for total RNA isolation. Northern blots were probed with cDNA for XBP-1, C/EBPβ, and the control gene β2M. Transfection of the HepG2 cell line was by calcium phosphate coprecipitation (Ausubel et al. 1987) using 1 μg of the α1AT (1.3 kb of the proximal promoter) or αFP (1.0 kb) luciferase reporter plasmids (gift of I. Stamenkovic, Harvard Medical School, Boston, MA) along with 3 μg of the XBP-1 expression constructs in the vector pcDNA-1. Transfection efficiencies were controlled for by a separate CMV-luciferase reporter construct. Luciferase activities from at least three experiments were used to calculate mean values and standard deviation.
Differential hybridization to microarray chips
RNA was isolated from +/+ and −/− E13.5 livers by lysis in guanidine and centrifugation through a CsCl cushion. Ten micrograms of total RNA were converted to double stranded cDNA using an oligo dT primer with a T7 RNA polymerase site on its 5′ end (5′-GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGG-3′). The cDNA was used directly in an in vitro transcription reaction in the presence of biotinylated nucleotides Bio-11-UTP and Bio-11-CTP (Enzo, Farmingdale, NY). To improve hybridization kinetics, the labeled antisense RNA was fragmented by incubating at 94°C for 35 min in 30 mm MgOAc, 100 mm KOAc. Hybridization to GenechipsTM (Affymetrix, San Jose, CA), displaying probes for 250 genes of immunological interest or 250 genes with roles in development was done at 40°C overnight in a mix including 10 μg of fragmented RNA, 6× SSPE, 0.005% Triton X-100, and 100 μg/ml herring sperm DNA in a total volume of 200 μl. Chips were washed, stained with phycoerythrin–streptavidin, and read using an Affymetrix GeneChip scanner and accompanying gene expression software. The software includes algorithms that determine whether a gene is absent or present and whether the expression level of a gene in the −/− sample was significantly increased or decreased relative to the +/+ sample.
Acknowledgments
We thank P. Fuschi for technical assistance, A. Yamada and H. Auchincloss for performing the hepatectomies, P. Zhang and D. Tenen for help with methylcellulose assays, T. Koh and T. Wang for help with BrdU assays, and I. Stamenkovic for gift of a plasmid. This work was supported by grants from the Arthritis Foundation and the NIH (A.M.R., L.H.G.), the Leukemia Society of America (A.P., M.J.G.), and a grant from the G. Harold and Leila Y. Mathers Charitable Foundation (L.H.G.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lglimche@hsph.harvard.edu; FAX (617) 432-0084.
References
- Andersson LO. Pharmacology of apolipoprotein A-I. Curr Opin Lipidol. 1997;8:225–228. doi: 10.1097/00041433-199708000-00006. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York, New York: Greene/Wiley Interscience; 1987. [Google Scholar]
- Beg AA, Sha WC, Bronson R T, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Chen BP, Wolfgang CD, Hai T. Analysis of ATF3, a transcription factor induced by physiological stresses and modulated by gadd153/Chop10. Mol Cell Biol. 1996;16:1157–1168. doi: 10.1128/mcb.16.3.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Egan JO, Chiu JF. Regulation and activities of alpha-Fetoprotein. Crit Rev Eukaryot Gene Exp. 1997;7:11–41. doi: 10.1615/critreveukargeneexpr.v7.i1-2.20. [DOI] [PubMed] [Google Scholar]
- Clauss IM, Chu M, Zhao J-L, Glimcher LH. The basic domain/leucine zipper protein hXBP-1 preferentially binds to and transactivates CRE-like sequences containing an ACGT core. Nucleic Acids Res. 1996;24:1855–1864. doi: 10.1093/nar/24.10.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss IM, Gravallese EM, Darling JM, Shapiro F, Glimcher MJ, Glimcher LH. In situ hybridization studies suggest a role for the basic region-leucine zipper protein hXBP-1 in exocrine gland and skeletal development during mouse embryogenesis. Dev Dyn. 1993;197:146–156. doi: 10.1002/aja.1001970207. [DOI] [PubMed] [Google Scholar]
- Faessler R, Meyer M. Consequences of lack of β1 integrin gene expression in mice. Genes & Dev. 1995;9:1896–1908. doi: 10.1101/gad.9.15.1896. [DOI] [PubMed] [Google Scholar]
- Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. New Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- Gualdi R, Bossard P, Zheng M, Hamada Y, Coleman JR, Zaret KS. Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control. Genes & Dev. 1996;10:1670–1682. doi: 10.1101/gad.10.13.1670. [DOI] [PubMed] [Google Scholar]
- Gunes C, Heuchel R, Georgiew O, Muller KH, Lichtlen P, Bluthmann H, Marino S, Aguzzi A, Schaffner W. Embryonic lethality and liver degeneration in mice lacking the metal-responsive transcriptional activator MTF-1. EMBO J. 1998;17:2846–2854. doi: 10.1093/emboj/17.10.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentsch B, Lyons I, Li R, Hartley L, Lints TJ, Adams JM, Harvey RP. Hlx homeo box gene is essential for an inductive interaction that drives expansion of embryonic liver and gut. Genes & Dev. 1996;10:70–79. doi: 10.1101/gad.10.1.70. [DOI] [PubMed] [Google Scholar]
- Hilberg F, Aguzzi A, Howells N, Wagner EF. c-Jun is essential for normal mouse development and hepatogenesis. Nature. 1993;365:179–181. doi: 10.1038/365179a0. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Iglesias A, Potocnik AJ, Hartmann U, Faessler R. Impaired migration but not differentiation of haematopoietic stem cells in the absence of β1 integrins. Nature. 1996;380:171–175. doi: 10.1038/380171a0. [DOI] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Johnson RS, van Lingen B, Papaioannou VE, Spiegelman BM. A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes & Dev. 1993;7:1309–1317. doi: 10.1101/gad.7.7b.1309. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Kokura K, Ohkawa N, Makino Y, Yoshida M, Hirohashi S, Niwa S, Muramatsu M, Tamura T. Enhanced expression of a new class of liver-enriched b-zip transcription factors, hepatocarcinogenesis-related transcription factor, in hepatocellular carcinomas of rats and humans. Cell Growth Differ. 1998;9:337–344. [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Liou H-C, Boothby MR, Finn PW, Davidon R, Nabavi N, Zeleznik-Le NJ, Ting JP, Glimcher LH. A new member of the leucine zipper class of proteins that binds to the HLA DRα promoter. Science. 1990;247:1581–1584. doi: 10.1126/science.2321018. [DOI] [PubMed] [Google Scholar]
- Maekawa T, Bernier F, Sato M, Nomura S, Singh M, Inoue Y, Tokunaga T, Imai H, Yokoyama M, Reimold A, et al. Mouse ATF-2 null mutants display features of a severe type of meconium aspiration syndrome. J Biol Chem. 1999;274:17813–17819. doi: 10.1074/jbc.274.25.17813. [DOI] [PubMed] [Google Scholar]
- Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- Mucenski ML, McLain K, Kier AB, Swerdlow SH, Schreiner CM, Miller TA, Pietryga DW, Scott WJ, Jr, Potter SS. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell. 1991;65:677–689. doi: 10.1016/0092-8674(91)90099-k. [DOI] [PubMed] [Google Scholar]
- Muller AM, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity. 1994;1:291–301. doi: 10.1016/1074-7613(94)90081-7. [DOI] [PubMed] [Google Scholar]
- Ono SJ, Liou HC, Davidon R, Strominger JL, Glimcher LH. Human X-box-binding protein 1 is required for the transcription of a subset of human class II major histocompatibility genes and forms a heterodimer with c-fos. Proc Natl Acad Sci. 1991;88:4309–4312. doi: 10.1073/pnas.88.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold AM, Grusby MJ, Kosaras B, Fries JW, Mori R, Maniwa S, Clauss IM, Collins T, Sidman RL, Glimcher MJ, Glimcher LH. Chondrodysplasia and neurological abnormalities in ATF-2-deficient mice. Nature. 1996a;379:262–265. doi: 10.1038/379262a0. [DOI] [PubMed] [Google Scholar]
- Reimold AM, Ponath PD, Li YS, Hardy RR, David CS, Strominger JL, Glimcher LH. Transcription factor B cell lineage-specific activator protein regulates the gene for human X-box binding protein 1. J Exp Med. 1996b;183:393–401. doi: 10.1084/jem.183.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph D, Tafuri A, Gass P, Hammerling GJ, Arnold B, Schutz G. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci. 1998;95:4481–4486. doi: 10.1073/pnas.95.8.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, Gherardi E, Birchmeier C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373:699–702. doi: 10.1038/373699a0. [DOI] [PubMed] [Google Scholar]
- Servillo G, Della Fazia MA, Sassone-Corsi P. Transcription factor CREM coordinates the timing of hepatocyte proliferation in the regenerating liver. Genes & Dev. 1998;12:3639–3643. doi: 10.1101/gad.12.23.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taub R. Transcriptional control of liver regeneration. FASEB J. 1996;10:413–427. [PubMed] [Google Scholar]
- Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, Kitamura N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373:702–705. doi: 10.1038/373702a0. [DOI] [PubMed] [Google Scholar]
- Wang LC, Kuo F, Fujiwara Y, Gilliland DG, Golub TR, Orkin SH. Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. EMBO J. 1997;16:4374–4383. doi: 10.1093/emboj/16.14.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]