

Abstract
We have examined the cellular function of Sgs1p, a nonessential yeast DNA helicase, homologs of which are implicated in two highly debilitating hereditary human diseases (Werner's and Bloom's syndromes). We show that Sgs1p is an integral component of the S-phase checkpoint response in yeast, which arrests cells due to DNA damage or blocked fork progression during DNA replication. DNA polε and Sgs1p are found in the same epistasis group and act upstream of Rad53p to signal cell cycle arrest when DNA replication is perturbed. Sgs1p is tightly regulated through the cell cycle, accumulates in S phase and colocalizes with Rad53p in S-phase-specific foci, even in the absence of fork arrest. The association of Rad53p with a chromatin subfraction is Sgs1p dependent, suggesting an important role for the helicase in the signal-transducing pathway that monitors replication fork progression.
Keywords: Sgs1p helicase, DNA replication, checkpoint, DNA damage, Bloom's syndrome, Werner's syndrome
As cells proliferate, the genome must be replicated and segregated with high fidelity. To ensure this event, DNA replication takes place in a defined temporal and spatial order. Temporally, DNA synthesis is restricted to S phase of the cell cycle, and spatially, synthesis is concentrated at distinct foci within the nucleus. Such foci are thought to ensure a high local concentration of the factors critical for DNA replication (for review, see Coverley and Laskey 1994; Pasero and Gasser 1998). Surveillance mechanisms, called checkpoints, detect damaged or replicating DNA and block cell cycle progression to allow adequate time to repair the damaged DNA or to finish DNA replication (Weinert 1998). In humans, deficiency for these checkpoints results in chromosome instability and a propensity for cancer, which underscores the fundamental importance that such surveillance mechanisms have for ensuring the integrity of the genome (Kastan 1997).
Several years ago, a novel gene was isolated from Saccharomyces cerevisiae which, when mutated, led to an increase in genome instability. This yeast gene, SGS1, encodes a 3′ to 5′ DNA helicase, a structural homolog of the RecQ helicase of Escherichia coli, which is implicated in DNA recombination processes (Gangloff et al. 1994; Watt et al. 1995). Mutation of SGS1 leads to an increase in mitotic recombination, particularly of repeated sequences, and elicits an increase in chromosome loss of 10- and 37-fold in mitosis and meiosis, respectively (Watt et al. 1996). Two-hybrid and in vitro binding assays suggest that Sgs1p interacts with DNA topoisomerases (topo) II and III, whereas genetic studies reveal synergism between Sgs1p and DNA topo I and topo III (Gangloff et al. 1994; Watt et al. 1995; Lu et al. 1996). Namely, an sgs1 top1 double mutant has a slower growth rate than either single mutant (Lu et al. 1996), whereas sgs1 mutations suppress the reduced growth rate provoked by loss of topo III, a single-strand, type-I topoisomerase (Gangloff et al. 1994). The physiological relevance of the interactions between Sgs1p with these enzymes is not yet clear, but it has been proposed that the combination of DNA helicase and topoisomerase activities could be used to facilitate aspects of DNA replication or recombination, by either introducing positive supercoils or helping to resolve intertwined strands where replication forks meet (Watt and Hickson 1996).
More recently, it has been shown that the life span of a yeast carrying a sgs1::HIS3 disruption is reduced by ∼60%, probably because of the accumulation of circular rDNA episomes, which result from enhanced recombination between direct rDNA repeats (Sinclair et al. 1997). The enhanced recombination rate may be an indirect product of Sgs1p inactivation, because this phenotype was at least partially independent of RAD52 and RAD1 (Watt et al. 1996), which contribute to most of the recombination events in yeast. Rather, it was proposed that mutation of the Sgs1 helicase leads to an accumulation of DNA damage or other structures that favor recombination between direct repeats.
DNA helicases that are structurally and, in some cases, functionally homologous to both the bacterial RecQ and yeast SGS1 gene products, have been characterized from many different organisms. These include products of the human Bloom's syndrome gene (BLM; Ellis et al. 1995), the Werner's syndrome gene (WRN; Yu et al. 1996), the murine BLM homolog (Chester et al. 1998), the human helicase RecQL (Puranam and Blackshear 1994), the Schizosaccharomyces pombe rqh1+ homolog (Stewart et al. 1997), a Drosophila melanogaster homolog DmBLM (Kusano et al. 1999), the Xenopus laevis DNA helicase FFA-1 (Yan and Newport 1995; Yan et al. 1998), as well as RecQ-like helicases from several bacterial species (Kusano et al. 1999). All except for RecQ and RecQL are fairly large proteins with a central DNA helicase domain and an acidic amino-terminal domain, and all share significant homology beyond the characteristic helicase motifs (Morozov et al. 1997; Kusano et al. 1999).
Patients that are homozygous for mutations in the Bloom's syndrome (BS) gene, have an increase in genomic abnormalities, including chromosome rearrangements and breakage, and sister chromatid exchange and recombination, leading to a dramatic predisposition to cancer (for review, see German 1993). Disruption of the murine BLM gene is embryonic lethal, yet embryonic fibroblasts have similar defects as human BS cells (Chester et al. 1998), arguing for a conservation of the BLM gene function. Humans deficient for the closely related WRN helicase age prematurely, manifesting the typical signs of being old before the age of 40 (for review, see Epstein et al. 1966). Strikingly, the genetic instability and reduced life-span phenotypes of sgs1 mutants in yeast are reminiscent of the phenotypes seen in BS and WRN patients, respectively (Sinclair and Guarente 1997). Furthermore, expression of either human gene in yeast is able to suppress the hyper-recombination phenotype of yeast lacking Sgs1p (Yamagata et al. 1998), demonstrating that these genes are not only structurally related, but are, at least in part, functional homologs. It is likely, therefore, that an elucidation of the Sgs1p function in yeast is directly relevant to these human diseases.
The fission yeast gene that is most homologous to SGS1, called variously rqh1+, hus2+, or rad12+, has also been studied genetically. Mutants show increased recombination, hypersensitivity to UV and ionizing radiation, and sensitivity to the inhibitor of deoxynucleotide biosynthesis, hydroxyurea (HU; Murray et al. 1997; Stewart et al. 1997; Davey et al. 1998). Overexpression, as well as deletion of rqh1+, appears to decrease the viability of cells as they recover from a cell cycle arrest provoked by HU (Stewart et al. 1997). These data do not directly implicate rqh1+ in a checkpoint response, but rather in the resumption of growth following genomic insult, perhaps defining a pathway that allows the replication fork to bypass DNA damage (Murray et al. 1997).
Here, we ask whether the S. cerevisiae Sgs1p helicase has a direct role in DNA replication and the cell cycle control of S-phase progression. Cellular responses of the sgs1 mutant in S phase are compared with those in G1 and G2. We find that the helicase activity is required for DNA checkpoint functions exclusively in S phase. We show that Sgs1p localizes to S-phase-specific nuclear foci and that a significant fraction colocalizes with ORC and with the signal-transducing kinase Rad53p. In a rad24 mutant, Sgs1p is required for Rad53p phosphorylation in response to replication fork arrest. This and the observation that Sgs1p is required for the association of Rad53p with an insoluble chromatin fraction, argues that Sgs1p interacts directly with the activated checkpoint kinase.
Results
Several lines of evidence suggest that the family of RecQ-like DNA helicases has a critical function in S phase in eukaryotic cells. First, cultured cells from BS and WRN patients have defects during S phase of the cell cycle, showing an abnormal profile of DNA replication intermediates (Lonn et al. 1990), retarded DNA chain growth rate in BS cells (Hand and German 1975), and an impaired S-phase progression in WRN cells (Poot et al. 1992). FFA-1, the Xenopus homolog, is essential for the formation of RP-A foci on demembranated sperm chromatin, possibly implicating FFA-1 in the organization of DNA replication foci prior to S phase (Yan and Newport 1995; Yan et al. 1998). A direct function of the Sgs1p helicase during DNA replication could explain the chromosome mis-segregation and hyper-recombination phenotypes (Watt et al. 1996). That is, aberrant DNA structures formed or stabilized during S phase might impair proper chromosome segregation or induce improper recombination events. On the other hand, if Sgs1p serves as a leading strand helicase during DNA replication, this activity must be redundant with other helicases, because the deletion of SGS1 in yeast, or of its homologs in human cells, is not a lethal event. In view of these considerations, we have examined in detail the S-phase function of the Sgs1p DNA helicase in yeast.
sgs1 cells are sensitive to HU
FACS analysis shows that the progression through S phase is not significantly altered in a S. cerevisiae strain lacking full-length Sgs1p (sgs1::LEU2, Gangloff et al. 1994), see also Figure 2, below, (sgs1–MMS). The mutation used in this study disrupts the SGS1 gene at amino acid 408, eliminating expression of its helicase and topoisomerase-binding domains (see Materials and Methods). This suggests that the Sgs1p helicase activity is not essential for S-phase progression in yeast. To examine whether S-phase checkpoints are intact in this mutant, we added HU to wild-type and sgs1 mutant yeast cells, prior to plating them on medium lacking the inhibitor. Only ∼50% of the sgs1 cells are viable after an 8-hr exposure to HU, whereas 90% of the isogenic wild-type cells survive (Fig. 1A,B). The sensitivity of the sgs1 strain is less pronounced than most bona fide DNA damage checkpoint alleles, but the strain is as sensitive as others that have defects in proteins that monitor replication fork arrest, such as those encoded by the temperature-sensitive alleles of pol2-11 or dpb11-1 (Araki et al. 1995; Navas et al. 1995). To identify the pathway affected by sgs1 mutations, we created double mutants of sgs1 with pol2-11 (Navas et al. 1995) and with a rad24 deletion (rad24::TRP1), which affects the integrity of the G1/S, the G2/M, and the intra-S DNA damage checkpoints (Lydall and Weinert 1997; Paulovich et al. 1997; de la Torre-Ruiz et al. 1998). The HU assay reveals no additive or synergistic effects between sgs1 and pol2-11 in the double mutant, either at permissive (25°C, Fig. 1A) or semipermissive (28°C, Fig. 1B) temperatures, whereas sgs1 and rad24 show additive sensitivity (Fig. 1C). This suggests that SGS1 and POL2 function in one epistasis group, whereas SGS1 and RAD24 do not. The simplest interpretation of this hypothesis is that Sgs1p functions on the same pathway as DNA polymerase ε (Pol2p), but in parallel to Rad24p.
Figure 2.
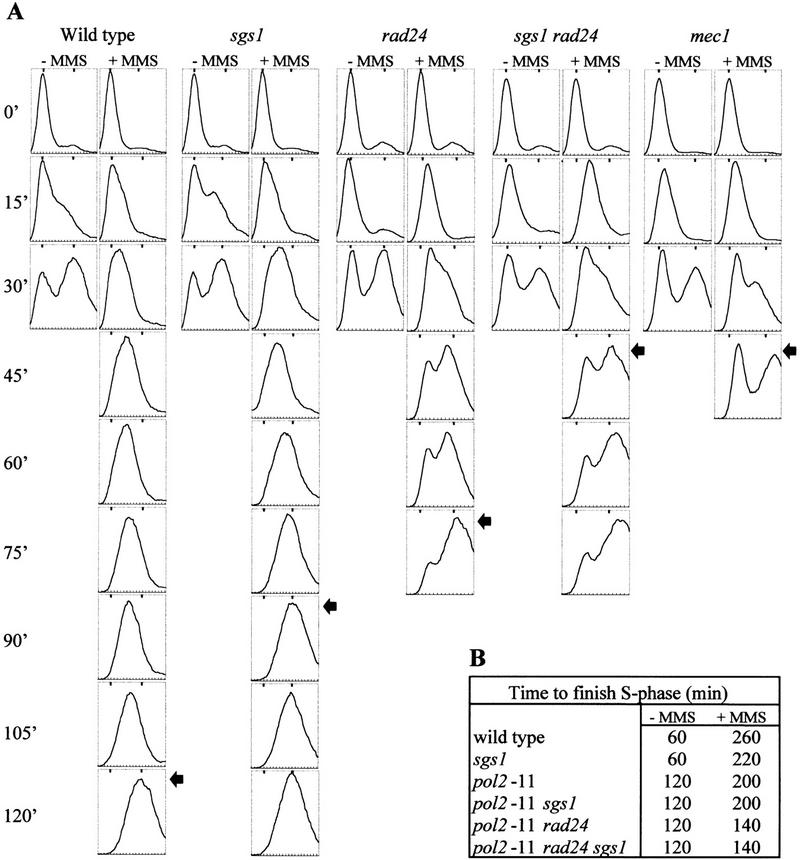
sgs1::LEU2 cells are partially defective for the intra-S checkpoint. (A) Exponentially growing cells were grown at 30°C and blocked in G1 with α-factor for two-thirds of the generation time. α-factor was washed away and the cells were incubated in fresh medium containing 0.033% MMS. Aliquots for FACS analysis were taken after the indicated time. The 1N and 2N DNA content peaks were taken from time point 0 and 30 min (without MMS) and indicated for every FACS sample as small bars at the top of each panel. In no case, were peaks other than 1N and 2N observed. Isogenic strains were as follows: wild type (GA-871); sgs1::LEU2 (GA-880); rad24::TRP1 (GA-872); rad24::TRP1 sgs1::LEU2 (GA-881); mec1 (mec1-1 sml1; GA-904); The end of S phase in the presence of MMS is indicated by an arrow. (B) The same as in A but all manipulations were done at 25°C, which allows growth of pol2-11. The time required to finish S phase as determined by a FACS scan is indicated. Note that the pol2-11 mutants traverse S phase more slowly than wild type in the absence of DNA damage (Budd and Campbell 1993). pol2-11 (GA-1021); pol2-11 sgs1::LEU2 (GA-1022); pol2-11 rad24::TRP1 (GA-1023), and pol2-11 rad24::TRP1 sgs1::LEU2 (GA-1024).
Figure 1.
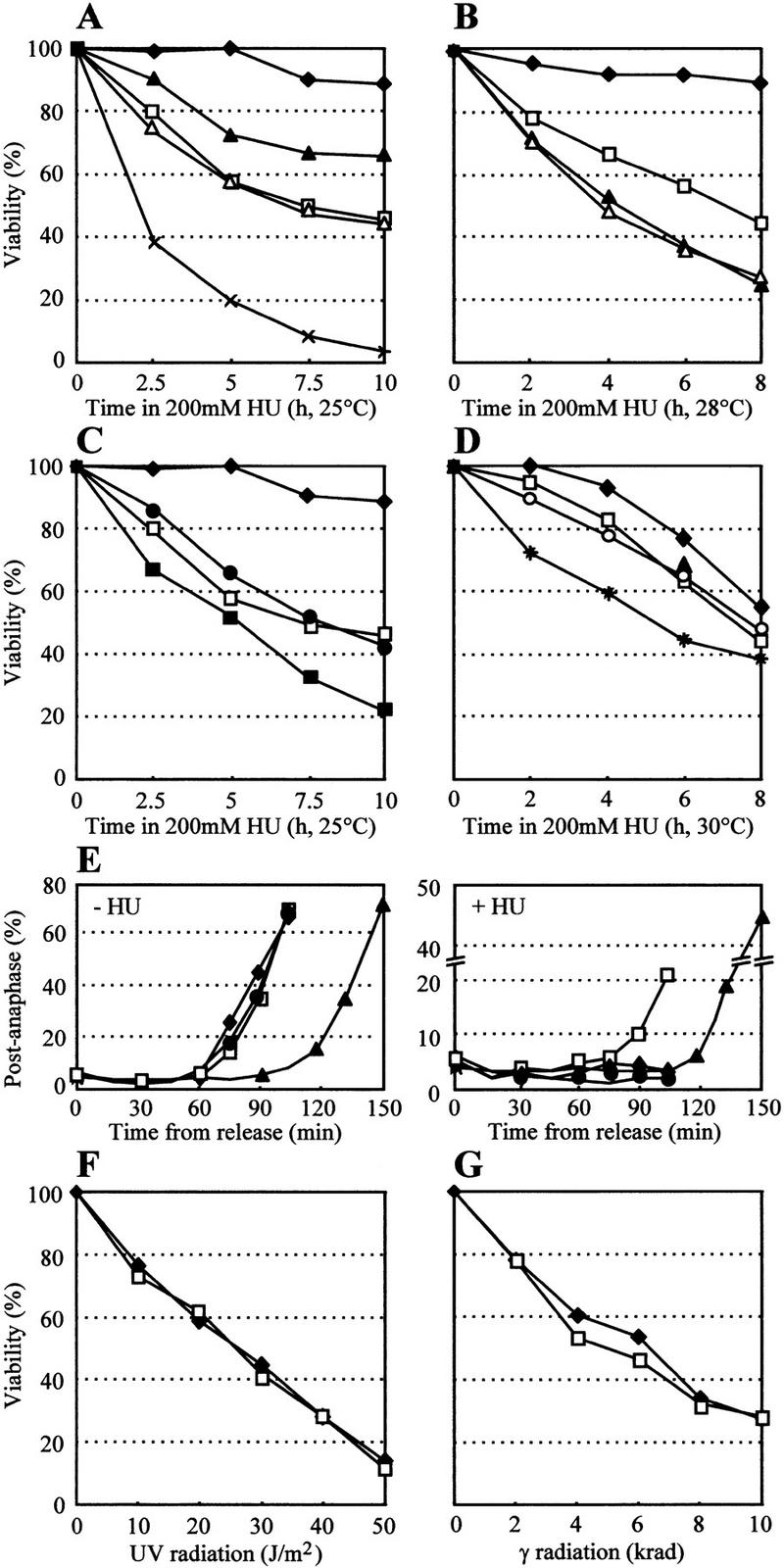
sgs1 cells are hypersensitive to HU but not to UV and γ radiation. (A–C) A total of 200 mm HU was added to exponentially growing cells and aliquots were washed and plated in triplicate on rich medium without HU at the indicated times. After 2–3 days, viability was scored. Standard deviation was <5%; for simplicity; error bars are not shown. All strains are isogenic to GA-871 carrying the following indicated mutations: (♦) wild type (GA-871); (□) sgs1::LEU2 (GA-880); (▴) pol2-11 (GA-1021); (▵) pol2-11 sgs1::LEU2 (GA-1022); (×) pol2-11 rad24::TRP1 (GA-1023); (●) rad24::TRP1 (GA-872); (█) rad24::TRP1 sgs1::LEU2 (GA-881). A and C were performed at 25°C (permissive temperature for the pol2-11 allele). B was performed as A, but the cells were incubated in HU at 28°C (semirestrictive temperature for pol2-11 allele), and plated at 25°C. (D) HU sensitivity of sgs1 cells depends on the helicase activity. sgs1::hisG (GA-737) were transformed with the empty vector (pRS415; □), pSGS1 (pJL31; ♦), psgs1-hd (helicase mutant; pJL37; ○) or psgs1-ct (carboxy-terminal truncation; pJM502; star). Plasmids are described in Lu et al. (1996). HU was added and viability was scored as above. (E) sgs1::LEU2 cells are partially defective for the S/M checkpoint. Cells were arrested in G1 with α-factor and released into S phase with (right) or without 200 mm HU (left). Aliquots were taken at the indicated times, stained with DAPI, and scored for cells that have undergone nuclear division. Staining of mitotic spindles with an antitubulin antibody gives an identical percentage of postanaphase cells (data not shown). FACS analysis shows that >98% of the cells fail to complete DNA replication during the course of the experiment (data not shown). Symbols and strains as in A–C. (F) sgs1::hisG cells are not hypersensitive to UV light. Cells were plated, UV radiated with the indicated dose, and plated on rich medium. (♦) SGS1 (GA-59); (□) sgs1::hisG (GA-737). (G) sgs1::hisG cells are not hypersensitive to γ radiation. Cells were plated and radiated with the indicated dose. All strains in D, F, and G are isogenic.
Passage through anaphase in HU
When DNA replication is blocked by HU, wild-type cells arrest cell cycle progression at G2/M until S phase is finished (S/M checkpoint). A strain defective in the S/M checkpoint cannot arrest and proceeds through the metaphase–anaphase transition (Weinert 1998). To see whether the HU sensitivity of the sgs1 mutant reflects a loss of the S/M checkpoint, cells were arrested in G1 with α-factor and released into S phase either in the presence or the absence of HU. Aliquots were removed, stained with DAPI, and cells were scored for nuclear division (chromosome segregation). In contrast to wild-type cells in which >98% of the cells are blocked in S phase by the presence of HU, ∼20% of the sgs1 cells fail to arrest the cell cycle and proceed to divide their chromatin, indicating a partial escape from the HU-induced S/M checkpoint (Fig. 1E). The pol2-11 mutant, as published previously (Navas et al. 1995), is almost completely defective for the S/M checkpoint, and most cells traverse G2/M. In contrast, rad24, a mutant that shows a similar HU sensitivity as sgs1, is blocked at G2/M as efficiently as a wild-type cell, consistent with previous data suggesting that Rad24p is not required for the S/M checkpoint (Weinert 1998). Thus, although the HU sensitivity of sgs1 is not completely penetrant, it suggests that the S/M checkpoint is impaired.
RAD53 encodes a signal-transducing kinase required for the S/M checkpoint and for all other DNA damage checkpoints identified so far. Previous studies indicate that there is no direct correlation between passage through anaphase and the percentage of rad53 cells that die in the presence of HU (Desany et al. 1998). Similarly, we show here that at 25°C, pol2-11 cells are less sensitive to HU than sgs1 cells, although it has a more pronounced effect on the S/M checkpoint. sgs1 and pol2-11 cells, released synchronously into HU, lose viability before they traverse anaphase (data not shown), suggesting that cell death is due to defects in the recovery from stalled replication forks, rather than from improper mitotic segregation events.
Sgs1p carboxy-terminal and helicase domains are both necessary for recovery from HU arrest
Published data suggest that the large polypeptide encoded by Sgs1p may have more than one function. One has been shown to require functional helicase motifs, whereas the other maps to the carboxy-terminal third of the protein, which lacks helicase function (Lu et al. 1996). To see which of these domains is sufficient to complement HU sensitivity phenotype, a helicase mutant (sgs1-hd; Lu et al. 1996), a carboxy-terminal truncation (sgs1-ct; Lu et al. 1996), and the full-length gene were expressed from low copy number plasmids introduced into a strain carrying a disrupted sgs1::hisG allele (see Materials and Methods). When HU sensitivity is compared with the strain carrying an empty vector, we see that the full-length gene is able to suppress the sensitivity of the sgs1 mutant in part (Fig. 1D). This incomplete suppression is not due to an additional mutation or a dominant effect of the sgs1 allele, because slightly higher expression of SGS1 (from a plasmid-borne Gal1 promoter on glucose) completely suppresses the HU sensitivity (data not shown). On the other hand, galactose-induced levels of Sgs1p again provoke hypersensitivity to HU, arguing that Sgs1p may function in a complex from which limiting factors can be titrated by an excess of Sgs1p, thereby impairing recovery from HU-mediated arrest (data not shown).
In Figure 1D, we show that the Sgs1p helicase activity is necessary for the checkpoint function, because low-level expression of the helicase mutant in the sgs1 strain does not enhance survival. Moreover, expression of the carboxy-terminally truncated form of Sgs1p actually increases HU sensitivity, although the protein made is still a functional helicase (Lu et al. 1996; Fig. 1D). This argues for the importance of the carboxy-terminal domain in Sgs1p function and is again consistent with a model in which truncated Sgs1p sequesters or interferes with other proteins involved in checkpoint function.
sgs1 mutants are not hypersensitive to UV or γ radiation, but are implicated in the intra-S DNA damage checkpoint
Mutant alleles of checkpoint genes are often hypersensitive to UV and ionizing radiation; mutant alleles of the S. pombe rqh1 gene show hypersensitivity to both of these types of DNA damage (Murray et al. 1997; Stewart et al. 1997; Davey et al. 1998). To see if this characteristic is shared by sgs1 cells, random cultures were treated with UV (Fig. 1F) or γ radiation (Fig. 1G). S. cerevisiae strains lacking Sgs1p are not more sensitive than the isogenic wild-type strain, arguing against a direct role for Sgs1p in nucleotide excision or double-strand break repair.
When DNA damage occurs during S phase, an intra-S checkpoint is activated (Paulovich and Hartwell 1995). This checkpoint blocks initiation and elongation of DNA replication by inhibiting polα/primase activity (Marini et al. 1997) and by modifying ssDNA-binding protein RP-A (Longhese et al. 1996). To test the integrity of the intra-S checkpoint in sgs1 cells, cultures were released from an α-factor block into rich medium containing low amounts of the DNA alkylating agent MMS. MMS was maintained at a concentration that precludes activation of the G1/S DNA damage checkpoint, but which is sufficiently high to activate the intra-S checkpoint in wild-type cells (Sidorova and Breeden 1997). FACS analysis shows that wild-type cells, as expected, slow their progression through S phase in the presence of MMS, requiring 2 hr to finish DNA replication as compared with 30 min without MMS (see arrows, Fig. 2A). In contrast, sgs1 cells complete S phase 30 min faster than the isogenic SGS1 cells in the presence of damaged DNA, and the rad24 deletion mutant completes replication 45 min faster than the wild-type strain (Fig. 2A). Combining the sgs1 and rad24 mutations is synergistic, and cells complete replication as rapidly as a strain mutant for mec1 or carrying the mec1 sgs1 double mutation (45 min, see Fig. 2A; data not shown). These results suggest that sgs1 cells are partially defective for the intra-S checkpoint, and that Sgs1p acts parallel to Rad24p, upstream of Mec1p, the central ATM1-like kinase of the yeast checkpoint pathways. The fact that a rad24 sgs1 double mutant completes S phase on MMS as fast as the mec1 mutant, argues that RAD24 and SGS1 define the two major epistasis groups upstream of MEC1 in the intra-S checkpoint. Because published data suggest that the remaining checkpoint activity in a rad24 mutant depends, at least in part, on RAD9 (Paulovich et al. 1997), the SGS1 and RAD9 epistasis groups may be partially redundant for the intra-S checkpoint, at least at the level of resolution achieved by FACS analysis.
Although DNA polε (Pol2p) was shown to function in the activation of Rad53p after DNA damage during S phase (Navas et al. 1996), there is no report of its involvement in the intra-S checkpoint. Performing time course experiments like those shown in Figure 2A, but at 25°C, we examined whether pol2-11 mutant is deficient in the intra-S checkpoint when released into S phase with low amounts of MMS. As shown in Figure 2B, pol2-11 cells complete replication 60 min faster than wild-type cells under identical conditions, implicating DNA polε, like Sgs1p, in the intra-S checkpoint. The pol2-11 sgs1 double mutant has no increased phenotype over the two single mutants, whereas the pol2-11 rad24 double mutant and the pol2-11 rad24 sgs1 triple mutant complete S phase almost as rapidly as pol2-11 cells in the absence of MMS (Fig. 2B). Again, this argues that DNA polε and Sgs1p function together in one epistasis group, whereas Rad24p acts on a parallel pathway. Similar data are obtaining with random cultures exposed to high levels (0.1%) of the alkylating agent MMS; sgs1 cells are slightly, but reproducibly, more sensitive than wild-type parental cells, and the combined effects of sgs1 and rad24 are additive (data not shown).
Sgs1p is not implicated in G2/M or G1/S DNA damage checkpoint pathways
The best-characterized DNA damage checkpoint during the yeast cell cycle is the G2/M checkpoint, which detects damaged DNA during G2 and blocks progression through metaphase by inhibiting Pds1p degradation (Cohen-Fix and Koshland 1997; Gardner et al. 1999). To see whether Sgs1p takes part in this checkpoint, wild-type and sgs1-disrupted cells were blocked in G2/M by nocodazole prior to the addition of MMS. They were subsequently released into fresh medium, and passage through the metaphase–anaphase transition was monitored by scoring post-metaphase segregation of the nuclei. Whereas all strains tested show identical kinetics for the recovery from the G2/M block in the absence of DNA damage, only the wild-type and sgs1 cells remain blocked for >100 min after nocodazole release in the presence of damage (Fig. 3A). The rad24 mutant and rad24 sgs1 double-mutant phenotypes are consistent with the characterized role for RAD24 in the G2/M DNA damage checkpoint (de la Torre-Ruiz et al. 1998).
Figure 3.
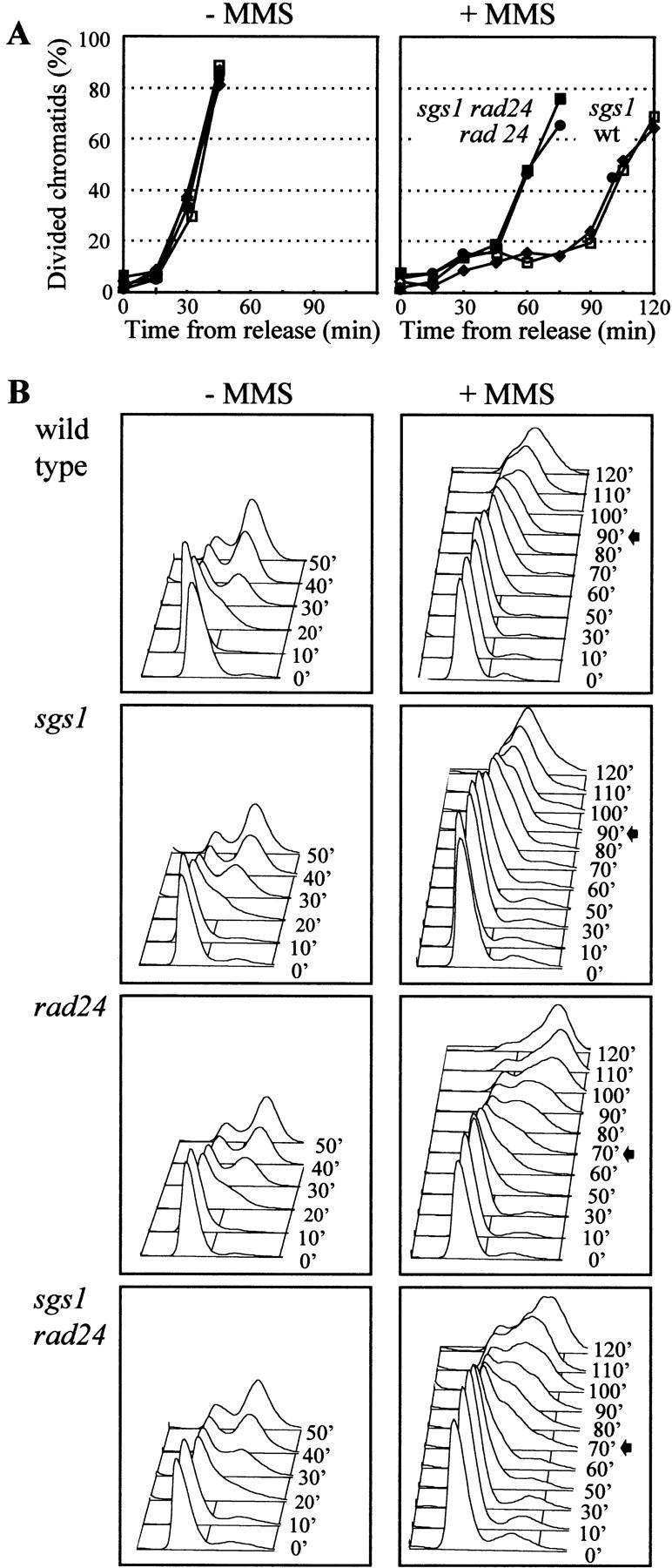
sgs1::LEU2 cells are proficient for the G2/M (A) and G1/S (B) DNA damage checkpoints. (A) Exponentially growing cells in YPAD were arrested at G2/M with 15 μg/ml nocodazole for 2 hr. For the last 30 min of the block, 0.15% MMS was added to one-half of the culture. MMS was then inactivated by the addition of 1 vol 10% Na-thiosulfate, cells were then washed and released in medium lacking nocodazole and MMS. After the indicated times, aliquots were removed, stained with DAPI, and cells of each strain were scored microscopically separated chromatids (post-anaphase). Strains isogenic to GA-871 were as follows: (♦) Wild type (GA-871); (□) sgs1::LEU2 (GA-880); (●) rad24::TRP1 (GA-872); (█) rad24::TRP1 sgs1::LEU2 (GA-881). (B) Exponentially growing cells were blocked in G1 with α-factor for 70 min and 0.2% MMS was added to one-half of the culture and incubated for the last 10 min of the α-factor block. The cells were then washed and released into medium lacking both α-factor and MMS. Aliquots were taken after the indicated time and FACS analysis was performed. Arrows indicate the aliquot when the cells enter S phase. These mutant strains are isogenic with wild type (GA-871) and contain full deletions or disruptions as described in A.
DNA damage that occurs during G1 also blocks cell cycle progression, impeding the G1/S transition (Sidorova and Breeden 1997; de la Torre-Ruiz et al. 1998). This situation is monitored by adding MMS to a pheromone-arrested culture. After release from the G1-phase block, the DNA damage induced by MMS normally arrests cells prior to S phase. Wild-type and sgs1 cells show identical patterns of recovery, remaining arrested until 90 min after the release from α-factor, followed by a slow S phase (Fig. 3B; Sidorova and Breeden 1997), whereas rad24 strains are partially defective in the G1/S checkpoint, entering S phase ∼70 min after α-factor release (Fig. 3B). Because the rad24 sgs1 double mutant is indistinguishable from the rad24 single mutant, we conclude that SGS1 is not involved in the G1/S DNA damage checkpoint.
Rad53p phosphorylation is compromised in a rad24 sgs1 double mutant
The genetic analysis described above suggests that Sgs1p functions together with DNA polε to generate the signal which activates Mec1p and Rad53p, that subsequently blocks cell cycle progression and induces repair of DNA damage. This signal may be provoked either by arrest of the replication fork (HU) or by other forms of DNA damage during S phase, and can be monitored by a mobility shift of Rad53p on an SDS gel due to its phosphorylation (Sanchez et al. 1996; Sun et al. 1996). To test directly whether Sgs1p functions upstream of the signal transducers Mec1p and Rad53p in the presence of HU, we screened for the activation of Rad53p by a MEC1-dependent phosphorylation event in sgs1 and wild-type strains. Cells were blocked in G1 by the addition of α-factor and released into S phase in medium containing 200 mm HU. Aliquots were taken at the indicated times and whole-cell extracts were analyzed by SDS-PAGE (Fig. 4A). Scoring for budding index ensures that cells entered S phase synchronously (Fig. 4C), and FACS analysis confirms that the cells do not progress beyond S phase during the experiment (data not shown). Cells lacking Sgs1p are still able to phosphorylate Rad53p, as do wild-type and rad24-deficient cells (Fig. 4A,B). However, the sgs1 rad24 double mutant strongly reduces the level of Rad53p phosphorylation. The simplest interpretation of these results, and the interpretation most consistent with our genetic analyses, is that both Rad24p and Sgs1p function upstream of Rad53p, independently of each other. The activation of the Rad24p group by replication fork arrest may be due to subsequent DNA strand breaks that are recognized as DNA damage. Thus, RAD24 may provoke Rad53p phosphorylation as a secondary response to HU, independent of the SGS1-dependent phosphorylation event, which is presumably elicited directly by polymerase arrest. The residual Rad53p phosphorylation in the sgs1 rad24 double mutant may depend on DBP11 and/or DRC1, as published in Wang and Elledge (1999).
Figure 4.

SGS1 and RAD24 are both required for Rad53p phosphorylation in HU. (A) Exponentially growing cells were blocked in G1 with α-factor for two-thirds of the generation time. α-factor was inactivated by the addition of pronase (50 μg/ml; Sigma) and HU was added to 200 mm. Aliquots were removed from the exponential growing cultures and at 0, 60, 80, and 100 min after release. Total cell extracts were prepared, run on a 7.5% SDS–polyacrylamide gel and blotted with the 9E10 antibody (α-Myc) for Rad53p–Myc. Wild-type extracts from an exponential culture and from cells 100 min after release were run on every gel and are shown in the last two slots (underlined). Isogenic strains with indicated mutations were as follows: wild type (GA-1040); sgs1::LEU2 (GA-1041); rad24::TRP1 (GA-1042); rad24::TRP1 sgs1::LEU2 (GA-1043); mec1-1 sml1 (GA-1048). The slower migrating band is the phosphorylated form of Rad53p. (B) Western blots were scanned and the percentage of phosphorylated Rad53p was quantified (Aida 200). (C) Aliquots were removed after the indicated time and after release into HU, fixed with Na-azide (0.0325%; Fluka), and budding index was scored under a light microscope. (♦) wild type; (□) sgs1::LEU2; (●) rad24::TRP1; (█) rad24::TRP1 sgs1::LEU2; (+) mec1-1.
Sgs1p levels peak in S phase
Because our data implicate SGS1 exclusively in S-phase checkpoint events, we examined whether Sgs1 protein levels fluctuate during the cell cycle. The endogenous copy of SGS1 under its own promoter was tagged with multiple Myc epitopes at its carboxyl terminus. Because the strain with the tagged allele retains wild-type sensitivity to HU (data not shown), we conclude that Sgs1p–Myc remains functional. To synchronize the culture, the cdc16-1 mutation was introduced into the Sgs1p–Myc strain, cells were blocked at metaphase–anaphase at restrictive temperature, and then released synchronously into G1 by a shift to permissive temperature. Aliquots were taken at regular intervals for 2 hr postrelease, and were probed by Western blot for Sgs1–Myc and tubulin, which serves as a loading control (Fig. 5A). To judge progression through the G1/S transition, bud emergence was scored microscopically. Normalized Sgs1p–Myc levels are very low during M and G1 phases of the cell cycle, and increase significantly as cells enter S phase, decreasing again in G2 (Fig. 5A). Cells released into HU, which gives an early S-phase block under these conditions, have a similar amount of Sgs1p as nonblocked early S-phase cells, suggesting that HU arrest does not induce Sgs1p transcripts nor stabilize the protein. These data are consistent with and extend microarray data that indicate that SGS1 transcripts peak in S phase (Cho et al. 1998).
Figure 5.

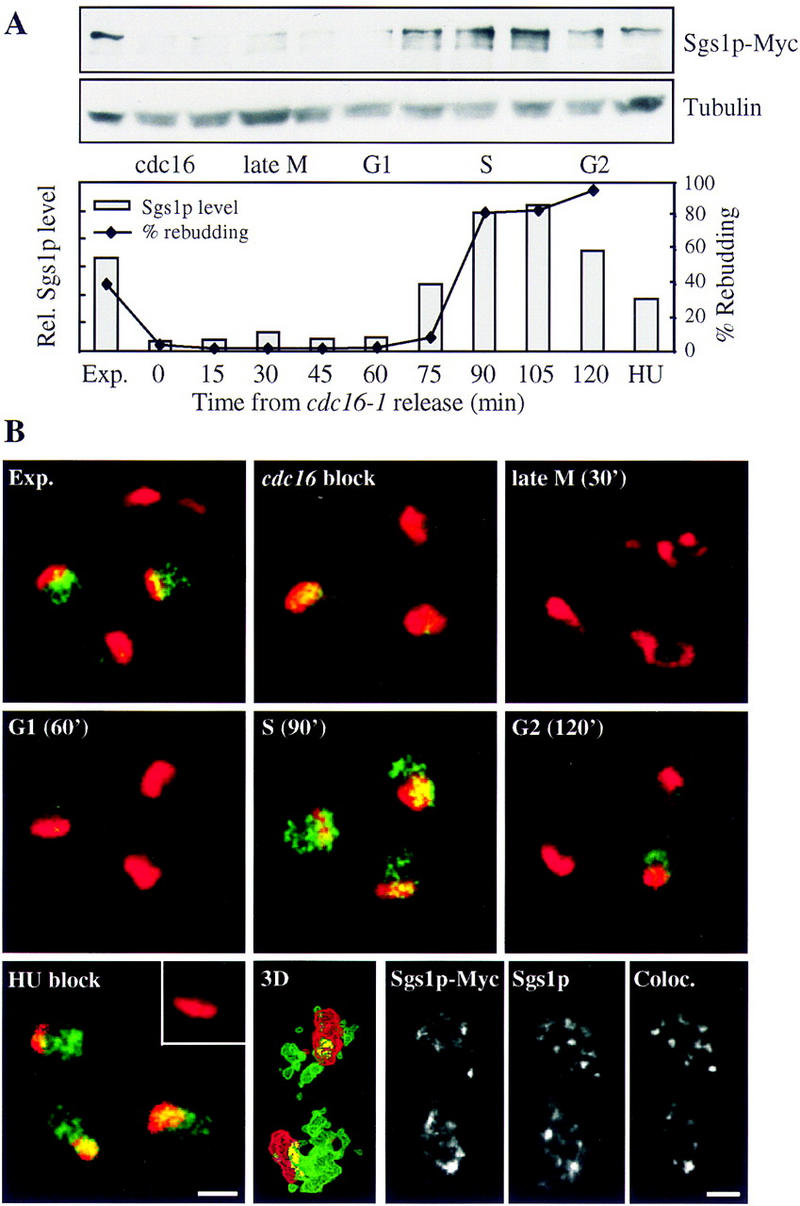
Sgs1p protein levels peak in S phase and Sgs1p localizes to nuclear foci. Diploid cdc16-1 SGS1-Myc strain (GA-1151) was blocked at the metaphase–anaphase transition by shifting to nonpermissive temperature (36°C) for 2 hr. Cells were released by shifting back to 25°C. Aliquots were removed at the indicated times postrelease for total cell extracts, scoring of the budding index (A), and for immunofluorescence (B). (A) Total cell extracts were run on an 8% SDS–polyacrylamide gel and blotted for Sgs1p–Myc. Sgs1p was quantified and normalized with tubulin. The budding index (♦) indicates re-entry into S phase. (B) Sgs1p–Myc was detected as described in Materials and Methods with 9E10 (green) and cells were counterstained for the abundant nucleolar antigen Nop1p (red; Gotta et al. 1997). A single 0.4 μm confocal microscope section is shown and colocalization is indicated in yellow. A control strain lacking the Myc epitope (GA-174) is shown for HU-arrested cells in the inset of HU blocked cells. For higher resolution analysis, 30–40 Z sections of 0.1-μm were captured on identical stained S-phase cells. Deconvolution was done with Huygans and the staining was reconstituted in 3 dimensions (Bitplane; see Materials and Methods). Colocalization is shown in yellow (bottom). Double labeling of Sgs1–Myc in the diploid strain GA-1151 was performed with anti-Myc (9E10) and an affinity-purified chicken anti-Sgs1p (Sinclair et al. 1997; bottom, last three pictures), with distinct fluorescent markers. Regions in which the two staining patterns coincide are shown in the bottom right panel. Bar, 2 μm.
Sgs1p is localized in S-phase-specific foci
To confirm the cell cycle-dependent fluctuation of Sgs1p, cells from the cdc16 release experiment carrying Sgs1–Myc were also processed for immunofluorescence (Fig. 5B). At the metaphase–anaphase transition (cdc16 block), at cytokinesis (30 min after release), and during G1 of the cell cycle (60 min release), Sgs1p is almost undetectable (shown in green). Because a similarly tagged Orc2p subunit, which is present in ∼400–500 copies per cell, is readily detected by immunofluorescence (data not shown), it is reasonable to suggest that the Sgs1p copy number drops significantly below this level in M and G1 phases. However, during S phase (90 min postrelease) Sgs1p was readily detected in the nucleus as a punctate or focal-staining pattern. This was not an artifact of the arrest/release protocol, as immunolabeling of a random culture revealed unbudded cells with very low levels immunostaining, and small- or medium-budded cells with a strong punctate staining pattern (data not shown). The foci of Sgs1p staining are lost as cells pass through G2 (120 min postrelease), although they are maintained in cells arrested by the addition of HU. These results suggest that Sgs1p localizes to nuclear foci during both a normal and an arrested S phase.
Earlier reports have suggested that Sgs1p is localized to the nucleolus, where it is thought to play a role in suppression of rDNA recombination (Sinclair et al. 1997). We therefore counterstained all our preparations with anti-Nop1p, an abundant nucleolar protein (Fig. 5B in red; Gotta et al. 1997). Contrary to results published by Sinclair et al. (1997), we do not find a significant enrichment of Sgs1p in the nucleolus. Careful quantitation of the overlap of the two signals in a single 0.4 μm focal section reveals that 8.5% of Sgs1p is localized to the nucleolus (Table 1). In addition to colocalization on a single confocal section, quantitation for the colocalization of Sgs1p–Myc and Nop1p was performed on three-dimensional reconstituted and deconvolved images (Imaris, Bitplane, see Material and Methods; Table 1; Fig. 5B). This technique is more accurate than the single confocal section, as out-of-plane fluorescence is removed and the entire nuclear space is evaluated rather than a single focal section. By quantifying the overlapping regions, 6% of the Sgs1p–Myc was found to colocalize with Nop1p (Table 1). The small amount of Sgs1p–Myc found in the nucleolus localizes to discrete S-phase-specific foci, probably representing sites of rDNA replication (see below). Because the nucleolus constitutes at least 15% of the nuclear volume, we conclude that Sgs1p–Myc is not enriched in the nucleolus.
Table 1.
Quantitation of two- and three-dimensional colocalization data
|
|
Rad53p
|
Orc1/2p
|
Nop1p
|
Rap1p
|
|---|---|---|---|---|
| 2D | ||||
| Sgs1p–Myc | 45.0 ± 6.5 | 49.0 ± 11.5 | 12.0 ± 4.0 | 13.5 ± 6.0 |
| 47.0 ± 7.5 | 42.0 ± 11.0 | 8.5 ± 4.0 | 9.5 ± 4.5 | |
| Rad53p–Myc | 66.0 ± 9.0 | 31.0 ± 7.5 | N.D. | N.D. |
| 60.0 ± 8.5 | 50.0 ± 5.0 | N.D. | N.D. | |
| 3D | ||||
| Sgs1p–Myc | 39.5 ± 8.0 | N.D. | 8.5 ± 3.5 | 4.5 ± 0.5 |
| 44.0 ± 13.0 | N.D. | 6.0 ± 2.5 | 1.5 ± 0.5 | |
| Rad53p–Myc | 54.0 ± 2.0 | 40.0 ± 15.0 | N.D. | N.D. |
| 74.5 ± 7.0 | 51.0 ± 4.0 | N.D. | N.D. |
Immunofluorescence labeling shown in Figs. 5 and 6 was digitalized by confocal microscopy, and the overlap of the two indicated staining patterns in a single 0.4-μm focal section (2D) was quantified using Adobe Photoshop (pixels × intensity). Three dimensional (3D) double-stained images were reconstituted and refined by deconvolution (see Materials and Methods). Three dimensional colocalization was quantified as voxel (pixel3) × density. For each pair of antigens, the top number indicates the amount of the protein indicated at top that colocalizes with the protein designated at left. The bottom number indicates the inverse. The number of cells analyzed is 13–20 for 2D and 4–8 for 3D. Numbers indicate percentages rounded to 0.5.
To rule out artifacts that might have arisen from use of a Myc-tagged Sgs1p, we obtained the previously published anti-Sgs1p serum (Sinclair et al. 1997) and used affinity-purified anti-Sgs1p antibodies to probe for their ability to colocalize with Myc-tagged Sgs1p. Double staining indicates that the two signals efficiently overlap (Fig. 5B), and again confirms that <10% of the nuclear Sgs1p is nucleolar.
Sgs1p colocalizes with Rad53p
We have shown above that Sgs1p is involved in the activation of Rad53p in cells blocked in S phase. To test whether the two proteins colocalize within the nucleus, double labeling was performed and analyzed as described above. Similar to Sgs1p, Rad53p is detected in nuclear foci (Fig. 6A) and a substantial fraction of the Sgs1p and Rad53p signals do overlap during a normal S phase. Quantitation of two-dimensional focal sections reveal that 47% of Sgs1p colocalizes with Rad53p and 45% of Rad53p with Sgs1p, respectively (Table 1). This is likely to be significant, for if two signals each stain between one-fourth and one-third of the volume of the nucleus, a random distribution of the two would result in <10% coincidence. As a control for the significance of this approach, a strain expressing the Rad53p–Myc tagged protein was labeled with both anti-Myc and an antibody specific to Rad53p (inset, Fig. 6A, gift of M. Schwartz, Yale University, New Haven, CT). In this case, 60%–66% of Rad53p–Myc is also detected by the anti-Rad53p antibody, indicating that the methods used for colocalization are ∼65% efficient. Thus, if we were to normalize to this value, the calculated overlap of Rad53p and Sgs1p would represent ∼70% of the Sgs1p pool.
Figure 6.
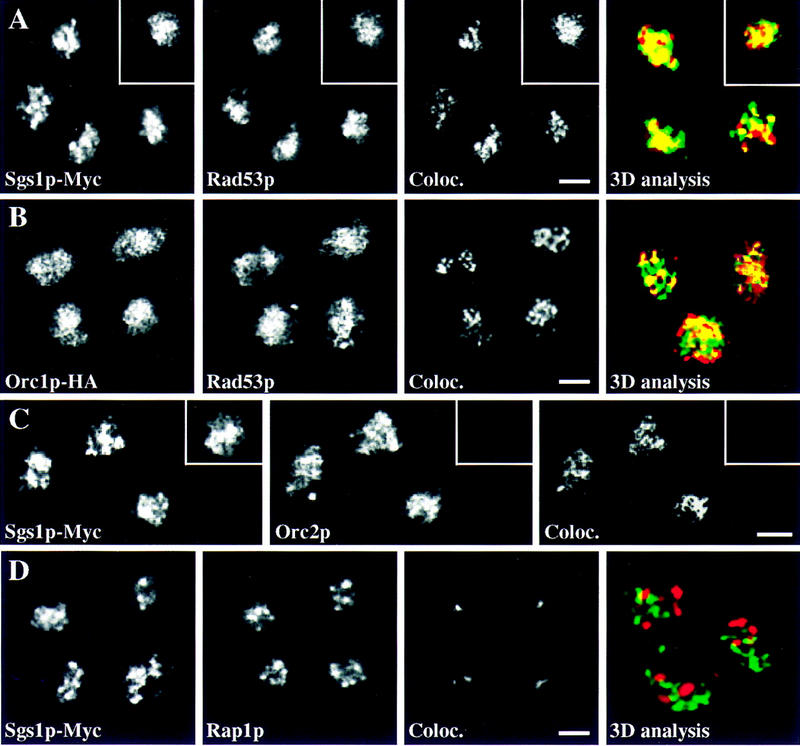
Sgs1p colocalizes with Rad53p and replication foci, but not with Rap1p. (A) A MATa/a diploid Sgs1p–Myc-tagged strain (GA-878) was blocked in G1 with α-factor and released into S phase. Immunofluorescence was performed as described in Materials and Methods by double labeling for Sgs1p–Myc (9E10) and Rad53p (rabbit anti-Rad53p). As a control, a diploid Rad53p–Myc-tagged strain (GA-1126) was double labeled with 9E10 (green) and affinity-purified rabbit anti-Rad53p (red), inset. The signal of Rad53p overlaps precisely with either Myc-tagged protein and is shown in the panel labeled Coloc. Z sections for three-dimensional reconstitution were taken and deconvolved as in Fig. 5B, but only one focal section of the reconstituted image is shown. In the merge of Sgs1p–Myc (9E10; green) and rabbit anti-Rad53p (red); the colocalization is yellow. (B) Temperature-sensitive diploid cdc4-3/cdc4-3 ORC1–HA/ORC1–HA cells (GA-1222) were blocked in G1 by shifting to 36°C for 2 hr. Cells were released by shifting back to permissive temperature (23°C) for 1 hr until cells entered S phase as judged by FACS analysis (data not shown). Double labeling was performed with HA.11 antibodies (Orc1–HA, green) and rabbit anti-Rad53p (red). (C) S-phase cells (as in A) were double labeled for Sgs1p–Myc (9E10) and goat anti-Orc2p (Santa Cruz, CA). Potential background staining by Orc2p antiserum was eliminated by a preincubation with the antigenic Orc2p peptide (inset). Because of the weak staining of the goat anti-Orc2p antiserum, we could not perform three-dimensional reconstitution. (D) Double labeling was performed with rabbit anti-Rap1p (red) and Sgs1p–Myc (9E10, green) in GA-878 released into S phase as in A. A single focal section of the deconvolved image is shown as in A. Overlapping regions are yellow. Bar, 2 μm.
Extending this analysis with the three-dimensional reconstitution and deconvolution method, we confirm that 44% of the Sgs1p–Myc signal colocalizes with Rad53p, an amount similar to the value obtained for two antibodies recognizing the same protein (Rad53p–Myc detected by both 9E10 and anti-Rad53p; Table 1). In contrast, after applying identical deconvolution programs to double labeling of Sgs1p–Myc and Rap1p, only 1.5%–4.5% of the signals coincide (Fig. 6D; Table 1). We conclude that there is a significant colocalization of Sgs1p and Rad53p, whereas Sgs1p is excluded from telomeres and is only minimally present in the nucleolus.
In mammalian cells as well as in yeast, DNA replication takes place in replication foci, which appear to contain between 30 and 300 replication forks in mammalian cells (Nakamura et al. 1986), or 20–30 in yeast (Pasero et al. 1997). Proteins that are directly involved in the process of DNA replication are usually concentrated at these sites, notably ORC, PCNA, and RP-A (Coverley and Laskey 1994; Pasero et al. 1999). To see whether the S-phase-specific Sgs1p foci are replication foci, we performed combined immunofluorescence for Sgs1p and Orc2p, as well as Rad53p and Orc1p. Orc1 and Orc2 proteins are components of the origin recognition complex (ORC), which remains DNA bound throughout the cell cycle, coinciding with sites of DNA synthesis in S phase (Pasero et al. 1999). Quantitation in S-phase cells shows that between 40% and 50% of the Sgs1p and Rad53p signals colocalizes with ORC subunits (Fig. 6B,C; Table 1). Thus, taken together, quantitative immunofluorescence data suggest that Sgs1p colocalizes with Rad53p at most, but perhaps not all, replication foci during a normal S phase.
Fractionation of Rad53p into an insoluble nuclear scaffold depends on SGS1
To reinforce the immunofluorescence data, a cell fractionation assay was used that permits isolation of a nuclear subfraction enriched in ORC and other proteins of the pre-replicative complex (Materials and Methods; Pasero et al. 1999). In brief, spheroplasted cells are lysed on ice by the addition of a nonionic detergent, and the lysate is centrifuged. The vast majority (85%–95%) of the total cellular protein remains in the supernatant (Sup) as soluble protein, whereas tightly associated chromosomal proteins sediment as a nuclear pellet, which contains all the genomic DNA as well as structural proteins of the nucleus. By digestion of the DNA with DNase I, this pellet can be further fractionated into a soluble chromatin (Chr) and an insoluble nuclear scaffold fraction (Sc). The soluble fraction contains most of the histones and nucleosome-associated enzymes, whereas the insoluble fraction is enriched for putative structural proteins such as topo II and ORC (Fig. 7A; Pasero et al. 1999).
Figure 7.
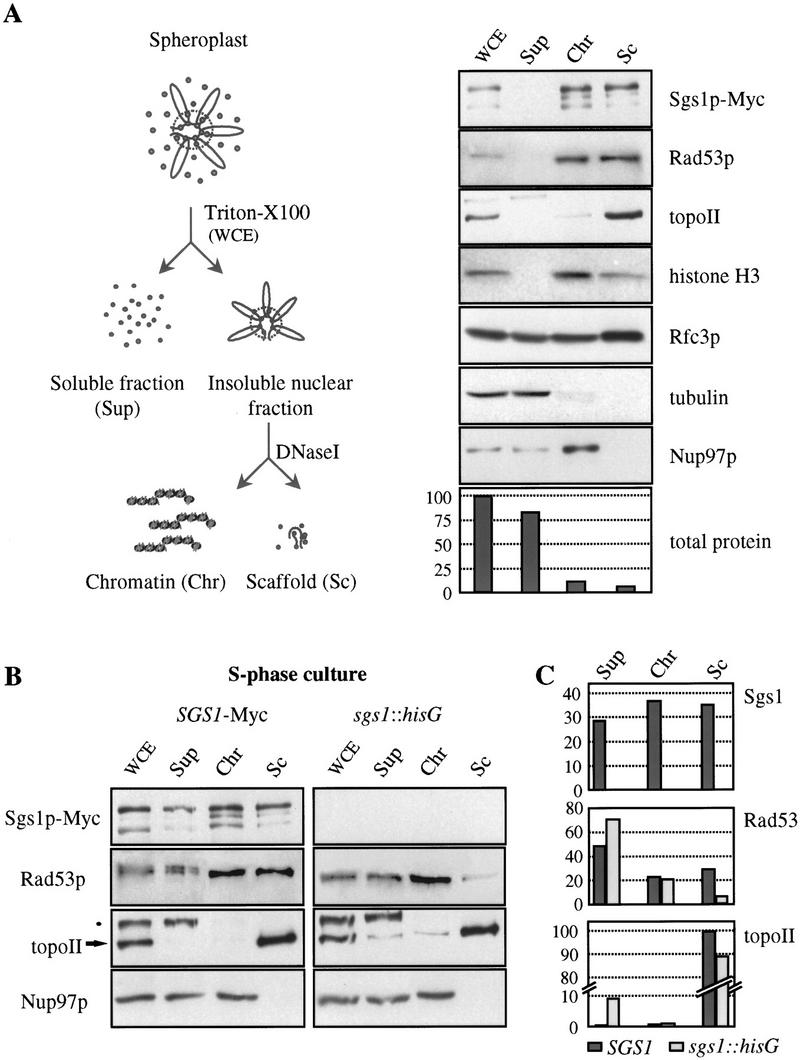
Rad53p cofractionates with Sgs1p in an insoluble chromatin fraction. (A) Scheme of the cell fractionation assay (left; see Materials and Methods for details). A random culture of GA-990 (Sgs1p–Myc) was used. One volume, representing the same number of starting cells for WCE and Sup samples, and fivefold equivalent volumes of Chr and Sc were used for Western blot analysis with different antibodies. Antibodies used were as follows: 9E10 for Sgs1p–Myc; anti-tubulin (gift of V. Simanis, ISREC) and mAb414 for Nup97 (BAbCO, Berkeley CA), while rabbit sera included, anti-Rad53p (gift of M. Schwartz), anti-topo II (Klein et al. 1992), anti-histone H3 (gift of D. Allis, University of Virginia), and anti-Rfc3p (gift of P. Burgers). All detections shown are performed sequentially on one membrane except histone H3, Rfc3p, and tubulin, which are from a parallel experiment. Bradford readings were taken to determine the protein concentration in each fraction, and are shown as percent of total protein per fraction. (B) SGS1–Myc (GA-990) and sgs1::hisG (GA-737) cells were blocked in G1 with α-factor and released into S phase. Cell fractionation was performed and blotted as in A. (Arrow) topo II; (●) an unidentified cross-reacting protein detected by the anti-topo II serum. (C) Western blots from B were quantified and normalized with pore staining and the distribution of each protein in the different fractions is shown as percent of total protein.
Samples from the chromatin fractionation assay performed on an unsynchronized population of wild-type cells were blotted for Sgs1p–Myc. Sgs1p is almost completely absent in the first supernatant, which is consistent with a tight association with chromatin. The efficiency of this binding is similar to that of topo II and ORC, and distinguishes it from other replication factors such as RF-C and RP-A, which are largely soluble in this assay (Fig. 7A; Pasero et al. 1999). Further processing of the first pellet into soluble and insoluble fractions reveals that roughly one-half of Sgs1p associates with the insoluble scaffold material. This is in contrast to histones or MCM proteins, which are released with solubilized nucleosomes (Fig. 7A; Pasero et al. 1999). By blotting the same fractions for Rad53p, we see a very similar fractionation pattern as that observed for Sgs1p. Rad53p is absent from the first supernatant and is found to distribute between soluble chromatin and the insoluble scaffold pellet (Fig. 7A). In view of the minor amount of total protein that is represented in this latter fraction (<10%), this indicates a considerable copurification of Sgs1p and Rad53p, and is consistent with the degree of colocalization detected by immunofluorescence.
Because the insolubility of Sgs1p precludes the use of coimmunoprecipitation assays to demonstrate protein–protein interactions, we asked whether the association of Rad53p with the insoluble chromatin fraction depends on Sgs1p. To test this hypothesis, isogenic wild-type and sgs1 mutant strains were synchronized in S phase and submitted to the fractionation protocol. A slightly larger subpopulation of Sgs1p and Rad53p proteins are released in the first fractionation step, probably reflecting the overall increase in Sgs1p and Rad53p protein levels in S phase. On the other hand, a large fraction of each protein still cofractionates with topo II in the insoluble scaffold fraction (Fig. 7B). The release of nuclear pore proteins and Mcm2p indicates that this pattern of fractionation is not due to a general insolubility of large complexes of protein. Strikingly, in the sgs1::hisG strain, Rad53p loses its association with the scaffold, whereas the recovery of topo II remains relatively stable (Fig. 7B). When quantified and normalized to a loading standard (Nup97p), the amount of Rad53p in the insoluble scaffold fraction is found to drop fourfold in the absence of Sgs1p (Fig. 7C). This suggests that the association of Rad53p with the scaffold fraction in S phase requires SGS1, consistent with the data presented above that Sgs1p is required for Rad53p activation in S phase.
We also note that although the vast majority of topo II is recovered in the insoluble scaffold fraction independent of the status of SGS1, there is a subpopulation of the enzyme that is reproducibly released to the supernatant in the sgs1::hisG strain (Fig. 7B,C). This may represent a fraction of topo II that, like Rad53p, is normally associated with the helicase (Watt et al. 1995).
Discussion
The survival of an organism depends on the proper duplication and segregation of its genome. Mutations in genes required for DNA replication and chromosome segregation generally lead to a rapid cell death. Those affecting DNA repair, recombination, or the genetic surveillance systems that monitor the integrity of the genome (DNA damage checkpoints) result in a more subtle genome instability, hastening senescence or provoking death indirectly through accumulated mutation. Several heritable human diseases, such as ataxia telangiectasia, xeroderma pigmentosum, the Nijmegen breakage syndrome, Fanconi anemia, Cockayne's, and Bloom's and Werner's syndromes (for review, see Auerbach and Verlander 1997), are due to defects in checkpoint or DNA damage repair functions. In most cases, humans affected with these diseases have a high incidence of cancer, most likely reflecting their inability to deal with genomic insult. SGS1, the S. cerevisiae homolog to the genes mutated in BLM and WRN syndromes, was isolated through its genetic interaction with yeast topo III (Gangloff et al. 1994) and its biochemical interaction with topo II (Watt et al. 1995). Cells lacking this 5′ to 3′ helicase are viable, but show an increase in DNA recombination and chromosome mis-segregation (Watt et al. 1996).
Here we present data implicating the Sgs1p DNA helicase directly in S-phase-specific checkpoints. We find that the protein is tightly cell cycle regulated, associating in S phase with foci that contain the ORC and Rad53p. Genetically, SGS1 and POL2 appear to form one epistasis group that acts upstream of Rad53p, a kinase essential for the cellular response to replication fork arrest and DNA damage. A significant fraction of Sgs1p colocalizes with Rad53p, and promotes the association of Rad53p with an insoluble nuclear fraction in S-phase cells. This suggests a direct role for Sgs1p in a checkpoint function. Possible roles for Sgs1p in DNA replication or in replication-linked repair are discussed below.
Sgs1p functions in S-phase-specific checkpoints
Strains lacking functional Sgs1p are partially deficient for S-phase checkpoints that respond to DNA damage and/or arrested replication forks. The defect may be at the level of detection of stalled replication forks or in the recognition or repair of resulting DNA damage. On the other hand, the G1/S and G2/M DNA damage checkpoints are fully functional in sgs1 cells. Consistent with an exclusive role in S phase, Sgs1p levels vary dramatically through the cell cycle, and the protein localizes to subnuclear foci during DNA replication (Fig. 5). These S-phase-specific foci label for both ORC and Rad53p, and analysis of the phosphorylation state of Rad53p in the presence of HU suggests that Sgs1p functions upstream of Rad53p, cooperating with Rad24p to stimulate Rad53p phosphorylation under conditions of fork arrest (Fig. 4). One means to function in this pathway would be to simply recruit Rad53p to its site of action. This is supported by a change in the fractionation properties of Rad53p in Sgs1p-deficient cells. However, recruitment is unlikely to be the only function of Sgs1p, because the helicase-deficient point mutant in sgs1-hd cannot rescue the HU sensitivity of the sgs1 disruption strain (Fig. 1D).
The localization and cell cycle regulation of Sgs1p are consistent with at least two models for Sgs1p function in S phase in addition to its potential role in Rad53p recruitment. In the first scenario, Sgs1p would be an integral replication fork helicase, whereas in the second, Sgs1p function would be restricted to surmounting impediments to fork progression (imposed by DNA damage, low nucleotide concentration, or proteins complexes) that might lead to single-strand gaps in the newly synthesized complementary strand (Bierne and Michel 1994). Sgs1p, in this situation, could function together with topo III or other topoisomerases to facilitate sister chromatid-mediated repair of gaps or deletions in the replicated DNA (for review, see Chakraverty and Hickson 1999).
Is Sgs1p part of the replication machinery?
It is striking that all proteins in the S/M replication checkpoint identified to date have an integral function in DNA replication. It has been proposed that the replication machinery signals continuously to Rad53p to delay M phase until replication is finished (Li 1995; Elledge 1996). The colocalization of both Rad53p and Sgs1p with ORC subunits, and their coincidence with sites of de novo DNA synthesis in isolated nuclei (data not shown; Pasero et al. 1999), fulfills the temporal and spatial requirements for a protein involved directly in DNA replication. However, as neither SGS1, nor its mammalian counterparts WRN (Yu et al. 1996) and BLM (Ellis et al. 1995), are essential genes, we must propose that either Sgs1p plays another role at these foci, or else it is redundant with other replication fork helicases.
As a partial test for a redundant function as the replication fork helicase, SGS1 was disrupted in strains carrying temperature-sensitive deficiencies in CDC47, a member of the MCM family that is itself a candidate for a replicative helicase (Aparicio et al. 1997), or DNA2, a helicase involved in Okazaki fragment processing (Budd and Campbell 1997). We detect no synergy between the sgs1 and cdc47-1 mutations, and in combination with the temperature-sensitive dna2-1 allele, we observe only a modest decrease in viability at semirestrictive temperatures (lethal at 28°C, rather than 32°C, data not shown). Although this may indicate a minor degree of synergy between Dna2p and Sgs1p functions, these are clearly not redundant enzymes. A definitive answer as to whether Sgs1p functions as a replication fork helicase might be obtained through a genetic screen for synthetic lethality with the sgs1 disruption and the identification of another redundant DNA helicase among the 40–50 DNA helicases encoded in the yeast genome.
Is Sgs1p involved in the repair of DNA damage provoked by DNA replication?
It has been proposed that low levels of DNA damage may occur as a result of normal replication fork movement, necessitating repair as S phase proceeds. In agreement with this, Ddc1p, a protein of the Rad24p DNA damage epistasis group, was observed to be phosphorylated and presumably activated, like Rad53p, during normal S-phase progression (Longhese et al. 1997). This suggests that normal DNA replication may be a discontinous process with replication forks being blocked either by limiting dNTP concentrations or by DNA-bound proteins. In E. coli, there is good evidence that a blocked replication fork induces DNA strand breaks (Bierne and Michel 1994; Kuzminov 1995); in yeast these could then be recognized as damaged DNA leading to complete Rad53p activation. Alternatively, the paused polymerase itself could send a signal to prevent cell cycle progression, consistent with roles assigned to POL2 and RFC in this event. In this model we propose that Sgs1p is important both for signal transmission to Rad53p and for recovery from replication fork arrest by helping in the repair of gaps, like those that are produced during normal DNA synthesis. For instance, Sgs1p may work together with DNA topoisomerases to resolve recombination events during S phase that allow sister chromatids to serve as templates for repair. Related functions have been proposed for the S. pombe homolog rqh1+ (Stewart et al. 1997).
Implications for Bloom's and Werner's syndromes
Perhaps the most significant new finding presented here is that both Rad53p phosphorylation and its subnuclear fractionation in S phase depend on Sgs1p. This provides a direct link between replication and/or repair proteins and a kinase that is essential for checkpoint function. This is consistent with the observation that one of the human homologs of Sgs1p, the BLM helicase, like the signal-transducing ATM1 kinase, colocalizes with Replication Protein A in meiotic mammalian cells (Plug et al. 1997, 1998; Walpita et al. 1999). It also is consistent with various replication defects associated with loss of the BLM helicase (Hand and German 1975; Lonn et al. 1990). A role for this class of helicase in checkpoint signaling is consistent with the cancer predisposition phenotypes associated with the BLM and WRN helicases, and underscores the importance of DNA replication-coupled damage surveillance mechanisms for the genetic integrity of a complex organism. However, it is less clear how the DNA damage checkpoint function might be responsible for the early aging and shortened life-span phenotypes found in Werner's syndrome patients and Sgs1p-deficient yeast cells (Salk et al. 1985; Sinclair et al. 1997).
Shortened life span in yeast is thought to be caused by the excision and accumulation of extrachromosomal circles containing rDNA repeats (Sinclair and Guarente 1997). However, in contrast to immunofluorescence data published by Sinclair et al. (1997), we do not find Sgs1p enriched in the nucleolus, but in S-phase-specific foci, of which a few localize to the nucleolus. We propose that the importance of Sgs1p for rDNA stability reflects a high level of gap-filling or sister-mediated recombinational repair found in the nucleolus (see also Chakraverty and Hickson 1999). Thus, the sgs1-induced shortening of life span would result from defects either in DNA replication or replication-associated checkpoints, rather than a specific nucleolar recombination defect. It remains unclear where the mammalian WRN helicase functions, as its reported localization to the nucleolus of mammalian cells has also been questioned (Marciniak et al. 1998; Shiratori et al. 1999). The work presented here raises the possibility that the WRN deficiency, like that of BLM, reflects loss of an S-phase surveillance mechanism, rather than loss of a specific nucleolar function.
Materials and methods
Strains and plasmids
All strains used are listed in Table 2. Unless stated otherwise, the disrupted allele sgs1-4 was used (sgs1::LEU2; Watt et al. 1995). This allele has the amino-terminal 408 amino acid intact, but the helicase and topo II interaction domains are downstream of the LEU2 insert. Expression of Sgs1p carboxy-terminal regions fused to Leu2p can be excluded because of stop codons (Watt et al. 1995), and Western blot analysis confirms that there is no translation of the carboxy-terminal 867 amino acid (data not shown). In GA-737, the LEU2 gene of sgs1::LEU2 was disrupted with hisG-URA-hisG. Counterselection to eliminate URA3 was performed on 5-FOA. Low-level expression of full-length SGS1 completely complements sgs1::LEU2 (data not shown), indicating that the truncated protein has no dominant phenotype. SS111-2-11 (pol2-11; Budd and Campbell 1993) was backcrossed three times to the A364a background (GA-871) to produce GA-1021–GA-1024. Sgs1p and Rad53p were tagged with 13 Myc epitopes at their carboxyl termini (Longtine et al. 1998), leaving them under the control of their endogenous promoters. Alleles of SGS1 carrying a helicase or a carboxy-terminal mutation are on a low-copy number plasmid (pRS415) under the control of the endogenous promoter. sgs1-hd (helicase mutant; Lu et al. 1996) has a mutation in the Walker A motif of the helicase domain and abolishes helicase activity in vitro. sgs1-ct is truncated at the carboxyl terminus downstream of the helicase domain (amino acid 1245; Lu et al. 1996).
Table 2.
Saccharomyces cerevisiae strains used in this study
| Strain
|
Genotype
|
Source
|
|---|---|---|
| GA-871 | MATa his3 leu2 ura3 trp1 (7830-2-4a) | Paulovitch et al. (1997) |
| GA-872 | GA-871 with rad24∷TRP1 (yMP10366a) | Paulovitch et al. (1997) |
| GA-904 | GA-871 with mec1-1 sml1 (yMP10847) | Paulovitch et al. (1997) |
| GA-880 | GA-871 with sgs1∷LEU2 | this study |
| GA-881 | GA-871 with rad24∷TRP1 sgs1∷LEU2 | this study |
| GA-1021 | GA-871 with pol2-11 | this study |
| GA-1022 | GA-871 with pol2-11 sgs1∷LEU2 | this study |
| GA-1023 | GA-871 with pol2-11 rad24∷TRP1 | this study |
| GA-1024 | GA-871 with pol2-11 sgs1∷LEU2 rad24∷TRP1 | this study |
| GA-1040 | GA-871 with RAD53–13Myc | this study |
| GA-1041 | GA-871 with sgs1∷LEU2 RAD53–13Myc | this study |
| GA-1042 | GA-871 with rad24∷TRP1 RAD53–13Myc | this study |
| GA-1043 | GA-871 with rad24∷TRP1 sgs1∷LEU2 RAD53–13Myc | this study |
| GA-1048 | GA-871 with mec1-1 sml1 RAD53–13Myc | this study |
| GA-1127 | MATa/α trp1/TRP1 ura3/URA3 leu2/LEU2 lys27LYS2 | this study |
| GA-1126 | GA-1127 with RAD53–13Myc/RAD53–13Myc | this study |
| GA-174 | MATa/α ura1/ura1 his7/his7 cdc16-1/cdc16-1 | L. Hartwell (University of Washington; Seattle) |
| GA-1151 | GA-174 with SGS1–13Myc/SGS1–13Myc | this study |
| GA-878 | MATa/a ade1/ade1 his2/his2 leu2/leu2 trp1/trp1 ura3/ura3 | this study |
| bar1/bar1 SGS1–13Myc/SGS1–13Myc | ||
| GA-59 | MATa leu2 ura3-52 trp1 prb1-1122 pep4-3 prc1-407 gal2 | E. Jones (Carnegie Mellon University, Pittsburgh, PA) |
| GA-737 | GA-59 with sgs1∷hisG | this study |
| GA-990 | GA-59 with SGS1–13Myc | this study |
| GA-1222 | MATa/α cdc4-3/cdc4-3 ade2/ade2 leu2/leu2 ura3/ura3 | this study |
| trp1/trp1 ORC1–3HA∷URA3/ORC1–3HA∷URA3 | ||
| SS111-2-11 | MATa trp1-289 ura3-1,2 ade2-101 gal2 can1 pol2-11 | Budd and Campbell (1993) |
HU, UV, and γ radiation assay
Where indicated, HU (Sigma) was added to 200 mm. Aliquots were removed and either plated in triplicate or fixed in formaldehyde and the concentration was determined with a cell counter (Schärfe System). Growing colonies versus plated cells were quantified. The 0-min time point was set as 100%. A UVC (254 nm) or a 137Cs 4000Ci source was used for UV or γ radiation, respectively. Viability was scored as with HU.
G1/S, G2/M, and intra-S checkpoint
MMS (Sigma) was added to create DNA damage and checkpoints were tested as in Sidorova and Breeden (1997) for G1/S; Xu and Norris (1998) for G2/M; and Paulovich and Hartwell (1995) for intra-S.
Immunofluorescence
Immunofluorescence was performed as described previously (Gotta et al. 1999) except that in all cases, cells were fixed with formaldehyde before spheroplasting. The polyclonal sera [chicken anti-Sgs1p, gift of D. Sinclair (MIT, Cambridge, MA); rabbit anti-Rad53p, gift of M. Schwartz; rabbit anti-Rap1p] were affinity purified. Goat anti-Orc2p (Santa Cruz) was blocked by incubation with the peptide against which the antibody was raised. Mouse anti-Nop1 and rabbit anti-Rap1 were described earlier (Klein et al. 1992; Gotta et al. 1997). The secondary antibodies labeled with the fluorochromes DTAF (green) and Cy-5 (red) show no crossreaction. Single focal sections were taken on a Zeiss LSM 410 confocal microscope and signal over background was determined with Adobe Photoshop. When double labeling was performed, only the areas in which the two signals perfectly coincide are shown and labeled Coloc.
Image analysis
Two-dimensional quantification was performed as described in Table 1 and in Gotta et al. (1996). For three dimensional reconstitution, 30–40 100 nm Z-sections were taken per nucleus with a Zeiss LSM 410 confocal microscope with a 63×/1.40 oil objective (Plan-Apochromat, Zeiss). Deconvolution done by Huygans with 10–12 iterations and overlapping regions was determined by Colocalization, quantified by VoxelShop Pro as mass (volume × density) and visualized by Imaris (Bitplane, Inc., Zürich, Switzerland).
Cell fractionation
One-half of an exponentially growing culture was blocked with α-factor and released into S phase. Random and S-phase cells were centrifuged, resuspended in 100 mm K-EDTA, 1 mm DTT, and incubated for 10 min at 30°C. Cells were recovered and spheroplasted in 1.2 m sorbitol in 0.4× YPAD at 30°C with gentle shaking. Cells were washed once in Sorbitol and once in wash buffer (5 mm Tris-Cl at pH 7.4, 20 mm KCl, 2 mm K-EDTA, 0.125 mm spermidine, 0.05 mm spermine, 0.4 m sorbitol, 1% thiodiglycol, 1% Trasylol, and 0.5 mm PMSF). A total of 100 μl of the cell pellet was resuspended in 500 μl of wash buffer and cells were lysed by the addition of 1 volume of lysis buffer (wash buffer plus 2% Triton X-100, 300 μg/ml benzamidine, 1 μg/ml pepstatin, 2 μg/ml anti-pain, 0.5 μg/ml leupeptin, 100 μg/ml TPCK, and 50 μg/ml TLCK). Lysis was performed on ice for 3–5 min and complete breakage was verified under a light microscope. An aliquot (WCE) was removed and the remaining cells were spun at 13,500 rpm at 4°C for 15 min, the Sup was saved and the pellet was resuspended and incubated at 4°C in 50 mm Tris-Cl at pH 7.4, 125 mm KCl, 7.5 mm MgCl2, 0.5% Triton X-100, with 7.5 μg DNase I and protease inhibitors. The supernatant, after centrifugation for 5 min at 13,500 rpm, was saved (soluble chromatin; Chr) and the pellet was resuspended in digestion buffer without DNase I (insoluble scaffold fraction; Sc). The protein concentration of each fraction was determined and SDS–polyacrylamide sample buffer was added. For Western blots, equivalent volumes, representing the same number of starting cells, are loaded for WCE and Sup, and five times this is used for Chr and Sc fractions. Nup97p (mAb 414; BAbCO) fractionates in a reproducible manner and is used as a loading control.
FACS analysis
Cells were fixed overnight in 70% ethanol at 4°C. After centrifugation, 200 μg/ml RNase A (Sigma) in 50 mm Tris-Cl at pH 7.4 was added for 2 hr at 37°C. Cells were spun and resuspended in 10 μg/ml propidium iodide (Sigma) in 50 mm Na-citrate (pH 7.0) and incubated overnight in the dark at 4°C. Samples were analyzed in a FACS scan.
Acknowledgments
We thank the following people for antibodies: M. Schwartz and D. Stern for anti-Rad53p, D. Sinclair and L. Guarente for chicken anti-Sgs1p, D. Allis for anti-histone H3, P. Burgers for anti-Rfc3p and V. Simanis for mouse anti-tubulin. We are grateful to L. Hartwell, M. Longtine, J. Lu, S. Brill, D. Sinclair, A. Sugino, P. Watt, and T. Weinert for yeast strains and plasmids. T. Laroche is acknowledged for his expert help with confocal microscopy and image analysis, B. Duncker for a critical reading of the manuscript. This research was founded by Swiss National Science Foundation and Swiss Cancer League grants to S.M.G.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL sgasser@eliot.unil.ch; FAX 41 21 652 6933.
References
- Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: Redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- Araki H, Leem SH, Phongdara A, Sugino A. Dpb11, which interacts with DNA polymerase II(ε) in S. cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc Natl Acad Sci. 1995;92:11791–11795. doi: 10.1073/pnas.92.25.11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach AD, Verlander PC. Disorders of DNA replication and repair. Curr Opin Pediatr. 1997;9:600–616. doi: 10.1097/00008480-199712000-00010. [DOI] [PubMed] [Google Scholar]
- Bierne H, Michel B. When replication forks stop. Mol Microbiol. 1994;13:17–23. doi: 10.1111/j.1365-2958.1994.tb00398.x. [DOI] [PubMed] [Google Scholar]
- Budd ME, Campbell JL. DNA polymerases δ and ε are required for chromosomal replication in S. cerevisiae. Mol Cell Biol. 1993;13:496–505. doi: 10.1128/mcb.13.1.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— A yeast replicative helicase, Dna2 helicase, interacts with yeast Fen-1 nuclease in carrying out its essential function. Mol Cell Biol. 1997;17:2136–2142. doi: 10.1128/mcb.17.4.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraverty RK, Hickson ID. Defending genome integrity during DNA replication: A proposed role for RecQ family helicases. BioEssays. 1999;21:286–294. doi: 10.1002/(SICI)1521-1878(199904)21:4<286::AID-BIES4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Chester N, Kuo F, Kozak C, O'Hara CD, Leder P. Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom's syndrome gene. Genes & Dev. 1998;12:3382–3393. doi: 10.1101/gad.12.21.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Koshland D. The anaphase inhibitor of S. cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc Natl Acad Sci. 1997;94:14361–14366. doi: 10.1073/pnas.94.26.14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coverley D, Laskey RA. Regulation of eukaryotic DNA replication. Annu Rev Biochem. 1994;63:745–776. doi: 10.1146/annurev.bi.63.070194.003525. [DOI] [PubMed] [Google Scholar]
- Davey S, Han CS, Ramer SA, Klassen JC, Jacobson A, Eisenberger A, Hopkins KM, Lieberman HB, Freyer GA. Fission yeast rad12+ regulates cell cycle checkpoint control and is homologous to the Bloom's syndrome disease gene. Mol Cell Biol. 1998;18:2721–2728. doi: 10.1128/mcb.18.5.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ruiz MA, Green CM, Lowndes NF. RAD9 and RAD24 define two additive, interacting branches of the DNA damage checkpoint pathway in budding yeast normally required for Rad53 modification and activation. EMBO J. 1998;17:2687–2698. doi: 10.1093/emboj/17.9.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes & Dev. 1998;12:2956–2970. doi: 10.1101/gad.12.18.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints—preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner's syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine. 1966;45:177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- Gangloff S, McDonald JP, Bendixen C, Arthur L, Rothstein R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: A potential eukaryotic reverse gyrase. Mol Cell Biol. 1994;14:8391–8398. doi: 10.1128/mcb.14.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R, Putnam CW, Weinert T. RAD53, DUN1, and PDS1 define two parallel G2/M checkpoint pathways in budding yeast. EMBO J. 1999;18:3173–3185. doi: 10.1093/emboj/18.11.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German J. Bloom syndrome: A mendelian prototype of somatic mutational disease. Medicine. 1993;72:393–406. [PubMed] [Google Scholar]
- Gotta M, Laroche T, Formenton A, Maillet L, Scherthan H, Gasser SM. The clustering of telomeres and colocalization with Rap1, Sir3, and Sir4 proteins in wild-type S. cerevisiae. J Cell Biol. 1996;134:1349–1363. doi: 10.1083/jcb.134.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotta M, Strahl-Bolsinger S, Renauld H, Laroche T, Kennedy BK, Grunstein M, Gasser SM. Localization of Sir2p: The nucleolus as a compartment for silent information regulators. EMBO J. 1997;16:3243–3255. doi: 10.1093/emboj/16.11.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotta M, Laroche T, Gasser SM. Analysis of nuclear organization in Saccharomyces cerevisiae. Methods Enzymol. 1999;304:663–672. doi: 10.1016/s0076-6879(99)04040-9. [DOI] [PubMed] [Google Scholar]
- Hand R, German J. A retarded rate of DNA chain growth in Bloom's syndrome. Proc Natl Acad Sci. 1975;72:758–762. doi: 10.1073/pnas.72.2.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB. Checkpoint controls and cancer. Cancer Surv. 1997;29:1–6. [PubMed] [Google Scholar]
- Klein F, Laroche T, Cardenas ME, Hofmann JF, Schweizer D, Gasser SM. Localization of RAP1 and topoisomerase II in nuclei and meiotic chromosomes of yeast. J Cell Biol. 1992;117:935–948. doi: 10.1083/jcb.117.5.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano K, Berres ME, Engels WR. Evolution of the RECQ family of helicases. A Drosophila homolog, DmBLM, is similar to the human Bloom's syndrome gene. Genetics. 1999;151:1027–1039. doi: 10.1093/genetics/151.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. Instability of inhibited replication forks in E. coli. BioEssays. 1995;17:733–741. doi: 10.1002/bies.950170810. [DOI] [PubMed] [Google Scholar]
- Li JJ. DNA replication. Once, and only once. Curr Biol. 1995;5:472–475. doi: 10.1016/s0960-9822(95)00094-7. [DOI] [PubMed] [Google Scholar]
- Longhese MP, Neecke H, Paciotti V, Lucchini G, Plevani P. The 70 kDa subunit of replication protein A is required for the G1/S and intra-S DNA damage checkpoints in budding yeast. Nucleic Acids Res. 1996;24:3533–3537. doi: 10.1093/nar/24.18.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese MP, Paciotti V, Fraschini R, Zaccarini R, Plevani P, Lucchini G. The novel DNA damage checkpoint protein Ddc1p is phosphorylated periodically during the cell cycle and in response to DNA damage in budding yeast. EMBO J. 1997;16:5216–5226. doi: 10.1093/emboj/16.17.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in S. cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lonn U, Lonn S, Nylen U, Winblad G, German J. An abnormal profile of DNA replication intermediates in Bloom's syndrome. Cancer Res. 1990;50:3141–3145. [PubMed] [Google Scholar]
- Lu JA, Mullen JR, Brill SJ, Kleff S, Romeo AM, Sternglanz R. Human homologues of yeast helicase. Nature. 1996;383:678–679. doi: 10.1038/383678a0. [DOI] [PubMed] [Google Scholar]
- Lydall D, Weinert T. G2/M checkpoint genes of S. cerevisiae: Further evidence for roles in DNA replication and/or repair. Mol & Gen Genet. 1997;256:638–651. doi: 10.1007/s004380050612. [DOI] [PubMed] [Google Scholar]
- Marciniak RA, Lombard DB, Johnson FB, Guarente L. Nucleolar localization of the Werner syndrome protein in human cells. Proc Natl Acad Sci. 1998;95:6887–6892. doi: 10.1073/pnas.95.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini F, Pellicioli A, Paciotti V, Lucchini G, Plevani P, Stern DF, Foiani M. A role for DNA primase in coupling DNA replication to DNA damage response. EMBO J. 1997;16:639–650. doi: 10.1093/emboj/16.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov V, Mushegian AR, Koonin EV, Bork P. A putative nucleic acid-binding domain in Bloom's and Werner's syndrome helicases. Trends Biochem Sci. 1997;22:417–418. doi: 10.1016/s0968-0004(97)01128-6. [DOI] [PubMed] [Google Scholar]
- Murray JM, Lindsay HD, Munday CA, Carr AM. Role of S. pombe RecQ homolog, recombination, and checkpoint genes in UV damage tolerance. Mol Cell Biol. 1997;17:6868–6875. doi: 10.1128/mcb.17.12.6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Morita T, Sato C. Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp Cell Res. 1986;165:291–297. doi: 10.1016/0014-4827(86)90583-5. [DOI] [PubMed] [Google Scholar]
- Navas TA, Zhou Z, Elledge SJ. DNA polymerase epsilon links the DNA replication machinery to the S phase checkpoint. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- Navas TA, Sanchez Y, Elledge SJ. RAD9 and DNA polymerase ε form parallel sensory branches for transducing the DNA damage checkpoint signal in S. cerevisiae. Genes & Dev. 1996;10:2632–2643. doi: 10.1101/gad.10.20.2632. [DOI] [PubMed] [Google Scholar]
- Pasero P, Gasser SM. New systems for replicating DNA in vitro. Curr Opin Cell Biol. 1998;10:304–310. doi: 10.1016/s0955-0674(98)80004-5. [DOI] [PubMed] [Google Scholar]
- Pasero P, Braguglia D, Gasser SM. ORC-dependent and origin-specific initiation of DNA replication at defined foci in isolated yeast nuclei. Genes & Dev. 1997;11:1504–1518. doi: 10.1101/gad.11.12.1504. [DOI] [PubMed] [Google Scholar]
- Pasero P, Duncker BP, Schwob E, Gasser SM. A role for the Cdc7 kinase regulatory subunit Dbf4p in the formation of initiation-competent origins of replication. Genes & Dev. 1999;13:2159–2176. doi: 10.1101/gad.13.16.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulovich AG, Hartwell LH. A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell. 1995;82:841–847. doi: 10.1016/0092-8674(95)90481-6. [DOI] [PubMed] [Google Scholar]
- Paulovich AG, Margulies RU, Garvik BM, Hartwell LH. RAD9, RAD17, and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics. 1997;145:45–62. doi: 10.1093/genetics/145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plug AW, Peters AH, Xu Y, Keegan KS, Hoekstra MF, Baltimore D, de Boer P, Ashley T. ATM and RPA in meiotic chromosome synapsis and recombination. Nat Genet. 1997;17:457–461. doi: 10.1038/ng1297-457. [DOI] [PubMed] [Google Scholar]
- Plug AW, Peters AH, Keegan KS, Hoekstra MF, de Boer P, Ashley T. Changes in protein composition of meiotic nodules during mammalian meiosis. J Cell Sci. 1998;111:413–423. doi: 10.1242/jcs.111.4.413. [DOI] [PubMed] [Google Scholar]
- Poot M, Hoehn H, Runger TM, Martin GM. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp Cell Res. 1992;202:267–273. doi: 10.1016/0014-4827(92)90074-i. [DOI] [PubMed] [Google Scholar]
- Puranam KL, Blackshear PJ. Cloning and characterization of RECQL, a potential human homologue of the Escherichia coli DNA helicase RecQ. J Biol Chem. 1994;269:29838–29845. [PubMed] [Google Scholar]
- Salk D, Bryant E, Hoehn H, Johnston P, Martin GM. Growth characteristics of Werner syndrome cells in vitro. Adv Exp Med Biol. 1985;190:305–311. doi: 10.1007/978-1-4684-7853-2_14. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- Shiratori M, Sakamoto S, Suzuki N, Tokutake Y, Kawabe Y, Enomoto T, Sugimoto M, Goto M, Matsumoto T, Furuichi Y. Detection by epitope-defined monoclonal antibodies of Werner DNA helicases in the nucleoplasm and their upregulation by cell transformation and immortalization. J Cell Biol. 1999;144:1–9. doi: 10.1083/jcb.144.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova JM, Breeden LL. Rad53-dependent phosphorylation of Swi6 and down-regulation of CLN1 and CLN2 transcription occur in response to DNA damage in Saccharomyces cerevisiae. Genes & Dev. 1997;11:3032–3045. doi: 10.1101/gad.11.22.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles–a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Mills K, Guarente L. Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science. 1997;277:1313–1316. doi: 10.1126/science.277.5330.1313. [DOI] [PubMed] [Google Scholar]
- Stewart E, Chapman CR, Al-Khodairy F, Carr AM, Enoch T. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 1997;16:2682–2692. doi: 10.1093/emboj/16.10.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Fay DS, Marini F, Foiani M, Stern DF. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes & Dev. 1996;10:395–406. doi: 10.1101/gad.10.4.395. [DOI] [PubMed] [Google Scholar]
- Walpita D, Plug AW, Neff NF, German J, Ashley T. Bloom's syndrome protein, BLM, colocalizes with replication protein A in meiotic prophase nuclei of mammalian spermatocytes. Proc Natl Acad Sci. 1999;96:5622–5627. doi: 10.1073/pnas.96.10.5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Elledge SJ. DRC1, DNA replication and checkpoint protein 1, functions with DPB11 to control DNA replication and the S-phase checkpoint in Saccharomyces cerevisiae. Proc Natl Acad Sci. 1999;96:3824–3829. doi: 10.1073/pnas.96.7.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt PM, Hickson ID. Genome stability—failure to unwind causes cancer. Curr Biol. 1996;6:265–267. doi: 10.1016/s0960-9822(02)00474-8. [DOI] [PubMed] [Google Scholar]
- Watt PM, Louis EJ, Borts RH, Hickson ID. Sgs1: A eukaryotic homolog of E. coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell. 1995;81:253–260. doi: 10.1016/0092-8674(95)90335-6. [DOI] [PubMed] [Google Scholar]
- Watt PM, Hickson ID, Borts RH, Louis EJ. Sgs1, a homologue of the Blooms and Werners Syndrome genes, is required for maintenance of genome stability in S. cerevisiae. Genetics. 1996;144:935–945. doi: 10.1093/genetics/144.3.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T. DNA damage checkpoints update: Getting molecular. Curr Opin Genet Dev. 1998;8:185–193. doi: 10.1016/s0959-437x(98)80140-8. [DOI] [PubMed] [Google Scholar]
- Xu Z, Norris D. The SFP1 gene product of S. cerevisiae regulates G2/M transitions during the mitotic cell cycle and DNA-damage response. Genetics. 1998;150:1419–1428. doi: 10.1093/genetics/150.4.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Kato J, Shimamoto A, Goto M, Furuichi Y, Ikeda H. Bloom's and Werner's syndrome genes suppress hyperrecombination in yeast sgs1 mutant: Implication for genomic instability in human diseases. Proc Natl Acad Sci. 1998;95:8733–8738. doi: 10.1073/pnas.95.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Newport J. FFA-1, a protein that promotes the formation of replication centers within nuclei. Science. 1995;269:1883–1885. doi: 10.1126/science.7569932. [DOI] [PubMed] [Google Scholar]
- Yan H, Chen CY, Kobayashi R, Newport J. Replication focus-forming activity 1 and the Werner syndrome gene product. Nat Genet. 1998;19:375–378. doi: 10.1038/1263. [DOI] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, et al. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]