

Abstract
We have uncovered a novel functional relationship between the hyaluronan receptor CD44, the matrix metalloproteinase-9 (MMP-9) and the multifunctional cytokine TGF-β in the control of tumor-associated tissue remodeling. CD44 provides a cell surface docking receptor for proteolytically active MMP-9 and we show here that localization of MMP-9 to cell surface is required for its ability to promote tumor invasion and angiogenesis. Our observations also indicate that MMP-9, as well as MMP-2, proteolytically cleaves latent TGF-β, providing a novel and potentially important mechanism for TGF-β activation. In addition, we show that MMP-9 localization to the surface of normal keratinocytes is CD44 dependent and can activate latent TGF-β. These observations suggest that coordinated CD44, MMP-9, and TGF-β function may provide a physiological mechanism of tissue remodeling that can be adopted by malignant cells to promote tumor growth and invasion.
Keywords: MMP-9, CD44, TGF-β, tumor angiogenesis, tumor invasion
The ability of tumor cells to establish a functional relationship with their host tissue microenvironment and to utilize the local resources to their advantage are key factors in determining tumor cell survival, local tumor development, and subsequent tumor invasion. Interaction between tumor cells and the host tissue stroma results in local tissue remodeling that, on the one hand, reflects a reaction of the tissue to the invading tumor cells and, on the other hand, serves to facilitate and promote tumor development. Similar to inflammation and repair, tumor-associated tissue remodeling relies on a complex interplay of at least three classes of molecules, that is, adhesion receptors that mediate physical interactions between tumor cells and host tissue stromal cells and extracellular matrix (ECM), matrix metalloproteinases (MMPs) that degrade ECM proteins, and cytokines/growth factors that promote tumor cell survival and growth.
Experimental evidence indicates that integrin (for review, see Varner and Cheresh 1996) and hyaluronan receptor CD44 (Bartolazzi et al. 1994; Gunthert et al. 1991; Sy et al. 1991)-mediated adhesion are required for tumor cell migration as well as for growth and dissemination of a variety of tumor types. Recent work suggests that signals transduced by integrins following engagement by ligands may provide tumor cells with critical survival signals within the host tissue microenvironment (Frisch and Ruoslahti 1997). Ligand engagement of CD44 can also promote survival of some types of tumor cells in vivo (Yu et al. 1997), and numerous studies have provided evidence that CD44 may play an important role in promoting tumor invasion and metastasis (Gunthert et al. 1991; Sy et al. 1991; Seiter et al. 1993; Bartolazzi et al. 1994; Lamb et al. 1997; Yu et al. 1997). Although the ability of CD44 to bind and internalize hyaluronan has been shown to correlate with tumor cell invasiveness (Bartolazzi et al. 1994; Culty et al. 1994; Yu et al. 1997), the mechanisms underlying CD44-mediated enchancement of tumor growth have remained largely obscure. We have shown recently that CD44 can localize proteolytically active matrix metalloproteinase-9 (MMP-9) to the surface of TA3 mammary carcinoma and MC melanoma cells (Yu and Stamenkovic 1999). Hyaluronan-mediated cross-linking of CD44 induces coclustering of CD44 and MMP-9 and promotes MMP-9 proteolytic activity, which correlates with TA3 cell invasiveness in vivo and in vitro and enhances tumor growth in vivo (Yu and Stamenkovic 1999). Compromising CD44 function by overexpression of a dominant-negative soluble CD44 disrupts cell surface CD44–MMP-9 cluster formation, reduces cell surface MMP-9 activity, and inhibits invasion and growth of the tumor cells (Yu and Stamenkovic 1999). Thus, one mechanism by which CD44 may promote tumor growth and invasion is by regulating MMP-9 function on the cell surface.
Matrix metalloproteinases are thought to play a key role in promoting tumor invasion and tissue remodeling by inducing proteolysis of several ECM components (Stetler-Stevenson et al. 1993; Werb 1997). Recent evidence indicates that MMPs may promote angiogenesis (Brooks et al. 1996; 1998; Vu et al. 1998), and augment genetic instability in tumor cells (Sternlicht et al. 1999). MMP expression is induced in stromal cells at sites of injury and repair as well as at sites of tumor invasion, and many tumors produce their own MMPs as a part of their invasive machinery. However, the mechanisms by which MMPs promote tumor invasion and angiogenesis are still poorly understood. For example, the role that MMP activity may play in the release and activation of ECM-sequestered growth and angiogenic factors, which may be essential for tumor development, is largely unknown. Furthermore, the majority of MMPs whose proteolytic activity is associated with tissue remodeling are soluble and are efficiently inhibited by tissue inhibitors of metalloproteinases (TIMPs), which are abundant in the ECM. Despite significant steps toward understanding the complexities of MMP activation (for review, see Nagase 1997), it remains unclear how MMPs might escape or overcome TIMP inhibition to promote ECM breakdown and tumor invasion.
In the present study, we show that cell surface localization of MMP-9 is an important factor in its ability to promote not only tumor invasion, but angiogenesis and growth as well. We also demonstrate that MMP-9, as well as its relative, MMP-2, cleave latent transforming growth factor-β (TGF-β), which constitutes a novel mechanism of TGF-β activation. Finally, we provide evidence that CD44-dependent cell surface localization of proteolytically active MMP-9, and the corresponding latent TGF-β activation, occur in normal as well as in malignant cells.
Results
CD44 is implicated in tumor angiogenesis
Using TA3 murine mammary carcinoma cells, which constitutively express several CD44 isoforms and exhibit CD44-dependent HA binding, up-take and degradation, we have shown previously that expression of dominant-negative soluble CD44 inhibits tumor invasion and growth in syngeneic mice (Yu and Stamenkovic 1999; Fig. 1 A–C). We now show that in addition to impaired invasiveness and reduced size, solid tumors derived from the soluble CD44-transfected TA3 cells (TA3sCD44) are less vascularized than tumors derived from TA3 cells transfected with expression vector only (TA3wt, Fig. 1A,B). To compare angiogenesis in the solid TA3wt and TA3sCD44 tumors, we performed immunohistochemistry on sections of tumors derived from subcutaneously injected TA3wt and TA3sCD44 cells using anti-von Willebrand factor antibody (anti-vWF), which stains vascular endothelial cells. Solid TA3sCD44 tumors were observed to display less vascular endothelial cell staining than their TA3wt counterparts (Fig. 1D,E). Blood vessel quantification by averaging the number of capillary blood vessels positively stained by anti-vWF antibody in 10 randomly picked microscopic fields revealed an approximately threefold reduction in the number of blood vessels in the solid tumors derived from TA3sCD44 cells compared with tumors derived from TA3wt cells (Fig. 1F). Thus, reduced angiogenesis in solid tumors derived from TA3sCD44 cells appeared to correlate with impaired CD44 function.
Figure 1.
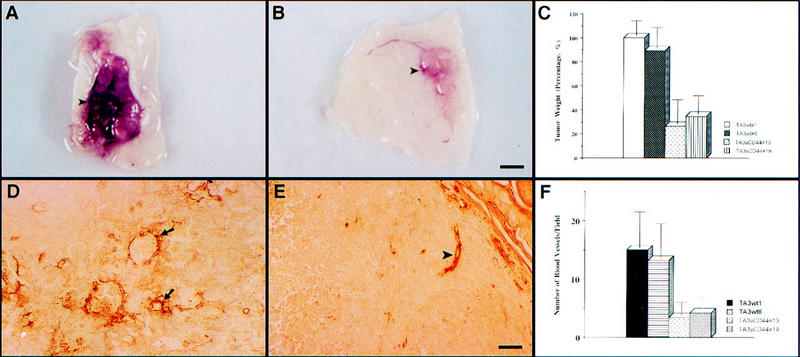
CD44 promotes tumor growth and angiogenesis. (A, B) Gross appearance of solid tumors derived from TA3wt (A) and TA3sCD44 cells (B). Bar, 7.5mm. (C) A total of 2 × 106 viable cells were injected subcutaneously into syngeneic A/Jax mice. The tumor specimens were collected 3 weeks after injection, and tumor weight was assessed. In each experiment, six mice were injected with each transfectant. Weight of tumors derived from TA3 transfectants is expressed relative to that of TA3wt 1 ±s.d. (D, E) Immunohistochemistry with anti-vWF antibody (Dako) on tissue sections of solid tumors derived from subcutaneously injected TA3wt (D) or TA3sCD44 cells (E). The average number of the capillary blood vessels, as indicated by anti-vWF antibody staining, per 10× power microscopic field from 10 randomly chosen fields is shown (F). Bar, 160 μm.
Localization of MMP-9 to the cell surface augments its ability to promote tumor invasion and angiogenesis
In previous studies we showed that CD44 localizes the proteolytic form of MMP-9 on the surface of TA3wt cells, and that expression of the CD44–MMP-9 complex correlates with TA3 cell invasiveness in vitro and in vivo (Yu and Stamenkovic 1999). To investigate whether the observed invasiveness is dependent on cell surface localization of the proteolytic activity of MMP-9, TA3sCD44 cells were stably transfected with a chimeric construct composed of sequences encoding MMP-9 and the transmembrane and intracellular domains of CD44 (MMP-9/CD44fp, Fig. 2A). On expression in mammalian cells, the resulting fusion protein was tethered to the cell surface. As a control, TA3sCD44 cells were transfected with carboxy-terminal v5-epitope-tagged soluble MMP-9 (MMP-9v5, Fig. 2A). Expression of endogenous, and soluble, transfected, CD44 were not affected by the introduction of MMP-9/CD44fp and MMP-9v5 cDNAs into TA3sCD44 cells, as assessed by Western blot analysis (Fig. 2Ba and b). Expression of soluble MMP-9v5 and the MMP-9/CD44 fusion protein were determined by gelatin zymography of the serum-free cell culture supernatants (Fig. 2B,c) and cell membrane lysates (Fig. 2B,d), respectively. The MMP-9/CD44 fusion protein displayed a molecular mass of ∼120 kD (Fig. 2B,d, lanes 5,6), compared with the ∼90 kD of MMP-9v5 (Fig. 2B,c, lanes 7–8). All of the transfectants expressed similar levels of endogenous MMP-9 of 83 kD. To compare the function of cell surface-tethered MMP-9–CD44fp with that of overexpressed soluble MMP-9v5, the transfectants were assessed for G8 myoblast monolayer invasion. Expression of the MMP-9–CD44 fusion protein was observed to restore the ability of TA3sCD44 cells to invade the G8 monolayer, whereas expression of soluble MMP-9v5 failed to do so (Fig. 2C).
Figure 2.

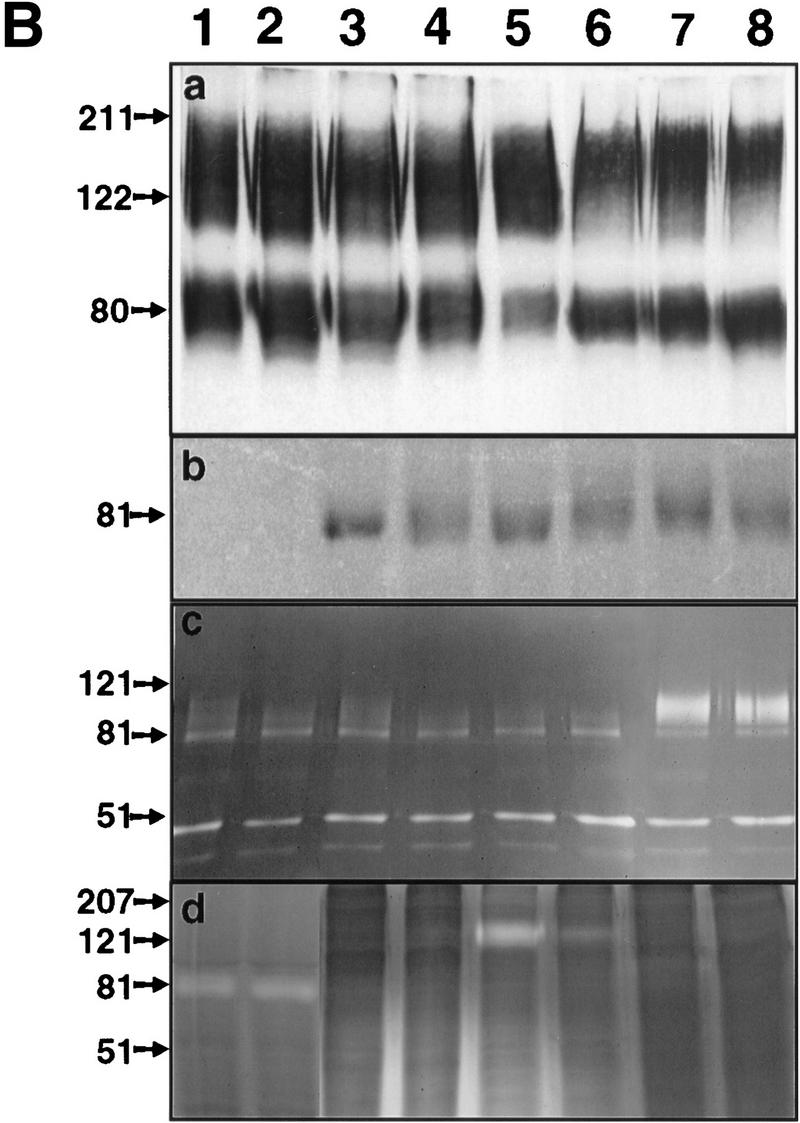
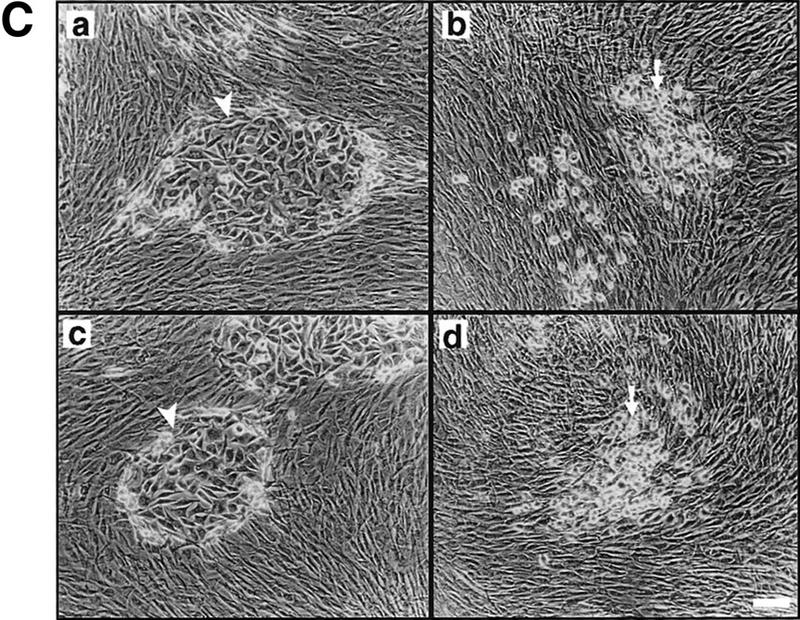
Characterization of the TA3 transfectants and G8 myoblast invasion assays. (A) Schematic representation of v5-tagged MMP-9 and the CD44–MMP-9 fusion protein. Amino acid sequences that form the fusion are indicated. (L) Leader peptide; (pro) prodomain; (Zn) zinc-binding domain (active site); (hemopex) hemopexin domain; (tm) transmembrane domain; (ic) intracellular domain; (v5) v5 peptide tag. (B) (a) Western blot of TA3 transfectant cell lysates and corresponding concentrated serum-free conditioned media (b), with anti-CD44 mAb IM7.8. Endogenous CD44 expression is comparable in all transfectants (a). Soluble CD44 expression is comparable in TA3sCD44, TA3sCD44mmp-9/CD44fp, TA3sCD44mmp-9v5 cells (b, lanes 3–8), whereas TA3wt cells do not express soluble CD44 (lanes 1,2). (c,d) Gelatin zymograms of (c) serum-free media and (d) crude membrane preparations of the transfected TA3 cells. Cell lysates (a), conditioned media (b,c), and crude membrane preparations (d) were from the following: (lanes 1, 2) TA3wt cells; (lanes 3,4) TA3sCD44 cells; (lanes 5,6) TA3sCD44mmp-9/CD44fp cells; (lanes 7,8) TA3sCD44MMP-9v5 cells. Molecular mass markers are indicated. (C) G8 myoblast monolayer invasion assay: Expression of the MMP-9–CD44 fusion protein by TA3sCD44mmp-9/CD44fp cells (a, arrowhead) rescues the ability of TA3sCD44 (b, arrow) to invade the G8 monolayer to an extent that is comparable with that of TA3wt cells (c, arrowhead), whereas overexpression of soluble MMP-9v5 has little effect (d, arrow). Bar, 80 μm.
The effect of the MMP-9–CD44 fusion protein and that of soluble MMP-9v5 expression on tumor growth, invasiveness, and angiogenesis in vivo were compared by injecting the respective TA3sCD44 cell transfectants subcutaneously into syngeneic A/Jax mice. Expression of the cell surface MMP-9/CD44 fusion protein in TA3sCD44 cells was found to restore both the invasiveness and growth of the resulting tumors in vivo, whereas overexpression of soluble MMP-9v5 had virtually no effect (Fig. 3 A,B). Similarly, expression of the MMP-9/CD44 fusion protein, but not soluble MMP-9v5, restored TA3sCD44 tumor angiogenesis, as assessed by the average number of anti-vWF-stained blood vessels per 10× microscopic field (Fig. 3C). Thus, localization of MMP-9 proteolytic activity to the cell surface, and not merely its secretion, is required to promote TA3 tumor invasion and angiogenesis in vivo.
Figure 3.
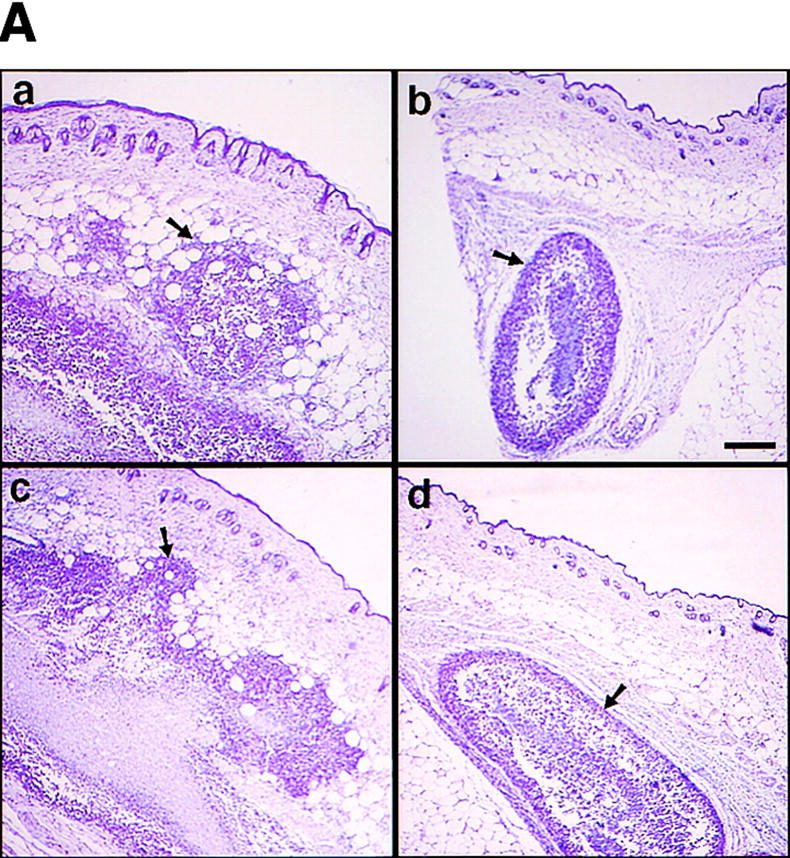


Expression of MMP-9–CD44 fusion proteins restores the invasiveness, growth, and angiogenesis of the TA3sCD44 cells in vivo. (A) Hematoxylin-stained sections of the representative tumors derived from the subcutaneously injected TA3wt (a), TA3sCD44 (b), TA3sCD44MMP-9/CD44fp (c), and TA3sCD44MMP-9v5 (d) cells. A total of 2 × 106 viable cells were injected, and the tumor specimens were collected 1 week after the injection. (B), In a separate set of animals, tumor weight was assessed 3 weeks after the injection. In each experiment, six mice were injected with each transfectant. Mean weight ±s.d. of tumors derived from the TA3 transfectants are shown. (C) Tumor angiogenesis was assessed and expressed as the average number of capillary blood vessels, as revealed by anti-vWF antibody, per 10× power microscopic field in the solid tumor sections derived from TA3 transfectants. The number ±s.d. represents the average blood vessel count per microscopic field from total counts in 10 randomly selected fields. Bar, 300 μm.
Vascular endothelial cell tubulogenesis is induced by TA3wt/G8 myoblast coculture-conditioned medium
To address the molecular mechanisms that underlie MMP-9-mediated tumor angiogenesis and to identify the putative substrate(s) of cell surface-localized MMP-9 that is(are) involved in promoting angiogenesis, we established a coculture system composed of wild-type or transfectant TA3 cells and fixed G8 myoblast monolayers, which approximates tumor and stromal cell interaction and up-regulates the expression of MMP-9 in TA3 cells (Yu et al. 1997; data not shown). The serum-free conditioned media derived from the cocultures were tested for their ability to stimulate formation of capillary tubes by bovine microvascular endothelial cells (BME) grown on collagen I gels. TA3 wild-type/G8 and TA3sCD44/MMP-9-CD44fp/G8 coculture conditioned media induced BME tubule formation (Fig. 4A,C), whereas TA3sCD44/G8 and TA3sCD44/MMP-9v5/G8 coculture conditioned media did not (Fig. 4B,D). Serum-free conditioned media derived from fixed G8 myoblasts or viable TA3wt cells alone had no tubulogenic effect on BME cells (data not shown). TA3 cell surface MMP-9 activity therefore appears to be required for the induction of BME tubulogenesis by the coculture conditioned medium.
Figure 4.

Endothelial cell tubule formation assay. BME cells (3 × 105/ml) were seeded onto type I collagen gels in medium supplemented with 10% calf serum. On the following day, the medium was aspirated and replaced with conditioned serum-free medium derived from cocultures of G8 myoblast monolayers and TA3wt (A) TA3sCD44 (B), TA3sCD44/MMP-9-CD44fp (C), or TA3sCD44/MMP-9v5 (D) cells. A total of 30 μg/ml of pan specific anti-TGF-β (E) or anti-bFGF (F) antibody were added to the TA3wt/G8-conditioned coculture medium prior to use in the assay. Tubules are indicated by arrows. Bar, 140 μm.
Activation of latent TGF-β underlies MMP-9-dependent tubulogenesis
Following a series of preliminary biochemical assays aimed at determining some of the properties of the BME tubulogenesis promoting factor(s), we found that the tubulogenic activity present in the TA3wt-G8 serum free coculture conditioned medium was not only resistant to heat (80°C, 5 min), but was activated by heat (data not shown). This observation led us to test the possibility that the putative vascular tubule-inducing factor might be TGF-β, which is highly heat resistant, can be activated from its latent form by heat and acid treatment (Roberts and Sporn 1993; Taipale et al. 1998) and can induce tubule formation by vascular endothelial cells grown in collagen gels (Sankar et al. 1996).
A neutralizing pan-specific anti-TGF-β antibody, added to the TA3/G8 coculture medium, blocked its ability to induce BME tubule formation, whereas immunodepletion of the conditioned medium of two major angiogenic factors, VEGF and b-FGF with anti-VEGF and anti-bFGF antibody, respectively, and verified by Western blot analysis, had no effect, similar to control rabbit IgG (Fig. 4E,F; data not shown).
Cell surface MMP-9 activates latent TGF-β2 and TGF-β3
To provide further evidence that the vascular tubule-inducing factor in the coculture media is TGF-β, we performed luciferase reporter assays using mink lung epithelial reporter cells (TMLC) stably transfected with luciferase cDNA downstream of a TGF-β-responsive portion of the plasminogen activator inhibitor 1 promoter (PAI-1, kindly provided by Dan Rifkin, New York University). Conditioned media from cocultures of several TA3 cell transfectants and G8 cells were applied to TMLCs and the corresponding luciferase activity was measured (Fig. 5). The results of these assays confirmed that cell surface proteolytic activity of MMP-9 is required for at least part of the observed activation of TGF-β in TA3 and G8 myoblast cocultures, because expression of soluble CD44 reduced TGF-β activation, presumably by disrupting endogenous CD44 aggregation and cell surface localization of proteolytically active MMP-9 (Fig. 5A). As expected, expression of the membrane-tethered MMP-9/CD44 fusion protein restored the ability of TA3sCD44 cells to induce activation of TGF-β when cocultured with G8 myoblast monolayers, whereas overexpression of soluble MMP-9v5 had only a minor effect (Fig. 5A). When a range of protease inhibitors were added to the TA3/G8 cocultures, and TGF-β activity of the corresponding conditioned media was determined in TMLC luciferase assays, activation of TGF-β was found to be blocked by the broad spectrum inhibitors of matrix metalloproteinases, 1, 10 phenanthroline, and MMPI, but not by a specific MMP-3 inhibitor peptide, the plasmin inhibitor aprotinin, or the cysteine protease inhibitors E64, leupeptin, and pepstain A (data not shown).
Figure 5.


CD44-anchored MMP-9 activates TGF-β. (A) The ability of TA3 transfectant-G8 myoblast coculture media to stimulate TMLC luciferase activity is shown. Conditioned coculture media tested are indicated. (B) Pan-specific TGF-β antibody (30 μg/ml) or specific antibodies against TGF-β1 (100 ng/ml), TGF-β2(100ng/ml), or TGF-β3(100 ng/ml) were used to determine the activity of TGF-β isoforms in the coculture media. One unit of luciferase activity corresponds to the activity produced by 5 pg of purified human TGF-β1 (R & D).
The pan anti-TGF-β-specific antibody used in the tubulogenesis experiments has a neutralizing effect on TGF-β1, TGF-β2, and TGF-β3. TA3 cells were found to express all three TGF-β isoforms, and TGF-β receptors type I, II, and III, as assessed by RT–PCR and Western blot analysis (data not shown). To determine whether cell surface MMP-9 activity preferentially activates any single TGF-β isoform, luciferase assays were performed with serum-free conditioned medium derived from cocultures of TA3wt cells and G8 monolayers in the presence of the individual anti-TGF-β1, TGF-β2, and TGF-β3-specific neutralizing antibodies. Anti-TGF-β2 antibody was observed to block most of the luciferase activity, whereas anti-TGF-β3 antibody partially blocked the luciferase activity. Anti-TGF-β1 and anti-bFGF antibodies had no blocking effect (Fig. 5B; data not shown).
Purified MMP-9 activates latent TGF-β isoforms
To determine whether MMP-9 can directly activate TGF-β, v5/histidine-tagged MMP-2, MMP-3, MMP-9, and TGF-β1, TGF-β2, and TGF-β3 cDNAs were overproduced by transient expression in COS cells and affinity purified with either protein A Sepharose-bound anti-v5 antibody or probond nickel-chelating resins for histidine epitope tags. Expression and purity of MMPs and TGF-β isoforms were determined by zymogram, Western blot, and Coumassie blue-stained SDS-PAGE analysis (data not shown). Concentrated serum-free supernatants from COS cells transiently transfected with cDNAs encoding TGF-β1, TGF-β2, or TGF-β3 were incubated with protein A Sepharose-bound, p-aminophenylmercuric acetate (AMPA; Nagase 1997)-activated MMP-2, MMP-3, and MMP-9 and applied to the TMLC luciferase assay. MMP-9 and MMP-2 activated latent TGF-β, whereas MMP-3 had only a marginal effect (Fig. 6A). Interestingly, TGF-β2 appeared to be the most sensitive of the three isoforms to MMP-9-mediated activation. These experiments were repeated with affinity-purified v5/his-tagged latent TGF-β2 and protein-A-Sepharose-bound AMPA-activated MMPs, and the results confirmed that latent TGF-β2 can be activated by purified active MMP-9 and MMP-2 (Fig. 6B). To obtain some indication as to the fraction of the latent TGF-β pool that is activated by MMP-9, we compared the TMLC luciferase activity induced by equal aliquots of heat and purified MMP-9-treated concentrated supernatants of TGF-β1, TGF-β2, and TGF-β3-transfected COS cells. Maximal luciferase induction was obtained when the supernatants were heated to 80°C for 5 min. Purified MMP-9 activated ∼12%, 11%, and 4% of the TGF-β2, TGF-β3, and TGF-β1 heat-activatable pool, as assessed by TMLC luciferase induction (data not shown).
Figure 6.




TGF-β is activated by purified MMP-9. (A) TMLC luciferase activity induced by concentrated COS cell supernatants containing the indicated latent TGF-β isoforms following incubation with the indicated purified AMPA-activated MMPs. Comparable amounts of latent TGF-β1, TGF-β2, and TGF-β3 were present in the supernatants as assessed by both Western blot analysis and TMLC-luciferase induction following heat treatment (80°C for 5min) (B) TMLC luciferase activity induced by affinity-purified TGF-β2 following incubation with the indicated purified activated MMPs. Activity is expressed in relative light units (RLU), in which 800 RLU corresponds to the luciferase activity generated by 1 pg of purified human TGF-β1 (R & D). (C) TGF-β is proteolytically cleaved by MMP-9 and MMP-2. Purified v5-tagged TGF-β2 (lane 1) was incubated with protein-A Sepharose-bound, AMPA-activated MMP-9 (lane 2), MMP-2 (lane 3), and MMP-3 (lane 4) for 90 min at 37°C. Following incubation, the supernatants were separated from the beads, subjected to SDS/12% PAGE, transferred to Hybond-C membranes, and blotted with anti-v5 antibody. (D) MMP-2/CD44 fusion protein expression promotes TGF-β activation in TA3sCD44 cells. TMLC luciferase assays were performed with serum-free coculture media from TA3sCD44 cells transiently transfected with the indicated cDNAs.
To determine whether TGF-β2 is cleaved by MMP-9 and MMP-2, concentrated supernatants from carboxy-terminal v5/histidine-tagged TGF-β2 cDNA-transfected COS cells were incubated with Protein A Sepharose-bound, AMPA-activated, MMP-9, MMP-2, or MMP-3 for 90 min at 37°C, and TGF-β cleavage was assessed by Western blot analysis with the anti-v5 antibody (Fig. 6C). Two TGF-β2 species, of ∼50 and 25 kD were detected in the conditioned medium of transfected COS cells, corresponding to proTGF-β2, containing the prodomain, or latency-associated protein (β-LAP) and mature TGF-β2, respectively (Fig. 6C, lane 1). MMP-9 and MMP-2 generated latent TGF-β cleavage products of ∼28 and 35/37 kD, respectively, whereas MMP-3, used as a control, did not induce detectable TGF-β cleavage. These observations suggest that MMP-9 and MMP-2 cleave latent TGF-β2 at different sites, which, because of the size of the cleavage products, are most likely located within the TGF-β latency-associated protein.
The observation that purified MMP-2 can proteolytically cleave and activate latent TGF-β in vitro, and the notion that MMP-2 is localized to the cell surface by αvβ3 integrin (Brooks et al. 1996), led us to test the possibility that cell surface-tethered MMP-2 might provide a cellular mechanism for latent TGF-β activation similar to that of MMP-9. TMLC luciferase-inducing ability could be restored in TA3sCD44 cells by expression of an MMP-2–CD44 fusion protein (Fig. 6D), providing evidence that MMP-2 localized to the cell surface can substitute for MMP-9 as an activator of latent TGF-β.
Expression of the CD44–MMP-9 complex on the surface of TA3 cells contributes to latent TGF-β activation by solid TA3 tumors
To determine whether the observations made with the cell culture systems reflect the in vivo situation in TA3 tumors, we measured TGF-β activity in solid tumor extracts derived from TA3wt, TA3sCD44, TA3sCD44/MMP-9-CD44fp, and TA3sCD44/MMP-9v5 cells using the TMLC luciferase assay. Subcutaneous tumors were removed 7 and 10 days following injection of tumor cells and equal protein amounts from the corresponding extracts were tested for their ability to induce TMLC luciferase activity. Lysates from TA3wt and TA3sCD44/MMP-9-CD44fp tumors were observed to induce significantly higher luciferase activity than those for TA3sCD44 and TA3sCD44/MMP-9 tumors (Fig. 7A).
Figure 7.




TGF-β activation by CD44-anchored MMP-9 is detected in tumor extracts and keratinocyte cultures. (A) Seven and 10-day solid tumors derived from TA3wt, TA3sCD44, TA3sCD44/MMP-9-CD44fp, and TA3sCD44/MMP-9v5 cells were extracted in 50 mm Tris-HCl, (pH7.5), 75 mm NaCl, 10 mm EDTA, containing AEBSF (1 μg/ml), aprotinin (0.05 units), leupeptin (1 μg/ml), pepstatin A (2 μg/ml) and E64 (1 μg/ml), and 100 μg of the extracted proteins were added to TMLCs, and resulting luciferase activity was determined. (B) Gelatin zymogram analysis of serum-free conditioned media (a) and crude cell membrane extracts (b) of wild-type (lane 1 in a and b) and CD44-null (lane 2 in a and b) keratinocytes. (C) TMLC luciferase assay with serum-free conditioned media from wild-type and CD44-null keratinocytes. (D) TMLC luciferase assay with serum-free conditioned media from CD44 null keratinocyte cultures transiently transfected with MMP-9/CD44fp, MMP-9v5, and sCD44 cDNAs. Luciferase activity is expressed in relative light units (RLU) in which 800 RLU correspond to the activity generated by 1 pg of purified human TGF-β1 (R & D).
CD44-dependent cell surface retention of proteolytic MMP-9 occurs in normal keratinocytes, and keratinocyte-derived MMP-9 activates latent TGF-β
To determine whether CD44-dependent MMP-9 localization to the cell surface and the corresponding latent TGF-β activation observed in TA3 cells reflect a physiological functional coordination among the three molecules, we compared cell surface MMP-9 activity in primary keratinocyte cultures of normal and CD44−/− mice. Keratinocytes from both wild-type and CD44-null mice were observed to secrete comparable amounts of MMP-9 and MMP-2, but only wild-type keratinocytes displayed cell surface MMP-9 activity (Fig. 7B). Consistent with the observations in TA3 cells, loss of CD44 expression resulted in reduction of keratinocyte ability to produce activated TGF-β, as assessed by the TMLC luciferase assay (Fig. 7C). Transient expression of the MMP-9/CD44 fusion protein restored production of activated TGF-β by CD44 null keratinocytes to the level produced by wild-type keratinocytes (Fig. 7D). Such reconstitution of activated TGF-β production could not be achieved by expression of soluble MMP-9v5 (Fig. 7D). Expression of soluble CD44 in CD44 null keratinocytes had no effect on TGF-β activation.
Discussion
In this work, we have uncovered an unexpected and potentially important functional relationship between the
cell surface hyaluronan receptor CD44, the metalloproteinase MMP-9, and the cytokine TGF-β in the control of tumor invasion, growth, and angiogenesis. By docking proteolytically active MMP-9 on the surface of tumor cells, CD44 helps enhance tumor invasion and angiogenesis as well as activation of latent TGF-β, which we have shown to be a substrate of both MMP-9 and MMP-2. Our discovery that MMP-9 induces TGF-β activation on the surface of normal keratinocytes as well as that of malignant cells, suggests that MMP-mediated TGF-β activation may play an important role in the control of tissue remodeling in both physiological and pathological situations.
The importance of cell surface localization of MMP-9 in invasion and angiogenesis
Similar to the majority of known MMPs, MMP-9 is secreted, yet recent evidence suggests that MMP-9 may play a major role in promoting tumor invasion (Kim et al. 1998). Our present observations indicate that localization of MMP-9 to the cell surface may be a key event in providing its proteolytic activity with the ability to promote tumor invasion and angiogenesis. TA3 cells in which cell surface MMP-9 activity had been abrogated by expression of soluble CD44 lost the ability to invade subcutaneous tissues in vivo and myoblast monolayers in vitro, and the corresponding tumors in syngeneic mice showed markedly reduced angiogenesis. Neither angiogenesis nor invasiveness could be adequately reconstituted by overexpression of soluble, v5-tagged, MMP-9 in these cells, whereas both properties could be restored by expression of an MMP-9–CD44 fusion protein designed to force MMP-9 localization to the cell surface. Consistent with these observations, integrin αvβ3-mediated cell surface localization of proteolytically active MMP-2 has been shown to promote angiogenesis and invasion in some tumor types (Brooks et al. 1996, 1998). Retention of MMPs on the cell surface by ECM adhesion receptors may therefore provide a general mechanism for cellular regulation of MMP activity with several important consequences. First, it may help concentrate the proteolytic activity at points of contact between the cell and the ECM, providing the cell with a measure of control over the manner in which substrate degradation occurs. Second, the local concentration of MMPs, resulting from their coclustering with the docking ECM receptor, may induce MMP autoactivation, consistent with the notion that at least some MMPs can initiate or maintain their own activation (Nagase 1997). Third, cell surface docking may provide a mechanism to protect MMP activity from inhibition by TIMPs. TIMPs are the major known MMP inhibitors and most of the soluble MMP activity is believed to be quenched as a result of TIMP binding to MMPs. However, TIMPs do not appear to be associated with the CD44–MMP-9 complex in TA3 cells, despite abundant intrinsic TIMP production, supporting the notion that the cell surface may provide a privileged site for MMP-9 activity (Q. Yu and I. Stamenkovic 1999, unpubl.). Whether CD44 blocks TIMP binding or whether other membrane-associated factors protect MMP-9 from TIMP-mediated inhibition remains to be determined.
MMP-9 and MMP-2 activate TGF-β
TGF-β1, TGF-β2, and TGF-β3 are closely related isoforms of a multifunctional cytokine involved in regulating embryonal development as well as repair and regeneration following tissue injury (Massague 1990; Roberts and Sporn 1993). All three isoforms are secreted in a latent, inactive complex composed of mature TGF-β and its prodomain, TGF-β latency-associated protein (β-LAP). Mature TGF-β is cleaved from its propeptide in the secretory pathway by a furin-like processing endoproteinase (Munger et al. 1997), but the propeptide remains associated with TGF-β by noncovalent interactions that confer latency on the complex (Munger et al. 1997). LAP is often linked to latent TGF-β-binding protein (LTBP) via a disulfide bond, which provides a mechanism for the deposition of TGF-β into the ECM, in which the bulk of TGF-β is sequestered. In physiological conditions, free active TGF-β is negligible compared with the LAP-bound latent form, and cannot initiate signal transduction by its specific receptors. Thus, release from the ECM and activation of the latent form are thought to be key events in the regulation of TGF-β function. Although the precise mechanism of TGF-β activation in vivo remains unknown, several candidate molecules, including plasmin, thrombospondin 1, and αvβ6 integrin have been found to activate latent TGF-β (Lyons et al. 1990; Sato et al. 1990; Schultz-Cherry et al. 1994; Crawford et al. 1998; Munger et al. 1999), either by proteolysis of the latent complex, in the case of plasmin, or by induction of conformational changes as a result of direct physical interaction. Our present observations indicate that MMP-9 and MMP-2 may provide alternative pathways for proteolytic activation of latent TGF-β in vivo. The potential physiological relevance of these pathways is supported by the notion that activation of latent TGF-β occurs at the cell surface, in the vicinity of betaglycan, which presents the ligand to its cell surface receptors (Taipale et al. 1998). Interestingly, MMP-9 and MMP-2 produce different TGF-β proteolytic cleavage products, as assessed by Western blot analysis, suggesting different recognition/cleavage sites. Whether the observed cleavage products themselves display activity or constitute inactive intermediates that are subsequently cleaved to yield active TGF-β, remains to be determined.
The basis for the difference in sensitivity to MMP-9-mediated activation among the three latent TGF-β isoforms is unclear at present, and its elucidation will require further work. One possible explanation may be that structural variations among the isoforms influence the accessibility of the cleavage site.
How might TGF-β activation by cell surface-localized MMPs be implicated in tumor-associated tissue remodeling and angiogenesis?
MMP-mediated activation of latent TGF-β may have different consequences in tissue remodeling by normal and malignant cells. In normal tissues, TGF-β promotes chemoattraction of leukocytes, stimulates synthesis and deposition of individual ECM components, decreases proteolytic activity of cells, inhibits epithelial cell proliferation and migration, and induces cell surface integrin expression that confers an adhesive phenotype on cells. (Border and Ruoslahti 1992; Pepper 1997; Taipale et al. 1998). Activation of TGF-β by the CD44–MMP-9 complex expressed on normal keratinocytes may therefore provide a potentially important mechanism for appropriate initiation of tissue repair.
Numerous tumor types and cultured tumor cells express TGF-β and its receptors, but are resistant to its antiproliferative effects. Current opinion holds that once tumor cells have escaped its proliferation inhibitory effects, TGF-β may enhance tumor growth by stimulating angiogenesis and desmoplasia, and suppressing immune surveillance. In several instances, TGF-β signals have been observed to induce an invasive and metastatic phenotype in tumor cells (Welch et al. 1990; Oft et al. 1996, 1998). It is conceivable, therefore, that CD44–MMP-9 complex-mediated TGF-β activation reflects a physiological event that may be adopted by tumor cells to enhance their ability to invade tissues and survive in a new microenvironment. Although it is unclear what proportion of malignant tumors rely on this mechanism, CD44-mediated cell surface localization of MMP-9 has been observed in mammary carcinomas unrelated to the one used in our study (Bourguignon et al. 1998).
Although there is strong evidence to support an active role for TGF-β in several aspects of vascular development (for review, see Pepper 1997), its role in angiogenesis is disputed. Nevertheless there are several arguments to suggest that TGF-β is implicated in angiogenesis. Targeted disruption of the TGF-β receptor endoglin results in perturbation of angiogenesis (Li et al. 1999). Exogenous TGF-β promotes angiogenesis in vivo (Roberts et al. 1986; Moses et al. 1990; Pepper 1997), and local administration of neutralizing anti-TGF-β antibodies has been observed to reduce capillary density in tumors derived from CHO cells stably transfected with TGF-β1 (Ueki et al. 1992). In light of these observations, it is tempting to speculate that latent TGF-β activation may constitute a part of the mechanism whereby MMP-9 and MMP-2 activity induce or promote angiogenesis.
Materials and methods
Cells and antibodies
G8 mouse fetal myoblasts and KM201 and IM7.8 hybridomas, which produce rat-anti-mouse CD44 mAb, were obtained from the American Type Culture Collection (ATCC, Rockville, MD), and cultured in DMEM with 10% FBS (Irvine Scientific, Santa Ana, CA). All TA3 transfectants were cultured in DMEM supplemented with 10% FBS and 0.5mg/ml G418 (GIBCO-BRL). Anti-MMP-9 antibody was from Oncogene (Cambridge, MA and Santa Cruz, CA) and anti-TGF-β1, TGF-β2, and TGF-β3 as well as pan-specific anti-TGF-β antibodies were from R & D Systems (Minneapolis, MN).
To obtain primary skin keratinocyte cultures, the skin of the newborn wild-type or CD44−/− mice was digested with 0.25% trypsin (GIBCO) overnight at 4°C. Epidermal layers of the digested skins were separated from dermal layers and cut into 1–2-mm slices and further digested in a trypsin solution. Dissociated cells were washed and 5 × 105 viable cells were seeded onto collagen-coated 60-mm dishes. The cells were allowed to grow to 80% confluence in keratinocyte serum-free medium (GIBCO). The serum-free conditioned media were collected and crude cell membrane extracts were prepared as described previously (Yu and Stamenkovic 1999) to assess gelatinase activity in both the media and the cell membranes and to measure their TGF-β activity with TMLCs.
Expression constructs and RT–PCR
Total RNA was isolated from mouse placenta and TA3 cells with Trizol reagent (GIBCO-BRL) according to the manufacturer's instructions. cDNA was synthesized from 5 μg total RNA with Superscript II RNase H reverse transcriptase (GIBCO-BRL). PCR was performed as described (Yu and Stamenkovic 1999). To generate the chimeric construct composed of mouse MMP-9 and the transmembrane and cytoplasmic domains of CD44, the corresponding cDNAs were amplified by PCR with appropriate synthetic oligonucleotide primer pairs. For MMP-9, the forward primer, 5′-CACGATAAGCTTATGAGTCCCTGGCAGCCCCTGCTC-3′ and the reverse primer, 5′-CACGATAGATCTAGGGCACTGCAGGAGGTCGTAGGT-3′ were used. For CD44, the primers were as follows: forward, 5′-CACGATAGATCTCCAGAATGGCTCATCATCTTGGCA-3′; reverse, 5′CACGATCTCGAGCTACACCCCAATCTTCATGTCCAC-3′. The two PCR-generated segments were digested with appropriate restriction enzymes, ligated together in frame, and inserted into pcDNA6/V5-His vector, which contains a blasticidine resistance gene (Invitrogene, Carlsbad, CA), allowing subsequent generation of double transfectants by use of cells into which plasmids containing the neomycin resistance gene had already been stably introduced. Soluble MMP-9 was generated by RT–PCR with the forward primer 5′-CACGACGATATCATGAGTCCCTGGCAGCCCCTGCTCCTG-3′ and the reverese primer, 5′-CACGATGATGGCGGCCGCAGGGCACTGCAGGAGGTCGTAGGT-3′, and was cloned into the pcDNA6/V5-His vector, engineered to generate carboxy-terminal v5 epitope tag fusions (Invitrogen). MMP-2, MMP-3, and TGF-β1, TGF-β2, and TGF-β3 cDNAs were generated by PCR with the appropriate oligonucleotide primers and inserted into pcDNA6/V5-His vectors. The primer pairs used in the PCR reactions correspond to the 30 nucleotides at the 5′ and 3′ extremities of the appropriate coding sequence in forward and reverse orientations respectively, the stop codon being omitted to allow fusion to the tag-encoding sequences. The cDNA sequences were derived from the GenBank under the accession numbers M84324 and X66402 for MMP-2 and MMP-3, respectively, and M13177, X57413, and M32745 for TGF-β1, TGF-β2, and TGF-β3, respectively. The MMP-2–CD44 fusion construct was generated in the same way as the MMP-9–CD44 chimera. Authenticity and correct orientation of the inserts were confirmed by DNA sequencing by the dideoxy chain termination method.
Transient and stable transfection
TA3 cells were transfected with Lipofectamine (GIBCO-BRL) with expression vectors containing cDNAs encoding soluble CD44, MMP-9–CD44 and MMP-2–CD44 chimeras, soluble MMP-9, and with expression vectors alone. Stable transfectants were selected for G418 (1.5 mg/ml) and blasticidine (10 μg/ml) resistance, and resistant colonies were picked after 2–3 weeks of growth in selection medium. The culture supernatants and lysates of transfectants were tested by ELISA, zymogram, and/or Western blot analyses for expression of the approrpriate gene products. To produce v5- and His-tagged TGF-β1, TGF-β2, and TGF-β3, and MMP-2, MMP-3, and MMP-9, COS cells were transiently transfected with the respective cDNAs with DEAE/Dextran as decribed (Aruffo et al. 1990). Primary keratinocytes from wild-type and CD44 null mice were transiensiently transfected with superfect (Qiagen) according to the manufacturer's recommendations. Assays using transient transfectants were performed 72 hr following tansfection.
Tumor growth and angiogenesis
Transfected TA3 cells (2 × 106 in 0.2 ml of Hank's balanced solution, HBSS, per mouse) were injected subcutaneously into male syngeneic A/Jax mice (Jackson Laboratory, Bar harbor, ME). At least two independent isolates of each transfectant were used. The animals were observed daily. Two mice injected with each transfectant were sacrificed 1 week after injection, and the tumors were fixed and sectioned to assess their invasiveness. Three weeks after injection, six mice injected with each transfectant were sacrificed , and the tumors were isolated, weighed, and sectioned to assess angiogenesis.
Histology and immunocytochemistry
Tumors from the experimental animals were dissected and fixed in 4% paraformaldehyde (Fisher) in PBS, washed with PBS, dehydrated through 30%, 70%, 95%, and 100% ethanol and xylene, and embedded in paraffin wax (Fisher). A total of 5–10 μm sections were cut, mounted onto slides, and stained with either Gill-2 Hematoxylin (Shandon) for histologic analysis, anti-WF (Dako) antibody to assess tumor angiogenesis, mAb IM7.8 to detect CD44 expression and localization, and anti-MMP-9 (Oncogene) to detect MMP-9 expression.
Invasion assays
G8 myoblast monolayers were prepared as described (Yu et al. 1997). Transfected TA3 cells were seeded onto the fixed G8 monolayers at 5 × 103 cells/well in 6-well plates. After incubation for 7–10 days, the invasiveness of the cells was documented by microscopy. All experiments were done in triplicate and at least two independent isolates of each transfectant were used.
Endothelial tubule formation assay
Endothelial tubulogenesis was performed as described by Montesano and Pepper (1993) with BME cells (kindly provided by Roberto Montesano). Briefly, BME cells were seeded onto type I collagen gels and cultured overnight. On the following day, the conditioned medium derived from cocultures of G8 myoblast monolayers and TA3 transfectants treated or without antibodies, protease inhibitors, heat, or acid, were added to the endothelial cells (detailed in figure legends). The cells were observed daily and endothelial tubule formation was documented by light microscopy 72 hr after addition of the coculture-conditioned media. The results shown are representative of at least three independent experiments.
Luciferase assay for TGF-β activity
To measure activity of TGF-β in serum-free cell culture media and in tumor extracts, mink lung epithelial cells transfected with TGF-β-responsive human PAI-1 promoter fused to the luciferase reporter gene (TMLC, kindly provided by Dr. Daniel Rifkin, Department of Cell Biology, NYU Medical Center, New York, NY), were used. TMLC were seeded onto wells of 24-well plates (2 × 105 cells/well/ml of 10% FBS DMEM) and allowed to attach for 3 hr. The cells were then washed and the different coculture conditioned medium and tumor extracts to be tested were added to the cells as detailed in the figure legends. The luciferase activities were measured 16 hr later by the Luciferase Assay System (Promega, Madison, MI) according to the manufacturer's instructions. All experiments were done in triplicate.
Sample preparation, zymogram, and Western blot analysis
Serum-free supernatants of cultured TA3 transfectants and keratinocytes derived from both wild-type and CD44 knock-out mice were collected, and attached cells were extracted as described (Yu and Stamenkovic 1999). The crude membrane preparations were lysed in RIPA buffer, 50 mm Tris-HCl (pH7.4) containing 150 mm NaCl, 5 mm EDTA, 1% Triton, 0.1% SDS, 2 mm PMSF, 2μg/ml leupeptin, and 0.05 U/ml aprotinin. Following removal of RIPA buffer-insoluble materials, the remaining supernatant was used in gelatin zymograms. A total of 50 μl of serum-free supernatant from the transfected TA3 cells and keratinocytes and 50 μg of proteins from crude membrane lysates of TA3 cells and keratinocytes were separated by 10% SDS-PAGE containing 1 mg/ml gelatin (Fisher, Columbia, MD). Following electrophoresis, gels were washed with 2.5% Triton X-100 to remove SDS, and incubated with 50 mm Tris-HCl (pH 8.0) containing 5 mm CaCl2, and 0.02% sodium azide at 37°C for 24 hr. Gelatin activity was visualized by staining the gels with 0.5% Coomassie blue. For Western blots, gels subjected to electrophoresis were blotted onto Hybond-ECL membranes (Amersham, Arlington Heights, IL). Monoclonal antibody IM7.8 (ATCC), anti-TGF-β1, TGF-β2, and TGF-β3 antibodies (R & D Systems), and anti-v5 epitope tag mAb (Invitrogen) were used to detect CD44, TGF-β1, TGF-β2, and TGF-β3, and v5 epitope-tagged proteins respectively.
Fusion protein purification, immunoprecipitation, and in vitro TGF-β activation and cleavage
Serum-free conditioned medium from COS cells transiently transfected with mouse TGF-β1, TGF-β2, or TGF-β3 cDNA containing plasmids, or expression vector alone, were concentrated with Centricon plus-80 (5000, NMWL, Amicon) and used directly in in vitro MMP-activation assays, or purified on probond nickel resins (Invitrogen) prior to application to the assay. The production and purity of the v5-tagged TGF-β were confirmed by Western blot analysis and SDS-PAGE staining with Coomassie Blue (Fisher). V5-tagged and COS cell-produced MMP-9, MMP-2, and MMP-3 were purified with mAb against the v5 peptide (Invitrogen) and protein A Sepharose beads (Amersham Pharmacia Biotech, Uppsala, Sweden). MMPs attached to protein A-coated beads were activated by incubating the beads with 1 mm p-Aminophenylmercuric acetate (Sigma) at 37°C for 2 hr. After extensive washes with PBS, the beads containing activated MMP-9, MMP-2, and MMP-3 were washed twice with 10 mm HEPES (pH 7.2), 150 mm NaCl, and 5 mm CaCl2. A total of 10 μl of the protein A beads containing ∼2 μg of activated MMPs, as judged by the enzymatic intensity of the purified MMPs on zymograms with commercially purified MMP-9 (Chemicon, Temecula, CA) as a standard, or protein A beads alone, were incubated with concentrated serum free media derived from COS cells transfected with TGF-β1, TGF-β2, TGF-β3 or expression vector alone at 37 °C for 2 hr. Purified TGF-β2 was treated under the same conditions as above. After incubation, the supernatants were collected after centrifugation of the beads, and either used to assess TGF-β activity in TMLC luciferase assays, or subjected to SDS/12% PAGE to address TGF-β cleavage by Western blot analysis with anti-v5 antibody. The beads were washed extensively, the bound proteins were eluted with SDS sample buffer, and loaded onto a gelatin or a β-casein gel to confirm the MMP activity.
Acknowledgments
We thank Tak Mak for the CD44-null mice, Roberto Montesano for BME cells, and Dan Rifkin for the TMLCs. This work was supported by NIH grant nos. CA55375 and GM48614. Q.Y. was supported by the NIH Training grant no. CA09216.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL stamenko@helix.mgh.harvard.edu; FAX (617) 726-5684.
References
- Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61:1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- Bartolazzi A, Peach R, Aruffo A, Stamenkovic I. Interaction between CD44 and hyaluronan is directly implicated in the regulation of tumor development. J Exp Med. 1994;180:53–66. doi: 10.1084/jem.180.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Border WA, Ruoslahti E. Transforming growth factor-β in disease: The dark-side of tissue repair. J Clin Invest. 1992;90:1–7. doi: 10.1172/JCI115821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguignon LYW, Gunja-Smith Z, Iida N, Zhu HB, Young LJT, Muller WJ, Ardiff RD. CD44v3, 8–10 is involved in cytoskeleton-mediated tumor cell migration and matrix metalloproteinase (MMP-9) association in metastatic breast cancer cells. J Cell Physiol. 1998;176:206–215. doi: 10.1002/(SICI)1097-4652(199807)176:1<206::AID-JCP22>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- Brooks PC, Silletti S, von Schalscha TL, Friedlander M, Cheresh DA. Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell. 1998;92:391–400. doi: 10.1016/s0092-8674(00)80931-9. [DOI] [PubMed] [Google Scholar]
- Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- Culty M, Shizari M, Thompson EW, Underhill CB. Binding and degradation of hyaluronan by human breast cancer cell lines expressing different forms of CD44: Correlation with invasive potential. J Cell Physiol. 1994;160:275–286. doi: 10.1002/jcp.1041600209. [DOI] [PubMed] [Google Scholar]
- Frisch S, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- Gunthert U, Hofmann M, Rudy W, Reber S, Zoller M, Haussmann I, Matzku S, Wenzel A, Ponta H, Herrlich P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. 1991;65:13–24. doi: 10.1016/0092-8674(91)90403-l. [DOI] [PubMed] [Google Scholar]
- Kim J, Yu W, Kovalski K, Ossowski L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell. 1998;94:353–362. doi: 10.1016/s0092-8674(00)81478-6. [DOI] [PubMed] [Google Scholar]
- Lamb RF, Hennigan RF, Turnbull K, Katsanakis KD, MacKenzie ED, Birnie GD, Ozanne BW. Ap-1 mediated invasion requires increased expression of the hyaluronan receptor CD44. Mol Cell Biol. 1997;17:963–976. doi: 10.1128/mcb.17.2.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, Boak BB, Wendel DP. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534–1537. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- Lyons RM, Gentry LE, Purchio AF, Moses HL. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J Cell Biol. 1990;110:1361–1367. doi: 10.1083/jcb.110.4.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. The transforming growth factor-β family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- Montesano R, Pepper MS, Orci L. Paracrine induction of angiogenesis in vitro by Swiss 3T3 fibroblasts. J Cell Sci. 1993;105:1013–1024. doi: 10.1242/jcs.105.4.1013. [DOI] [PubMed] [Google Scholar]
- Moses LH, Yang EY, Pietenpol JA. TGF-beta stimulation and inhibition of cell proliferation: New mechanistic insights. Cell. 1990;63:245–247. doi: 10.1016/0092-8674(90)90155-8. [DOI] [PubMed] [Google Scholar]
- Munger JS, Harpel JG, Gleizes P-E, Mazzieri R, Nunes I, Rifkin DB. Latent transforming growth factor-β: Structural features and mechanisms of activation. Kidney Internat. 1997;51:1376–1382. doi: 10.1038/ki.1997.188. [DOI] [PubMed] [Google Scholar]
- Munger JS, Huang XZ, Kawakatsu H, Griffiths MJD, Dalton SL, Wu J, Pittet J-F, Kaminski N, Garat C, Matthay MA, et al. The integrin αvβ6 binds and activates latent TGFβ1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- Nagase H. Activation mechanisms of matrix metalloproteinases. Biol Chem. 1997;378:151–160. [PubMed] [Google Scholar]
- Oft M, Peli J, Rudaz C, Schwartz H, Beug H, Reichmann E. TGF-β1 and Ha-Ras collaborate in modulating the phenotype plasticity and invasiveness of epithelial tumor cells. Genes & Dev. 1996;10:2462–2477. doi: 10.1101/gad.10.19.2462. [DOI] [PubMed] [Google Scholar]
- Oft M, Heider KH, Beug H. TGF-beta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- Pepper MM. Transforming growth factor-beta: Vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine & Growth Factor Rev. 1997;8:21–43. doi: 10.1016/s1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- Roberts A, Sporn MB. Physiological actions and clinical applications of transforming growth factor-β (TGF-β) Growth Factors. 1993;8:1–9. doi: 10.3109/08977199309029129. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, Fauci AS. Transforming growth factor type β: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankar S, Mahooti-Brooks N, Bensen L, McCarthy TL, Centrella M, Madri JA. Modulation of transforming growth factor b receptor levels of microvascular endothelial cells during in vitro angiogenesis. J Clin Invest. 1996;97:1436–1446. doi: 10.1172/JCI118565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Tsuboi R, Moses H, Rifkin DB. Characterization of the activation of latent TGF-β by co-cultures of endothelial cells and pericytes or smooth muscle cells: A self-regulating system. J Cell Biol. 1990;111:757–763. doi: 10.1083/jcb.111.2.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Cherry S, Lawler J, Murphy-Ullrich JE. The type 1 repeats of thrombospondin 1 activate latent transforming growth factor-beta. J Biol Chem. 1994;269:26783–26788. [PubMed] [Google Scholar]
- Seiter S, Arch R, Reber S, Komitowski D, Hofmann M, Ponta H, Herrlich P, Matzku S, Zoller M. Prevention of tumor metastasis formation by anti-variant CD44. J Exp Med. 1993;177:443–455. doi: 10.1084/jem.177.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. The stromal proteinase MMP3/stromelysin-1 promotes mammarycarcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler-Stevenson WG, Aznavoorian S, Liotta LA. Tumor cell interactions with the extracellular matrix duing invasion and metastasis. Annu Rev Cell Biol. 1993;9:541–573. doi: 10.1146/annurev.cb.09.110193.002545. [DOI] [PubMed] [Google Scholar]
- Sy MS, Guo Y-J, Stamenkovic I. Distinct effects of two CD44 isoforms on tumor growth in vivo. J Exp Med. 1991;174:859–866. doi: 10.1084/jem.174.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Saharinen J, Keski-Oja J. Extracellular matrix-associated transforming growth factor-β: Role in cancer cell growth and invasion. Adv Cancer Res. 1998;75:87–134. doi: 10.1016/s0065-230x(08)60740-x. [DOI] [PubMed] [Google Scholar]
- Ueki N, Nakazato M, Ohkawa T, Ikeda T, Amuro Y, Hada T, Higashino K. Excessive production of transforming growth factor-β1 can play an important role in the development of tumorigenesis by its action for angiogenesis: Validity of neutralizing antibodies to block tumor growth. Biochim Biophys Acta. 1992;1137:189–196. doi: 10.1016/0167-4889(92)90201-l. [DOI] [PubMed] [Google Scholar]
- Varner JA, Cheresh DA. Integrins and cancer. Curr Op Cell Biol. 1996;8:724–730. doi: 10.1016/s0955-0674(96)80115-3. [DOI] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/Gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch DR, Fabra A, Nakajima M. Transforming growth factor β stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc Natl Acad Sci. 1990;87:7678–7682. doi: 10.1073/pnas.87.19.7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werb Z. ECM and cell surface proteolysis: Regulationg cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 (MMP-9) to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes & Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Toole BP, Stamenkovic I. Induction of apoptosis of metastatic mammary carcinoma cells in vivo by disruption of tumor cell surface CD44 function. J Exp Med. 1997;186:1985–1196. doi: 10.1084/jem.186.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]