

Abstract
Methadone is a long-acting opioid used in the treatment of various pain states and substitution therapy in heroin addiction. Extensive behavioral characterization has been carried out utilizing the racemate, but limited investigation has been performed with the individual isomers. While the l-isomer is a potent opioid agonist, the d-isomer has weak μ opioid activity and has also been shown to possess N-methyl-d-aspartate (NMDA) antagonist properties in vitro. The acute antinociceptive effects of the isomers were evaluated in rats using a warm water tail withdrawal procedure at two stimulus intensities (50° and 55° C). Increasing dose ratios of d- to l-methadone were administered chronically to determine the ability of the d-isomer to modulate antinociceptive tolerance to the l-isomer. Acutely, both l- (0.1-5.6 mg/kg, sc) and d- (3.0-56.0 mg/kg, sc) methadone produced antinociception though the efficacy of the d-isomer was limited at 55° C. These effects were dose-dependently blocked by naltrexone (0.01-1.0 mg/kg, sc). Administered chronically, d-methadone (1.7-10 mg/kg, sc) dose-dependently blocked tolerance development to the l-isomer (1.7 mg/kg, sc). These findings support the antinociceptive effects of the isomers being opioid receptor mediated with the l-isomer functioning as a full efficacy agonist whereas the d-isomer appears to have lower efficacy. The ability of nonracemic doses of the d-isomer to prevent tolerance development to the l-isomer may be attributed to partial μ agonist activity however NMDA antagonist activity cannot be discounted.
Keywords: Methadone, Isomers, Opioid, NMDA, Rat, Antinociception, Tolerance, Warm water tail withdrawal, rat
Introduction
Methadone (6-Dimethylamino-4,4-diphenyl-3-heptanone) is a synthetic opioid of the diphenylmethane class originally characterized as an analgesic (Scott and Chen, 1946). Dole and Nyswander first proposed methadone's primary clinical use as a substitution therapy in heroin addiction in 1965. This was the only approved use of methadone in the U.S. until 1976 when it was also approved for use as an analgesic (Schmidt, 1976). Since this time methadone has continued to be used in opioid dependence therapy and for the treatment of chronic pain conditions such as those associated with neuralgia and cancer (for reviews see Layson-Wolf et al., 2002 and Nicholson, 2007). Reasons for its continued clinical success include high oral availability and a long half-life, both of which make fewer daily doses possible (Gourlay et al., 1986).
In clinical use, methadone is available as the racemic mixture of the d- and l- isomers. The analgesic effects of methadone were ascribed to activity at opioid receptors as evidenced by high affinity for the mu (μ) subtype (Ki = 5.6 nM in human and 19 nM in rat μ receptor expressing COS-7 cells, Raynor et al., 1995). Furthermore, these effects were attributed primarily to the l- isomer which has 25-fold greater affinity for the μ receptor than the d-isomer (Terenius, 1974). In addition, both isomers have affinity for delta (δ) opioid (IC50 = 9.532 and 0.371 μM for d- and l-, respectively), alpha3beta4 nicotinic acetylcholine (IC50 = 2.5 and 2.0μM), and N-methyl-d-aspartate (NMDA) glutamate (Ki = 7.4 and 3.4 μM) receptors (Gorman et al., 1997; Kristensen et al., 1995; Xiao et al., 2001). Many studies have demonstrated the ability of antagonists of the NMDA receptor to alter the effects of opioids including: potentiation of opioid analgesic activity; attenuation, prevention, and reversal of opioid tolerance; and attenuation of opioid physical dependence (Trujillo and Akil, 1991; Popik et al., 2000; Kozela et al., 2001; Zhu and Barr, 2001; Kozela et al., 2003). In vitro data show both isomers of methadone bind with low affinity within the NMDA channel and functionally inhibit channel currents (Ebert et al., 1995; Gorman et al., 1997; Callahan et al., 2004). Consistent with the actions of other NMDA antagonists, in vivo studies have shown that the d-isomer is able to block morphine antinociceptive tolerance in the tail-flick test and NMDA-induced hyperalgesia as assessed by thermal paw withdrawal latency (Davis and Inturrisi, 1999). Thus, it is possible that the d-isomer of methadone may modify the opioid receptor mediated effects of the l-isomer when clinically used as the racemate. Indeed, it has been proposed that this characteristic of the methadone isomers is responsible for production of analgesia by methadone in patients no longer responsive to other opioids (Morley, 1998) and the slower development of tolerance to methadone relative to morphine (Inturrisi et al., 1990).
Other studies, however, suggest that the effects produced by d-methadone are due to its weak opioid activity. When applied to dorsal horn neurons d-methadone was able to inhibit NMDA-induced activity but this was prevented by pretreatment with the opioid antagonist naloxone (Chizh et al., 2000). C-fibre activity, which is predominantly μ-opioid and not NMDA receptor mediated, was inhibited by d-methadone, an effect which was reversed by naloxone (Carpenter et al., 2000). In behavioral experiments, d-methadone is able to produce antinociception that is naloxone reversible in the Randall-Selitto model of inflammation (Chizh et al., 2000) and in acetic acid-induced stretching (Smits and Myers, 1974).
Given the conflicting reports regarding the primary mechanism of action of d-methadone we have conducted a series of experiments to further investigate the behavioral effects produced by the individual isomers. In the first set of experiments the acute analgesic effects of the individual isomers were determined using the warm water tail withdrawal (WWTW) measure of antinociception in rats. The ability of naltrexone to antagonize antinociceptive effects produced by the isomers was evaluated in the same subjects. Subsequently, the development of tolerance to the antinociceptive effects of the l-isomer following chronic administration of different dose ratios of the d- and l- isomers was evaluated using the WWTW procedure in separate groups of rats.
Methods
All studies were performed at Virginia Commonwealth University (VCU). VCU is accredited by the American Association for the Accreditation of Laboratory Animal Care. All laboratory practices and animal care were consistent with current NIH guidelines and all experimental protocols were approved by the VCU Institutional Animal Care and Use Committee (IACUC).
Subjects
A total of 58 male Sprague-Dawley rats weighing 250g upon arrival were obtained from Charles River (Raleigh, N.C.). Animals were housed individually in the University vivarium and maintained on a 12:12 h light:dark cycle and allowed free access to food and water.
Apparatus
Experiments were conducted in flat-bottomed restraint tubes (model FB-Large) obtained from Braintree Scientific (Braintree, MA). The clear plastic semi-cylindrical tubes measured 3.375″ × 8.5″ with a guillotine-style closure in the caudal end which had a central opening at the bottom allowing protrusion of the tail.
Procedure
The experimental design was based on procedures used by Cook et al. (2000). Briefly, animals were habituated to restraint by placing them in the restraint tubes with their tails freely extending out of the guillotine closure. During habituation and all testing an opaque cloth cover was extended above the restraint tubes. This served not only to enhance habituation due to the restricted light exposure, but also to minimize the impact of any ambient drafts on the tail withdrawal. Time spent in the restraint tubes was gradually increased from 20 minutes to 90 minutes over the five days prior to testing. A mark was placed on each rat's tail 5 cm from the distal end. On test days, rats were brought to the laboratory and left undisturbed for 10 minutes prior to being placed into the restraint tubes for an additional10 minutes. Subsequently, rats underwent a qualifying test to identify any subjects that would exhibit tail withdrawal under non-noxious conditions and thus were ineligible for testing. In the qualifying tests, the distal 5 cm of each rat's tail was immersed in water at 40°C contained in an insulated thermos and the latency to tail withdrawal was manually recorded. To qualify for continued testing during the same session, latency to tail withdrawal had to equal the designated cut-off time (15 seconds) for at least 2 of 3 trials separated by 2 minutes. The cut-off time of 15 seconds was used to prevent tissue damage.
Acute drug administration
Immediately following qualifying tests, all rats meeting criteria were given an injection of vehicle (saline). Fifteen minutes post injection, tail-withdrawal latencies were recorded at water temperatures of 50°C and 55°C. Testing with the two temperatures was separated by 2 minutes and the order of presentation randomized. During the 2-minute separation time, in both qualifying and testing trials, the rats' tails were dried with Delicate Task Kimwipes (Kimberly-Clark, Roswell, GA). After determination of the saline control withdrawal times the rats were administered cumulative doses of l-methadone (0.1-5.6 mg/kg, n = 8) or d-methadone (3.0-56.0 mg/kg, n = 8) every 15 min and tested for withdrawal latencies using the same procedure. Incremental dosing and testing ended when the average latency for tail withdrawal reached 80% maximum possible effect (MPE) for tail withdrawal from the 55°C stimulus. All latencies were measured manually. The testing procedure was repeated at most once per week to avoid the development of any tolerance to or physical dependence on the isomers of methadone. In subsequent testing, various doses of naltrexone (0.01-1.0 mg/kg) were given in lieu of saline to evaluate the impact of opioid (μ) receptor blockade on the antinociceptive effects of the isomers. On the final test day, the highest dose of naltrexone necessary to completely block antinociception was administered followed by 4 injections of saline to control for any effect of naltrexone administration.
Chronic Administration
Similar procedures were used in individual groups of rats (n = 5-7 per group) to evaluate the development of tolerance to the antinociceptive effects of l-methadone following chronic treatment with various ratios (0:1, 1:1, 3:1, 6:1, 10:1, or 6:0) of d- to l-methadone doses. These ratios corresponded to administration of the following doses of d-methadone with 1.7 mg/kg l-methadone: 0 mg/kg (saline); 1.7 mg/kg; 5.1 mg/kg, 10 mg/kg; and 17 mg/kg. In addition, a control group was administered 5.1 mg/kg d-methadone with 0 mg/kg l-methadone (saline) repeatedly providing our 6:0 dose ratio group. On the first day (day 0) an acute l-methadone dose-effect curve was generated using the WWTW procedure as described above. Rats were then assigned to one of 6 chronic treatment groups and administered twice daily injections on days 1-7. Each group received an injection of saline or d-methadone followed 30 minutes later by an injection of 1.7 mg/kg l-methadone or saline. This combination of injections was repeated every 12 hours. On day 8 an l-methadone cumulative dose-effect curve was regenerated in the morning. Twelve hours later, the rats received a final set of d- and l-methadone injections. On the morning of day 9, a morphine cumulative dose-effect curve was generated. The morphine data were compared to results from a separate group of rats that had completed an acute morphine dose-effect curve only.
Drugs
d- and l-Methadone HCl were obtained through the National Institute on Drug Abuse Drug Supply Program (Bethesda, MD). Morphine sulfate and naltrexone HCl were obtained from Mallinckrodt Pharmaceuticals (St. Louis, MO). All drugs were dissolved in physiological saline (0.9% NaCl) to achieve the desired concentration for the individual doses. d- and l-methadone and morphine were administered s.c. and naltrexone was administered i.p. in a volume of 1 ml/kg.
Data Analysis
Percent maximum possible effect (% MPE) was calculated for each subject at each test dose using the following formula: [(Test latency – Baseline latency) / (Cut-off time – Baseline latency)] × 100. Each data point represents the mean %MPE for all subjects tested in each condition. For both the acute and chronic experiments, full antinociception was defined as ≥ 80% MPE. ED50 values for antinociception were calculated using regression analysis of the linear portions of the dose-effect curves after a log10 transformation of the dose (Tallarida and Murray, 1987). The ED50 potency ratios and 95% confidence limits (C.L.) were calculated for each treatment condition comparison. If in each of these comparisons the upper and lower potency ratio 95% confidence limits did not encompass a value of 1, the ED50 values between the dose effect curves compared were considered significantly different (Bliss, 1967).
Results
Figure 1 shows the results for testing antinociception following administration of acute l-methadone and d-methadone alone and in combination with varying doses of naltrexone at two stimulus intensities. At both water temperatures, l-methadone produced full antinociception (100 and 96% MPE). Naltrexone pretreatment dose-dependently blocked the antinociception produced by l-methadone at both stimulus intensities. For the 50°C stimulus, d-methadone dose-dependently produced antinociception with the highest dose (56 mg/kg) producing full antinociception (82% MPE). Naltrexone pretreatment dose-dependently attenuated the antinociceptive effects produced by d-methadone. At the 55°C stimulus, d-methadone produced only partial antinociception (maximum of 57% MPE) when dosed up to 56 mg/kg. When tested in combination with increasing doses of naltrexone (0.01, 0.1, and 0.3 mg/kg) the antinociceptive effects of d-methadone were dose-dependently blocked at both the 50° and 55°C stimuli. When administered alone, naltrexone did not produce any antinociceptive or hypernociceptive effects in either group (data not shown). For each isomer, the ED50 values for production of antinociception and the ED50 potency ratios between stimulus intensities and between isomers are presented in Table 1. When comparing the ED50 values of the individual isomers at the two different water temperatures, l-methadone was approximately 3-fold and d-methadone was approximately 6-fold more potent at producing antinociceptive effects at the 50°C stimulus than the 55°C stimulus. Comparing ED50 values between the isomers, l-methadone was approximately 30- and 70-fold more potent than d-methadone at the 50°C and 55°C water temperatures, respectively. ED50 values could not be calculated for d-methadone following pretreatment with the 0.1 and 0.3 mg/kg doses of naltrexone nor for l-methadone pretreated with 1 mg/kg naltrexone because these pretreatments blocked antinociception resulting in % MPE values well below 50%.
Fig. 1.
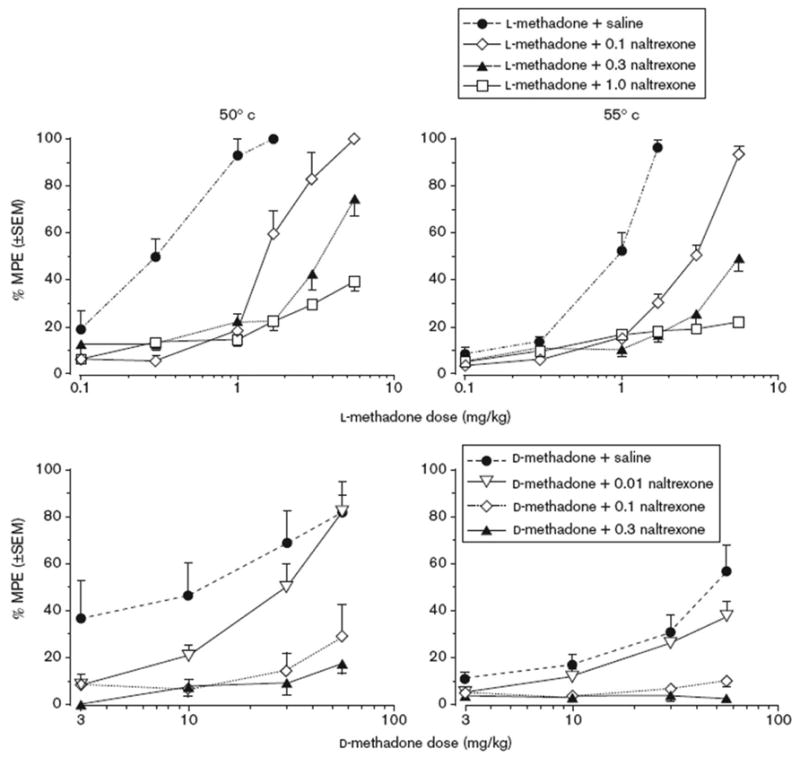
Acute antinociceptive effects of l-methadone (l-MTD) and d-methadone (d-MTD) alone and in combination with various doses of naltrexone (NTX) at 50°C (left panel) and 55°C (right panel). Each data point is based on the mean value from 8 rats (±SEM) in the warm water tail withdrawal procedure.
Table 1. ED50 values and their potency ratios for individual methadone isomers in rats tested in the warm water tail withdrawal procedure at 50°C and 55°C water temperatures.
| Dosing condition | ED50 mg/kg (95% CL) at 50°C | ED50 mg/kg (95% CL) at 55°C | ED50 potency ratio (55/50°C) |
|---|---|---|---|
| l-methadone | 0.27 (0.2–0.4) | 0.73 (0.6–0.9) | 2.70 |
| d-methadone | 8.64 (3.4–21.7) | 51.32 (32.4–81.2) | 5.94 |
| ED50 potency ratio (d/l-isomer) | 32.00 | 70.30 | – |
CL, confidence limits.
The antinociceptive effects of l-methadone before and after chronic administration of increasing ratios of d:l-methadone dosage combinations are shown in Figure 2. Before chronic dosing, l-methadone dose-dependently produced full antinociception (>80% MPE) at both stimulus intensities in all subjects. Following 7 days of twice daily dosing with 1.7 mg/kg l-methadone pretreated with saline, l-methadone produced full antinociception at both the 50°C and 55°C water temperatures (100 and 85% MPE, respectively), however, there was a rightward shift in the dose effect curves. As shown in Tables 2 and 3, the ED50 values for antinociception at 50°C and 55°C (0.22 and 0.79 mg/kg, respectively) following chronic dosing were significantly (p < 0.05) greater than the initial ED50 values (2.7-fold increase at 50°C and 3.4-fold increase at 55°C). For each of the different dose ratio combinations of the isomers administered chronically (1:1, 3:1, 6:1 and 10:1 d- to l-methadone), l-methadone was able to produce full antinociception in the WWTW procedure before and after chronic dosing as shown in Figure 2. As the ratio of d:l-methadone was increased, the rightward shifts in the dose effect curves determined prior to and after chronic dosing became less pronounced. When comparing ED50 values (Tables 2 and 3), it can be seen that the potency ratios diminished when increasing doses of d-methadone were administered chronically in combination with 1.7 mg/kg l-methadone indicating a dose-dependent blockade of tolerance development to l-methadone. When evaluated at the 50°C stimulus intensity, the 3:1 ratio of isomers (5 mg/kg d-methadone:1.7 mg/kg l-methadone) prevented any significant potency change for l-methadone (potency ratio of 1.04). At the 55°C stimulus intensity, the development of tolerance was no longer seen following chronic administration of the 6:1 ratio (10 mg/kg d-methadone:1.7 mg/kg l-methadone) isomer combination (potency ratio1.35). The effects of chronic d-methadone administration only (10 mg/kg d-methadone combined with saline) on tolerance development to l-methadone are shown in Figure 3. Chronic exposure to d-methadone did not alter the antinociceptive effects of acute l-methadone at either stimulus intensity as evidenced by no significant difference between pre- and post-chronic ED50 values (Tables 2 and 3).
Fig. 2.
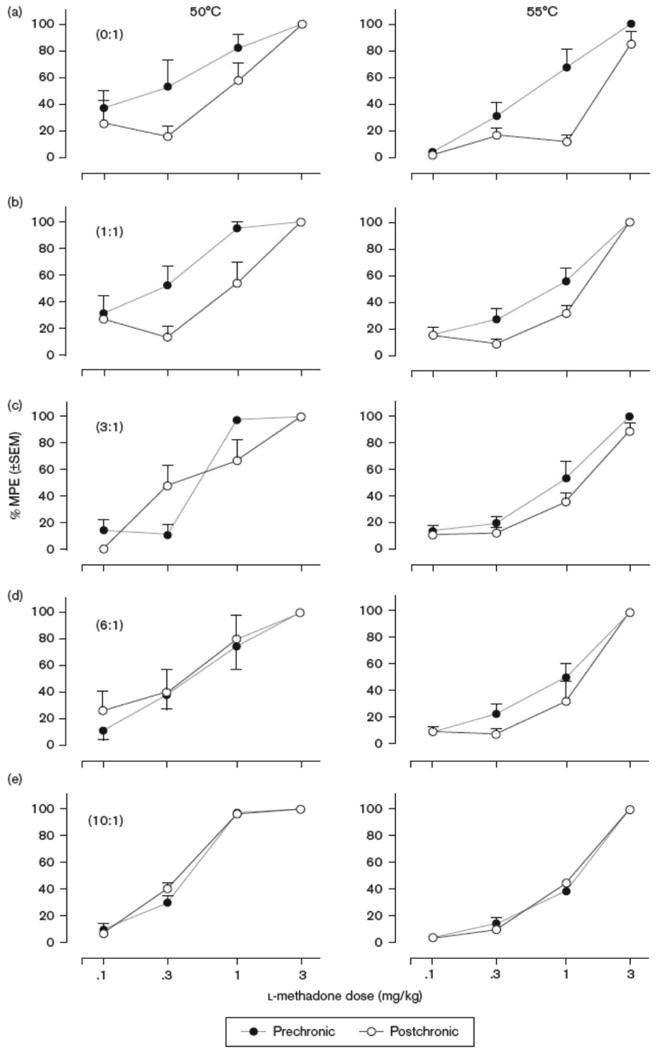
Antinociceptive effects of l-methadone as assessed by warm water tail withdrawal before and after chronic treatment with increasing dose ratios of d:l-methadone at both 50°C (left column) and 55°C (right column) water temperatures. A) 0:1 d- to l-isomer, n= 5, B) 1:1, n=6, C) 3:1, n=7, D) 6:1, n=5, E) 10:1, n=6.
Table 2. Relative antinociceptive potency of l-methadone and ED50 potency ratios before and after chronic administration of various dose ratios of d:l-methadone in the WWTW procedure at 50°C.
| Dosing condition | Prechronic ED50 mg/kg (95% CL) | Post chronic ED50 mg/kg (95% CL) | ED50 potency ratio (95% CL) |
|---|---|---|---|
| 0:1 | 0.22 (0.14–0.34) | 0.79a (0.65–0.96) | 2.70 (1.22–6.80) |
| 1:1 | 0.22 (0.13–0.37) | 0.83a (0.64–1.09) | 3.50 (1.94–6.46) |
| 3:1 | 0.52 (0.46–0.58) | 0.37 (0.05–3.01) | 1.04 (0.53–1.88) |
| 6:1 | 0.44 (0.28–0.68) | 0.33 (0.18–0.62) | 0.80 (0.32–1.89) |
| 10:1 | 0.35 (0.29–0.42) | 0.33 (0.29–0.36) | 0.94 (0.80–1.10) |
| 6:0 | 0.32 (0.26–0.39) | 0.31 (0.26–0.38) | 0.98 (0.77–1.24) |
CL, confidence limits.
Postchronic ED50 significantly different from prechronic at P<0.05.
Table 3. Relative antinociceptive potency of l-methadone and ED50 potency ratios before and after chronic administration of various dose ratios of d:l-methadone in the WWTW procedure at 55°C.
| Dosing condition | Prechronic ED50 mg/kg (95% CL) | Postchronic ED50 mg/kg (95% CL) | ED50 potency ratio (95% CL) |
|---|---|---|---|
| 0:1 | 0.54 (0.39–0.75) | 1.78a (1.48–2.15) | 3.41 (2.04–5.76) |
| 1:1 | 0.69 (0.58–0.83) | 1.06a (0.88–1.28) | 1.49 (1.15–2.02) |
| 3:1 | 0.78 (0.66–0.92) | 1.09a (0.88–1.34) | 1.40 (1.03–1.95) |
| 6:1 | 0.76 (0.64–1.91) | 1.04 (0.75–1.44) | 1.35 (0.89–2.13) |
| 10:1 | 0.94 (0.80–1.12) | 0.94 (0.85-1.03) | 0.99 (0.88–1.12) |
| 6:0 | 0.63 (0.55–0.71) | 0.68 (0.57–0.81) | 1.08 (0.94–1.23) |
CL, confidence limits.
Postchronic ED50 significantly different from prechronic at P<0.05.
Fig. 3.
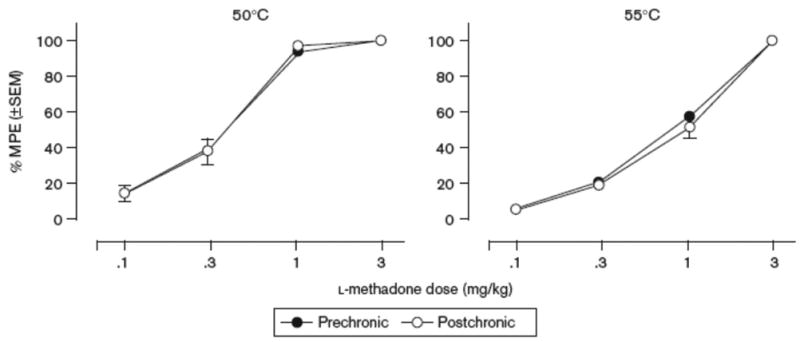
Antinociceptive effects of l-methadone as assessed by warm water tail withdrawal before and after chronic dosing with 10 mg/kg d-methadone at both 50°C (left column) and 55°C (right column) water temperatures, n=6
On day 9, a morphine dose-effect curve was generated for each group dosed chronically with different dose ratios of d- to l-methadone to assess the development of opioid cross-tolerance (Figure 4). Data from these curves were compared to a control group of animals that were tested acutely with morphine. In the latter group, the ED50 values for morphine antinociception at 50°C and 55°C were 1.25 and 5.13 mg/kg, respectively. Repeated administration of 1.7 mg/kg l-methadone with saline resulted in a rightward shift in the morphine dose effect curve at both stimulus intensities. Addition of d-methadone during chronic dosing showed a dose-dependent attenuation of the rightward shift (Figure 4). The ED50 potency of morphine was significantly different between the morphine-only group and the 0:1 and 1:1 d- to l-methadone treatment groups (p < 0.05) at both temperatures suggesting the development of cross-tolerance (Table 4). In the groups receiving higher chronic doses of d- and l-methadone (3:1, 6:1, and 10:1) dose combinations and d-methadone alone, ED50 potencies for the antinociceptive effects of morphine were not significantly different from the control group that had received morphine only at either temperature, indicating a lack of cross-tolerance (Table 4).
Fig. 4.
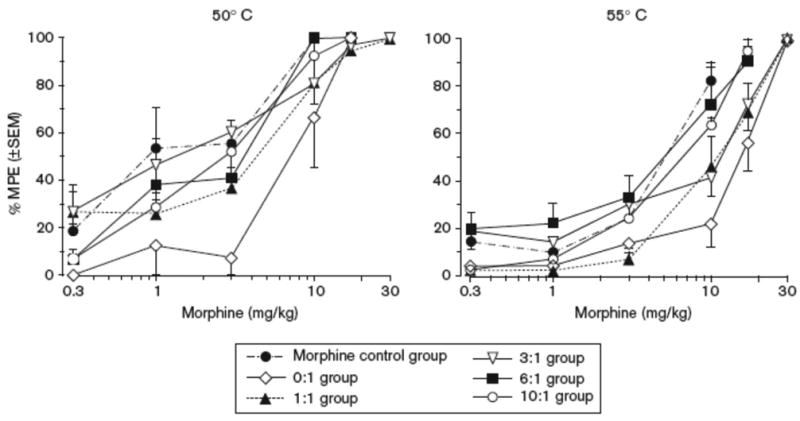
Antinociceptive effects of morphine as assessed by warm water tail withdrawal in a control group (n=8) that received no chronic dosing as well as separate groups of rats before and after chronic dosing with increasing dose ratios of d:l- methadone at 50°C (left column) and 55°C (right column) water temperatures (0:1 d- to l-isomer n= 5; 1:1 d- to l-isomer n=6; 3:1 d- to l-isomer n=7; 6:1 d- to l-isomer n=5; and 10:1 d- to l-isomer n=6).
Table 4. Relative antinociceptive potency of morphine and ED50 potency ratios for a morphine control group (no chronic treatment) compared to groups following chronic administration of various dose ratios of d:l-methadone as assessed in the WWTW procedure.
| Dosing condition | Morphine ED50 mg/kg (95% CL) at 50° | ED50 potency ratio (95% CL) | Morphine ED50 mg/kg (95% CL) at 55° | ED50 potency ratio (95% CL) |
|---|---|---|---|---|
| Morphine control | 1.25 (0.66–2.37) | – | 5.13 (4.05–6.51) | – |
| 0:1 | 6.87a (5.13–9.19) | 4.44 (1.90–10.28) | 15.18a (13.02–17.69) | 2.84 (2.14–3.75) |
| 1:1 | 3.27a (1.97–5.41) | 2.45 (1.04–5.63) | 10.05a (7.98–12.67) | 1.94 (1.31–2.84) |
| 3:1 | 1.31 (0.67–2.56) | 1.18 (0.44–3.01) | 9.39 (3.65–24.19) | 2.05 (0.64–3.88) |
| 6:1 | 2.00 (1.02–3.93) | 1.59 (0.64–4.50) | 3.98 (2.55–6.20) | 0.83 (0.45–1.52) |
| 10:1 | 2.12 (1.80–2.49) | 1.68 (0.99–3.02) | 5.21 (4.43–6.12) | 1.04 (0.79–1.39) |
| 6:0 | 1.09 (0.92–1.29) | 0.87 (0.48–1.71) | 3.98 (3.43–4.61) | 0.81 (0.61–1.08) |
CL, confidence limits.
Significantly different from morphine control at P< 0.05.
Discussion
The behavioral effects produced by l- and d-methadone in the present study suggest that the primary site of action for the acute effects of both isomers is at the μ-opioid receptors. In the WWTW procedure, as expected, l-methadone produced dose-dependent antinociception that was antagonized by naltrexone at both stimulus intensities (Figure 1). Similarly, d-methadone produced full antinociception at the 50°C water temperature reaching 82% MPE at the 56 mg/kg dose (Figure 1). The d-isomer also produced a dose-dependent antinociception at the 55°C stimulus, however, it only produced a maximum of 57% MPE at the highest dose tested. Greater doses of d-methadone to determine its ability to produce full antinociception at the higher stimulus intensity were not tested due to toxic side effects. The antinociceptive effects of the d-isomer were blocked by pretreatment with naltrexone lending further evidence that d-methadone prevents noxious stimulus transmission primarily via agonist actions at the μ-opioid receptor. Our results are comparable to those seen when testing morphine, meperidine, etorphine, or buprenorphine with naloxone or naltrexone in similar models of acute or physiological pain (Leander, 1979; Walker et al., 1994). The inability of the d-isomer to produce full antinociception at the higher stimulus intensity suggests it functions as a partial μ agonist. When tested at varying water temperatures, partial agonists such as butorphanol, buprenorphine, and pentazocine show full antinociceptive efficacy at low but not high stimulus intensities (Morgan et al., 1999a,b; Terner et al., 2003).
Potency differences between the isomers are highlighted by the l-methadone ED50 value being 32-fold and 70-fold more potent than that for d-methadone at 50°C and 55°C, respectively (Table 1). The ED50 potency difference for the isomers is consistent with their relative in vitro measurements of μ opioid receptor binding. The d-isomer was 25 fold less potent than the l-isomer in displacement of [3H]-dihydromorphine (IC50 = 1×10-7 vs. 4×10-9 M, respectively; Terenius, 1974). Similarly, Horng and colleagues (1976) investigated 3H-dihydromorphine and 3H-naloxone displacement by the isomers of methadone and showed d-methadone to have approximately 30–fold lower affinity for μ opioid receptors than l-methadone. Our results are consistent and remarkably similar to these in vitro findings (Table 1). The greater than predicted potency difference based on binding affinity at the higher stimulus intensity could reflect the d-isomer acting as a lower efficacy agonist relative to the l-isomer. This is consistent with whole cell current- and voltage-clamp recordings which showed a greater intrinsic activity for l-methadone relative to d-methadone (Matsui and Williams, 2010). However, further testing with combinations of the two isomers will be required to demonstrate definitively that, consistent with these in vitro assays, d-methadone functions as a partial efficacy μ agonist in vivo. An additional consideration is the two d-methadone metabolites, α-l-methadol and α-l-dinormethadol, which have also been shown to have opioid receptor affinity similar to or greater than the parent compound (Horng et al., 1976). Previous studies have found that metabolites of d-methadone produce antinociception (Sullivan et. al., 1972; Terenius, 1974; Sullivan et. al., 1975) which was attributed primarily to formation of the α-l-normethadol metabolite. α-l-Methadol has been found in rat lung tissue one hour after administration of d-methadone (Sullivan et. al., 1975). Therefore it is very possible that the metabolites also contribute to d-methadone's antinociceptive effects in the current study.
The majority of previous studies evaluating d-methadone have found evidence of μ-opioid receptor mediated antinociceptive effects. In contrast, Shimoyama et al. (1997) reported that intrathecal d-methadone produced no antinociception in a radiant heat tail flick test and was ineffective in phase one but effective, and naloxone irreversible, in phase 2 of the formalin test. The latter results suggest that the primary action of d-methadone is through a non-opioid mechanism and are consistent with actions as an NMDA antagonist. Additionally, Leander & McCleary (1982) found that decreases in keypecking behavior elicited by d-methadone in pigeons was unaffected by concurrent naloxone treatment. The present results are more similar to those of Smits and Myers (1974) and Chizh et. al. (2000) who found that d-methadone produced naloxone-reversible antinociception in acetic acid-induced stretching (ED50 = 6.1 mg/kg) and the Randall-Selitto model of inflammatory pain (ED50 = 6.6 mg/kg), respectively. Furthermore, Chizh et al. (2000) also found that naloxone reversed d-methadone inhibition of noxious stimulation of single motor units. Similarly, the ability of d-methadone to inhibit C-fibre evoked activity can be prevented by naloxone (Carpenter et. al., 2000). An explanation for discrepancies between the behavioral results in the different experiments could be the doses of drugs used, the pain model employed, animal species used, and/or the route of administration. Shimoyama et al. (1997) reported testing only one dose of d-methadone and naloxone was administered intrathecally. It is possible that the dose of naloxone (30μg) given was insufficient to displace d-methadone (250μg) from a sufficient number of opioid receptors to reverse any opioid mediated effects. Alternatively, the contribution of NMDA antagonism versus μ-opioid receptor activation may vary dependent upon the pain model employed and the route of administration (i.e., intrathecal versus systemic). The relevance of the pain model employed is supported by testing of racemic methadone in a neuropathic pain model. In the control subjects, methadone-induced antinociception was shown to be μ opioid mediated whereas in subjects experiencing neuropathic pain, a component of methadone's antinociceptive effects were not naloxone reversible and were attributed to NMDA antagonist action (Sotgiu et al., 2009). Similarly, under the current testing system evaluating antinociception in a spinally-mediated model of acute pain, both the d- and l-isomers appear to produce antinociception via opioid receptor-mediated mechanisms.
The ability of d-methadone to block development of opioid antinociceptive tolerance to a fixed dose of l-methadone (1.7 mg/kg) given for 7 days was demonstrated. Combinations of 5.1, 10.0, and 17.0 mg/kg d-methadone with 1.7 mg/kg l-methadone blocked tolerance at 50°C while only 10.0 and 17.0 mg/kg d-methadone doses combined with 1.7 mg/kg l-methadone blocked tolerance development when evaluated at 55°C (Figure 2, Tables 2 and 3). After chronic administration, it was also demonstrated that d-methadone (5.1, 10.0, and 17.0 mg/kg, Table 4) combination treatment with l-methadone was also able to block cross-tolerance to morphine-induced antinociception. Additionally, another group was repeatedly administered only the d-isomer (10 mg/kg) for 7 days to determine its effects on acute administration of l-methadone and morphine. This moderate dose of d-methadone was chosen because it was able to block tolerance when combined with the l-isomer. This treatment did not result in any change in the antinociceptive potency of l-methadone or morphine indicating that d-methadone itself does not induce tolerance to opioids nor result in a hyperalgesic state at this dose. It also demonstrated that chronic administration of the d-isomer does not cause an altered response to acute administration of the potent opioid agonists that might enhance their effects and thus appear as a blockade of tolerance (Figure 2, Tables 2, 3, and 4). It is possible that higher doses of d-methadone could produce tolerance due to a greater stimulation of opioid receptors as has been seen with chronic administration of partial agonists (Walker and Young, 2001; Grecksch et al., 2006).
Similar blockade of opioid tolerance development has been found using an acute pain model with d-methadone given in combination with morphine for 5 days (Davis and Inturrisi, 1999). Based on the current chronic administration study and previous studies, it is possible that blockade of opioid tolerance by coadministration of d-methadone was due to action of the isomer as an antagonist of NMDA receptors. However, it is also possible that the current results were due to the d-isomer acting as a lower affinity, lower efficacy agonist at μ-opioid receptors. In rats administered morphine chronically, pretreatment with low doses of an opioid antagonist (naloxone and naltrexone) or with a low efficacy opioid agonist (nalbuphine) was able to block the development of antinociceptive tolerance (Lee et. al., 1997; Powell et. al., 2002; Jang et. al., 2006). This suggests that concurrent opioid receptor blockade or occupancy by a low efficacy agonist during repeated exposure to a high efficacy agonist can prevent the development of tolerance.
The relative binding potency of the isomers to μ-opioid receptors versus NMDA channel sites in vitro provides further support for opioid-mediated antinociception and tolerance attenuating behavioral effects for the d-isomer (Ebert et. al., 1995; Gorman et. al., 1997; Callahan et. al., 2004). d-Methadone is approximately 5-fold more potent at activating μ-opioid than inhibiting NMDA receptors (EC50 = 0.6 μM vs. IC50 = ∼3.5 μM, respectively; Matsui and Williams, 2010). This comparison seems particularly relevant since the doses attenuating tolerance development were similar to or lower than those showing naloxone-antagonized antinociceptive effects. Clinically, plasma levels achieved in methadone-maintained patients approximates the levels associated with opioid receptor activation (Kreek, 1973) whereas doses associated with plasma levels in the NMDA receptor IC50 range have been associated with overt toxicity (Perret et al., 2000). Thus, with the much greater affinity for μ-opioid than NMDA receptors, the role of NMDA antagonism in the antinociceptive and tolerance blocking actions of d-methadone could be questioned. It must be noted that the level of NMDA receptor blockade (i.e., percent of receptors blocked) necessary to block tolerance development is unknown therefore it cannot be ruled out that a very low level of NMDA channel blockade did contribute to the tolerance attenuation. In addition, binding studies show the density of NMDA receptors is much greater than that of μ-opioid receptors in brain tissue thus providing more targets for the d-isomer to exert its glutamatergic effects (Goodman and Pasternak, 1985; Monaghan and Cotman, 1985). Interestingly, l-methadone has slightly higher affinity for binding within NMDA channels than d-methadone (Gorman et. al., 1997). However, l-methadone is approximately 600-fold more potent (IC50 = 5.0 nM vs. 3.4 μM, respectively) in binding to μ-opioid receptors relative to NMDA channel binding. Therefore, the very potent effects of the l-isomer at μ-opioid receptors prevent in vivo administration of doses which would produce CNS concentrations sufficient to block NMDA receptor channels.
In conclusion, both d- and l-methadone isomers produce naloxone-reversible antinociception after acute administration, although the d-isomer was less efficacious at higher levels of noxious stimuli, and the l-isomer was significantly more potent under all conditions. The d-isomer was also shown to block antinociceptive tolerance development following chronic l-methadone administration as well as cross-tolerance to morphine without having any effects on tolerance when administered alone. The acute antinociceptive effects appear to be the result of d-methadone acting as a partial μ-opioid agonist. While the current results following chronic administration cannot conclusively be attributed solely to μ-opioid receptor activity, the role of NMDA antagonism to account for these findings can be questioned. Further studies to investigate the potential utility of varying combinations of d- and l-methadone could be informative for the clinical use of methadone as an analgesic with less susceptibility for the development of tolerance. Methadone is currently available in the U.S. is a 1:1 ratio of the d- and l-isomers. This ratio of the isomers would not be expected to modify the actions of the l-isomer based on the current results. However, it is possible that the use of higher ratios of d:l methadone clinically could delay or prevent the development of analgesic tolerance.
Acknowledgments
The authors would like to thank Dr. Robert L. Balster for his intellectual advice and support. This work was made possible by NIDA grants DA01442 and DA007027.
References
- Bliss CI. Statistics in biology. New York: McGraw Hill; 1967. [Google Scholar]
- Callahan RJ, Au JD, Paul M, Liu C, Yost CS. Functional inhibition by methadone of N-methyl-d-aspartate receptors expressed in Xenopus oocytes: stereospecific and subunit effects. Anesth Analg. 2004;98:653–659. doi: 10.1213/01.ane.0000099723.75548.df. [DOI] [PubMed] [Google Scholar]
- Carpenter KJ, Chapman V, Dickenson AH. Neuronal inhibitory effects of methadone are predominantly opioid receptor mediated in the rat spinal cord in vivo. Eur J Pain. 2000;4:19–26. doi: 10.1053/eujp.1999.0147. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Schlutz H, Scheede M, Englberger W. The N-methyl-d-aspartate antagonistic and opioid components of d-methadone antinociception in the rat spinal cord. Neurosci Lett. 2000;296:117–120. doi: 10.1016/s0304-3940(00)01638-4. [DOI] [PubMed] [Google Scholar]
- Cook CD, Barrett AC, Roach EL, Bowman JR, Picker MJ. Sex-related differences in the antinociceptive effects of opioids: importance of rat genotype, nociceptive stimulus intensity, and efficacy at the mu opioid receptor. Psychopharmacology (Berl) 2000;150:430–442. doi: 10.1007/s002130000453. [DOI] [PubMed] [Google Scholar]
- Davis AM, Inturrisi CE. d-Methadone blocks morphine tolerance and N-methyl-d-aspartate -induced hyperalgesia. J Pharmacol Exp Ther. 1999;289:1048–1053. [PubMed] [Google Scholar]
- Dole VP, Nyswander M. A medical treatment for diacetylmorphine (heroin) addiction. A clinical trial with methadone hydrochloride. JAMA. 1965;193:646–650. doi: 10.1001/jama.1965.03090080008002. [DOI] [PubMed] [Google Scholar]
- Ebert B, Andersen S, Krogsgaard-Larsen P. Ketobemidone, methadone and pethidine are non-competitive N-methyl-d-aspartate (NMDA) antagonists in the rat cortex and spinal cord. Neurosci Lett. 1995;187:165–168. doi: 10.1016/0304-3940(95)11364-3. [DOI] [PubMed] [Google Scholar]
- Goodman RR, Pasternak GW. Visualization of mu1 opiate receptors in rat brain by using a computerized autoradiographic subtraction technique. Proc Natl Acad Sci USA. 1985;82:6667–6671. doi: 10.1073/pnas.82.19.6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman AL, Elliott KJ, Inturrisi CE. The d- and l- isomers of methadone bind to the non-competitive site on the N-methyl-d-aspartate (NMDA) receptor in rat forebrain and spinal cord. Neurosci Lett. 1997;223:5–8. doi: 10.1016/s0304-3940(97)13391-2. [DOI] [PubMed] [Google Scholar]
- Gourlay GK, Cherry DA, Cousins MJ. A comparative study of the efficacy and pharmacokinetics of oral methadone and morphine in the treatment of severe pain in patients with cancer. Pain. 1986;25:297–312. doi: 10.1016/0304-3959(86)90234-4. [DOI] [PubMed] [Google Scholar]
- Grecksch G, Bartzsch K, Widera A, Becker A, Holt V, Koch T. Development of tolerance and sensitization to different opioid agonists in rats. Psychopharmacology (Berl) 2006;186:177–184. doi: 10.1007/s00213-006-0365-8. [DOI] [PubMed] [Google Scholar]
- Horng JS, Smits SE, Wong DT. The binding of the optical isomers of methadone, α-methadol, α -acetylmethadol and their N-demethylated derivatives to the opiate receptors of rat brain. Res Commun Chem Pathol Pharmacol. 1976;14:621–629. [PubMed] [Google Scholar]
- Inturrisi CE, Portenoy RK, Max MB, Colburn WA, Foley KM. Pharmacokinetic-pharmacodynamic relationships of methadone infusions in patients with cancer pain. Clin Pharmacol Ther. 1990;47:565–577. doi: 10.1038/clpt.1990.77. [DOI] [PubMed] [Google Scholar]
- Jang S, Kim H, Kim D, Jeong MW, Ma T, Kim S, et al. Attenuation of morphine tolerance and withdrawal syndrome by coadministration of nalbuphine. Arch Pharm Res. 2006;29:677–684. doi: 10.1007/BF02968252. [DOI] [PubMed] [Google Scholar]
- Kozela E, Danysz W, Popik P. Uncompetitive NMDA receptor antagonists potentiate morphine antinociception recorded from the tail but not from the hind paw in rats. Eur J Pharmacol. 2001;423:17–26. doi: 10.1016/s0014-2999(01)01084-6. [DOI] [PubMed] [Google Scholar]
- Kozela E, Pilc A, Popik P. Inhibitory effects of MPEP, an mGluR5 antagonist, and memantine, an N-methyl-D-aspartate receptor antagonist, on morphine antinociceptive tolerance in mice. Psychopharmacology (Berl) 2003;165:245–251. doi: 10.1007/s00213-002-1287-8. [DOI] [PubMed] [Google Scholar]
- Kreek MJ. Plasma and urine levels of methadone. Comparison following four medication forms used in chronic maintenance treatment. N Y State J Med. 1973;73:2773–2777. [PubMed] [Google Scholar]
- Kristensen K, Christensen CB, Christup LL. The mu1, mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. 1995;56:PL45–50. doi: 10.1016/0024-3205(94)00426-s. [DOI] [PubMed] [Google Scholar]
- Layson-Wolf C, Goode JC, Small RE. Clinical use of methadone. J Pain Palliat Care Pharmacother. 2002;16:29–59. [PubMed] [Google Scholar]
- Leander JD. Comparison of morphine, meperidine, anileridine, and alphaprodine on schedule-controlled responding and analgesia. Pharmacol Biochem Behav. 1980;214:16–23. doi: 10.1016/0091-3057(80)90168-9. [DOI] [PubMed] [Google Scholar]
- Leander JD, McCleary PE. Opioid and nonopioid behavioral effects of methadone isomers. J Pharmacol Exp Ther. 1982;220:592–596. [PubMed] [Google Scholar]
- Lee SC, Wang JJ, Ho ST, Tao PL. Nalbuphine coadministered with morphine prevents tolerance and dependence. Anesth Analg. 1997;84:810–815. doi: 10.1097/00000539-199704000-00021. [DOI] [PubMed] [Google Scholar]
- Matsui A, Williams JT. Activation of μ-opioid receptors and block of KIR 3 potassium channels and NMDA receptor conductance by l- and d-methadone in rat locus coeruleus. Br J Pharmacol. 2010;161:1403–13. doi: 10.1111/j.1476-5381.2010.00967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan DT, Cotman CW. Distribution of N-methyl-d-aspartate -sensitive l-[3H]glutamate-binding sites in rat brain. J Neurosci. 1985;5:2909–2919. doi: 10.1523/JNEUROSCI.05-11-02909.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Cook CD, Smith MA, Picker MJ. An examination of the interactions between the antinociceptive effects of morphine and various μ -opioids: the role of intrinsic efficacy and stimulus intensity. Anesth Analg. 1999a;88:407–413. doi: 10.1097/00000539-199902000-00035. [DOI] [PubMed] [Google Scholar]
- Morgan D, Cook CD, Picker MJ. Sensititivity to the discriminative stimulus and antinociceptive effects of μ opioids: role of strain of rat, stimulus intensity, and intrinsic efficacy at the μ opioid receptor. J Pharmacol Exp Ther. 1999b;289:965–975. [PubMed] [Google Scholar]
- Morley JS. Opioid rotation: does it have a role? Palliat Med. 1998;12:464–466. doi: 10.1177/026921639801200611. [DOI] [PubMed] [Google Scholar]
- Nicholson A. Methadone for cancer pain. Cochrane Database Syst Rev. 2007;4:CD003971. doi: 10.1002/14651858.CD003971.pub3. [DOI] [PubMed] [Google Scholar]
- Perret G, Deglon JJ, Kreek MJ, Ho A, La Harpe R. Lethal methadone intoxications in Geneva, Switzerland, from 1994 to 1998. Addiction. 2000;95:1647–1653. doi: 10.1046/j.1360-0443.2000.951116475.x. [DOI] [PubMed] [Google Scholar]
- Popik P, Kozela E, Danysz W. Clinically available NMDA receptor antagonists memantine and dextromethorphan reverse existing tolerance to the antinociceptive effects of morphine in mice. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:425–432. doi: 10.1007/s002109900205. [DOI] [PubMed] [Google Scholar]
- Powell KJ, Abul-Husn NS, Jhamandas A, Olmstead MC, Beninger RJ, Jhamandas K. Paradoxical effects of the opioid antagonist naltrexone on morphine analgesia, tolerance, and reward in rats. J Pharmacol Exp Ther. 2002;300:588–596. doi: 10.1124/jpet.300.2.588. [DOI] [PubMed] [Google Scholar]
- Raynor K, Kong H, Mestek A, Bye LS, Tian M, Liu J, et al. Characterization of the cloned human mu opioid receptor. J Pharmacol Exp Ther. 1995;272:423–428. [PubMed] [Google Scholar]
- Schmidt AM. Restrictions on distribution of methadone. Federal Register. 1976;41:28261–28264. [Google Scholar]
- Scott CC, Chen KK. The action of l,l-diphenyl-l-(dimethylaminoisopropyl)-butanone-2, a potent analgesic agent. J Pharmacol Exp Ther. 1946;87:63–71. [PubMed] [Google Scholar]
- Shimoyama N, Shimoyama M, Elliott K, Inturrisi CE. d-Methadone is antinociceptive in the rat formalin test. J Pharmacol Exp Ther. 1997;283:648–652. [PubMed] [Google Scholar]
- Smits SE, Myers MB. Some comparative effects of racemic methadone and its optical isomers in rodents. Res Commun Chem Pathol Pharmacol. 1974;7:651–662. [PubMed] [Google Scholar]
- Sotgiu ML, Valente M, Storchi R, Caramenti G, Biella GE. Cooperative N-methyl-D-aspartate (NMDA) receptor antagonism and mu-opioid receptor agonism mediate the methadone inhibition of the spinal neuron pain-related hyperactivity in a rat model of neuropathic pain. Pharmacol Res. 2009;60:284–290. doi: 10.1016/j.phrs.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Sullivan HR, Smits SE, Due SL. Metabolism of d-methadone: isolation and identification of analgesically active metabolites. Life Sci. 1972;11:1093–1104. [Google Scholar]
- Sullivan HR, Due SL, McMahon RE. The difference in activity between (+)- and (-)-methadone is intrinsic and not due to a difference in metabolism. J Pharm Pharmacol. 1975;27:728–732. doi: 10.1111/j.2042-7158.1975.tb09391.x. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ, Murray RB. Manual of pharmacological calculation with computer programs. New York: Springer-Verlag; 1987. [Google Scholar]
- Terenius L. A rapid assay of affinity for the narcotic receptor in rat brain: Application to methadone analogues. Acta Pharmacol Toxicol. 1974;34:88–91. doi: 10.1111/j.1600-0773.1974.tb02016.x. [DOI] [PubMed] [Google Scholar]
- Terner JM, Barrett AC, Cook CD, Picker MJ. Sex differences in (-)-pentazocine antinociception: comparison to morphine and spiradoline in four rat strains using a thermal nociceptive assay. Behav Pharmacol. 2003;14:77–85. doi: 10.1097/00008877-200302000-00008. [DOI] [PubMed] [Google Scholar]
- Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- Tung AS, Yaksh TL. The antinociceptive effects of epidural opiates in the cat: studies of the pharmacology and the effects of lipophilicity in spinal analgesia. Pain. 1982;12:343–356. doi: 10.1016/0304-3959(82)90179-8. [DOI] [PubMed] [Google Scholar]
- Walker EA, Young AM. Differential tolerance to antinociceptive effects of mu opioids during repeated treatment with etonitazene, morphine, or buprenorphine in rats. Psychopharmacology (Berl) 2001;154:131–142. doi: 10.1007/s002130000620. [DOI] [PubMed] [Google Scholar]
- Walker EA, Makhay MM, House JD, Young AM. In vivo apparent pA2 analysis for naltrexone antagonism of discriminative stimulus and analgesic effects of opiate agonists in rats. J Pharmacol Exp Ther. 1994;271:959–968. [PubMed] [Google Scholar]
- Xiao Y, Smith RD, Caruso FS, Kellar KJ. Blockade of rat alpha3beta4 nicotinic receptor function by methadone, its metabolites, and structural analogs. J Pharmacol Exp Ther. 2001;299:366–371. [PubMed] [Google Scholar]
- Zhu H, Barr GA. Inhibition of morphine withdrawal by the NMDA receptor antagonist MK-801 in rat is age-dependent. Synapse. 2001;40:282–293. doi: 10.1002/syn.1051. [DOI] [PubMed] [Google Scholar]