

Abstract
Despite their monophyletic origin, mitochondrial (mt) genomes of plants and animals have developed contrasted evolutionary paths over time. Animal mt genomes are generally small, compact, and exhibit high mutation rates, whereas plant mt genomes exhibit low mutation rates, little compactness, larger sizes, and highly rearranged structures. We present the (nearly) whole sequences of five new mt genomes in the Beta genus: four from Beta vulgaris and one from B. macrocarpa, a sister species belonging to the same Beta section. We pooled our results with two previously sequenced genomes of B. vulgaris and studied genome diversity at the species level with an emphasis on cytoplasmic male-sterilizing (CMS) genomes. We showed that, contrary to what was previously assumed, all three CMS genomes belong to a single sterile lineage. In addition, the CMSs seem to have undergone an acceleration of the rates of substitution and rearrangement. This study suggests that male sterility emergence might have been favored by faster rates of evolution, unless CMS itself caused faster evolution.
Keywords: mitochondrial genomes, comparative genomic, rearrangements, phylogeny, Beta vulgaris
Introduction
Despite their monophyletic origin (Gray et al. 1999), mitochondrial (mt) genomes of plants and animals have developed contrasted evolutionary paths over time. Animal mt genomes are generally compact and small (around 16 kb) and exhibit a high mutation rate, whereas plant mt genomes exhibit a low mutation rate, little compactness, subsequent larger sizes (from 200 to 900 kb, for whole sequenced genomes; Alverson et al. 2010), and highly rearranged structures (Palmer and Herbon 1988; see review in Kubo and Newton 2008). This variation in size and structure is also observable at the species level, as illustrated by the recent study in maize, where five genomes were totally sequenced (Allen et al. 2007). Among the sequenced mt genomes in maize, genomes called C, S, and T are associated with cytoplasmic male sterility (CMS) genomes, two are considered fertile. Maize mt genome size varies from 535,825 to 739,719 bp, mainly due to large duplications (up to 120 kb). The genomes are highly rearranged when compared with one another, although the level of substitution among species is low, even at the genus level (Darracq et al. 2010).
CMS is an interesting case of nuclear–cytoplasmic interaction. Described in several crop species (Chase 2007), it is commonly found in wild populations. Species that bear CMS and therefore exhibit sexual polymorphism, that is, hermaphrodites and females, are called gynodioecious. Wild beet, Beta vulgaris ssp. maritima, the wild relative of sugar beet, is one of these species. Within the species, at least four CMSs (Owen, CMS-E, CMS-G, and CMS-H) have been described out of a total of 20 mt types through the use of restriction fragment length polymorphism markers (Cuguen et al. 1994). Theoretical models suggest that the maintenance of male-sterile plants in populations implies the relative fitness advantage of being a female, for example, by producing more seeds through the economy of pollen production, consequently inducing a selection of CMS in populations. In wild beet, the study of gynodioecy occurrence in populations suggests a balancing selection dynamics that favors CMSs when they are rare. When CMS becomes common, it creates a selection pressure that favors the recruitment of the corresponding nuclear restorer alleles, restoring pollen production and generating restored hermaphrodite (Dufaÿ et al. 2007, 2009). This evolutionary dynamics of gynodioecy is expected to leave a signature in mt gene nucleotide diversity (Charlesworth 2002; Touzet and Delph 2009). In addition, it raises the question of the events and the evolutionary forces that led to the emergence of sterilizing genes. In beet, a phylogeny built from chloroplastic polymorphism has suggested that all four CMSs are independent and derived from an ancestral fertile cytoplasm (Fénart et al. 2006). This was corroborated by the polymorphism at an mt intergenic sequence (Nishizawa et al. 2007). One CMS (TK81-MS which corresponds to Owen) and one non-CMS (TK81-O) mt genome have previously been sequenced (Kubo et al. 2000; Satoh et al. 2004). They exhibited a large variation in size and gene order. In the present study, we sequenced five additional mt genomes, two non-CMSs (A and B), two CMSs (CMS-E and CMS-G), and one mt genome in B. macrocarpa, a sister species belonging to the same taxonomic section. We describe the diversity of the mt genome in size, content, and structure in this eudicot species. We then show that, contrary to what was assumed previously, all three CMS genomes belong to a single sterile lineage. In addition, the CMSs seem to have undergone an acceleration of the rates of substitution and rearrangement.
Materials and Methods
mtDNA Preparation
For mitogenomes of B. v. ssp. maritima, maternal progenies were collected in populations from free pollinated plants. Maternal plants were characterized for their mt type according to Cuguen et al. (1994). Mitogenomes A and B are fertile cytoplasms, whereas CMS-E and CMS-G are sterilizing ones. As an outgroup, we used B. macrocarpa, which belongs to the same Beta section (accession from Morocco, IDBBNR 8549). mtDNAs were extracted from roots for B. v. ssp. maritima and from leaves for B. macrocarpa using procedures described in Scotti et al. (2001).
Library Construction, Sequencing, and Finishing
To generate random fragments, the mtDNAs were mechanically sheared and 5 kb generated inserts were cloned into pcDNA2.1 plasmid vector (Invitrogen). Vector DNAs were purified and end sequenced using dye terminator chemistries on ABI3730 sequencers until an average of 12-fold coverage for each genome. A pre-assembly was made without repeat sequences as described by Vallenet et al. (2008) using Phred/Phrap/Consed software package (www.phrap.com). The finishing steps were achieved through primer walking, transposition (using TGS II KIT and FINNZYMES), and polymerase chain reaction (PCR) amplifications.
Annotation
A local database was built to facilitate genome annotation. The annotation of genes, tRNAs, and rRNAs of newly sequenced genomes was referenced on the whole sequences of mt genomes from beet (TK81-O [GenBank: BA000009] and TK81-MS [GenBank: BA000024]), Arabidopsis thaliana (GenBank: Y08501), and tobacco (GenBank: BA000024), as well as whole sequences of chloroplastic (cp) genomes of tobacco (GenBank: Z00044) and spinach (GenBank: AJ400848). Edited sites on genes were determined using the annotation of TK81-O, which has been experimentally validated (Mower et al. 2006). Annotated tRNAs were validated using the software tRNAScan-SE version 1.23 (Mower and Palmer 2006).
Open Reading Frames (ORFs) with a minimum size of 300 bp were scanned for and first compared with known genes of the reference genomes of the database. If they did not match with a known gene in the database, Blast analyses were conducted on GenBank nonredundant database (April 2010). In order to find chimeric ORFs, ORFs were compared with genes, tRNAs, and rRNAs from TK81-0 using YASS software (Noe and Kucherov 2005) with an E value of 0.1. Only matches of 100% were considered. In addition, Blast analyses were conducted with a word size of 16 and an E value >0.1.
Beta vulgaris ssp. maritima annotated sequences were deposited in EMBL under accession numbers FP885845 (A), FP885834 (B), FQ014226 (CMS-E circular contig), FQ014231 (CMS-E linear contig), FP885871 (CMS-G circular contig), and FP885876 (CMS-G linear contig) and B. macrocarpa under accession number FQ378026.
Plastid Sequences Contained in mt Genomes
Each genome was analyzed through YASS against the spinach chloroplast genome (E value of 1 × 10−01; with a score of +1 for matches and −3 for substitutions). We kept sequences longer than 30 bp and with less than 10% of substitutions and insertions/deletions (indels). Note that with this criteria, cp-tRNA–like sequences were included for the calculation of the ratio of plastid sequences in the genome.
Chloroplastic DNA Isolation and Sequencing
In order to compare chloroplastic and mt genome evolution, we sequenced chloroplastic fragments. Total genomic DNA was isolated from 10–15 mg of dried leaf tissue using Nucleospin Plan Kit (Macherey-Nagel) for genomes A, B, CMS-E, CMS-G, and macrocarpa. TK81-O and TK81-MS DNAs were kindly provided by Dr Kubo, Hokkaido University, Japan. Thirty-one cpDNA regions were selected for sequencing (supplementary table S1, Supplementary Material online). PCR amplification was performed in a 25-μl mix containing 25 ng of DNA template, 3 mM of MgCl2, 2.5 μl of Buffer 10× (PerkinElmer, Norwalk, CT), 0.2 μM of each primer, 200 μM of each dNTP, and 1.25 U/μl of hot start Taq polymerase (AmpliTaq Gold, PerkinElmer). PCR mixture underwent the following conditions on a Mastercycler ep (Eppendorf): 5 min denaturing at 95 °C, 35 cycles of 40 s denaturing at 95 °C, 45 s annealing at Annealing temperature (supplementary table S1, Supplementary Material online), and from 1 to 2 min extension (depending on the fragment length) at 72 °C and a final extension step at 72 °C for 10 min, after 35 cycles. The PCR products were then purified using a Nucleospon 96 Extract kit (Macherey-Nagel) and directly sequenced with an ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit (PerkinElmer). Sequence data were obtained on a 3130xl Genetic Analyzer (Applied Biosystems).
Genome Complexity
In order to establish the genome complexity defined as the complete sequence information found in a given genome keeping only one copy of each duplicate (>500 bp), a YASS analysis was conducted on each genome in order to detect any duplication of length longer than 500 bp following an E value <1 × 10−30 (with a score of +1 for matches and −3 for substitutions) and with less than 5% of substitutions and insertions/deletions (indels). In order to establish genome complexity on two contig mt genomes, we concatenated their contig sequences.
Backbone Sequences
In order to compare mitogenomes at the sequence level, all common sequences between the genome complexities were sought out using Mauve (seed = 9, minimum island = 15, maximum backbone gap size = 15, minimum backbone gap size = 50) (Darling et al. 2004). This resulted in sets of homologous fragments common to all genomes. For each genome, the corresponding DNA fragments of each set output by Mauve were concatenated following mt genome A order for phylogenetic analyses. The resulting concatenation for each genome constitutes its backbone sequence.
Synteny Anchors
From the sets of common fragment output by Mauve, we computed the synteny anchors (SAs), that is, sequences of contiguous common fragments to all mitogenomes. After that, duplicated parts were reintroduced. Each SA was then assigned a number, the same number being used in case of duplicates. This resulted in sequences of numbers for each mitogenome. These sequences were used to study genomic rearrangements.
Rearrangement Distance
We calculated a distance between genomes based on the concept of generalized gene adjacencies introduced in Zhu et al. (2009). In this concept, neighboring SAs define a distance between two genomes. Let d be an integer, two SAs are d related only if they are separated by at most d-1 SAs. Sets of SAs were computed for each pair of genomes. The choice of d value is discussed by Yang and Sankoff (2009) and must be greater than three for our set of genomes. We chose three as long as greater values produced the same results. The resulting distance between two genomes is defined as:
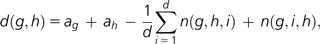 |
where g and h are the two SA sequences, ag is the number of adjacencies in g (1-related couple of genes), and n(g,h,i) is the number of couples of SAs that are i related in g and adjacent in h. Two contig mt genomes were considered as multichromosomal genomes. Because the distance measure is based on the neighborhood of genes, the overall organization of the genomes does not really affect the distance. Hence, even if some genomes are in two contigs or alternative genomic configurations, this will not forbid to use this distance as a measure of the synteny.
Phylogenetic Analyses
Neighbor-Joining (NJ) analyses were performed using BIONJ (Gascuel 1997) with SplitsTree4 software (Huson and Bryant 2006) on backbone sequences for mt analyses and on the concatenations of sequenced fragments for chloroplastic analyses. Parameters used are bootstrap 1000× and Kimura-2 parameters for correction. Maximum likelihood (ML) analyses were conducted with Tree-Puzzle (Schmidt et al. 2002) using the general time reversible (GTR) nucleotide model. To investigate the robustness of the topologies, the same analyses were conducted on the alignments with only the variable sites. We also checked the GTR + Gamma model for potential violations induced by the data using posterior predictive tests implemented in PhyloBayes 3.3 (Lartillot et al. 2009). For rearrangement phylogeny, we used the distance matrix computed with the above formula with SA sequences and BIONJ to obtain a phylogenetic tree.
Data Analyses
On concatenated 29 protein coding sequences (consensus sequence of 29,220 bp when considering the longest CMS-G-cox1), summary statistics on nucleotide diversity and divergence were calculated using DnaSP version 4.10.9 (Rozas et al. 2003): πs, the average number of pairwise synonymous differences per synonymous site; πa, the average number of pairwise nonsynonymous differences per nonsynonymous site; and Ks and Ka, respectively, the number of synonymous and nonsynonymous nucleotide substitutions from the outgroup B. macrocarpa. In the alignment, the edited sites that were not experimentally confirmed in TK81-O by Mower and Palmer (2006) were transformed into T.
We used HyPhy (Kosakovsky Pond et al. 2004) to test for variation in synonymous substitution rate, using the GY94_3×4 substitution model, and the tree topology based on backbone sequences obtained from BIONJ. The likelihoods of two models were compared with a null model with an identical synonymous substitution rate across all branches of the tree. In the first model, we allowed CMS and non-CMS clades to have different synonymous substitution rates. In the second model, we restricted to the variation of dS to the TK81-MS and CMS-G clades only.
The same analyses were conducted on two additional data sets: 2) alignment with the shorter G-cox1 (28,812 bp) and 3) alignment with the shorter G-cox1 and without the presequence of atp6 found in CMS-E and TK81-MS (27,639 bp).
Estimated size of CMS-G-cox1 was calculated from Expasy's compute pI/MW program (http://scansite.mit.edu/cgi-bin/calcpi). Correlation analyses were conducted with R using Mantel test with Spearman method.
Results
Genome Size and Composition
Using a shotgun-sequencing strategy similar to the one that successfully sequenced all mt genomes in maize (Allen et al. 2007), we were able to entirely sequence two wild fertile mt genomes A and B of B. vulgaris, an mt genome of B. macrocarpa, a sister species, and nearly complete the sequencing of CMS-E and CMS-G. A master circle was reconstructed for A, B, and macrocarpa, whereas CMS-E and CMS-G genomes were reconstructed in two contigs (one circular and one linear) (fig. 1). Total genome sizes varied from 341,257 bp for CMS-G to 385,220 bp for B. macrocarpa, which are closer to the size of the previously sequenced fertile mt genome TK81-O (368,801 bp) than to CMS TK81-MS (501,020 bp). Table 1 summarizes features of the five newly sequenced genomes and the two previously described ones. Over the seven mt genomes, there was a ratio of 1.47 between the longest and the shortest genomes. The median GC content was 43.89%, similar to that in other plant mt genomes (Allen et al. 2007). The large variation of size was partly due to duplications that represent from 4.55% for CMS-G to 29.98% for TK81-MS (median = 9.30%) (Spearman coefficient of correlation r = 0.75, P = 0.03). Genome complexity, which is the nonredundant genetic content of each genome, was less variable among genomes, from 325,716 bp for CMS-G to 357,125 bp for CMS-E (size ratio of 1.10 between the largest and the shortest) with a median value of 335,262 bp. Consequently, genome complexity was not correlated with genome size (Spearman coefficient of correlation r = 0.32, P = 0.24).
FIG. 1.—

Beta mt genome representations. Beta mitogenomes are represented in linear form. Internal boxes represent duplicate regions, and external boxes represent genes. CMS-E1 and CMS-G1 correspond to circular contigs, and CMS-E2 and CMS-G2 correspond to linear contigs.
Table 1.
Portions of Beta mt Genomes Present as Genes and ORFs (Macro Corresponds to Beta macrocarpa)
| Genomes |
|||||||
| Genome Features | TK81-O | TK81-MS | A | B | CMS-E | CMS-G | Macro |
| Genomes | |||||||
| Total genome size (bp) | 368,801 | 501,020 | 364,950 | 367,943 | 378,457 circular: 268,616 linear: 109,841 | 341,257 circular: 269,136 linear: 72,121 | 385,220 |
| % GC | 43.86 | 43.89 | 43.91 | 43.89 | 43.88 | 43.92 | 43.89 |
| Total repeated sequence (bp) | 34,313 | 150,214 | 29,688 | 37,461 | 21,332 | 15,541 | 37,595 |
| Total repeated sequence, % total genome | 9.30 | 29.98 | 8.13 | 10.18 | 5.64 | 4.55 | 9.76 |
| Genome complexity (bp) | 334,488 | 350,806 | 335,262 | 330,482 | 357,125 | 325,716 | 347,625 |
| Genes | |||||||
| Protein genes (bp) | 27,693 | 37,485 | 27,693 | 28,593 | 28,845 | 28,118 | 27,693 |
| Single-copy protein genes (bp) | 27,693 | 28,839–29,142a | 27,693 | 27,693 | 28,845 | 28,118 | 27,693 |
| Cis introns (bp) | 18,727 | 30,641 | 18,749 | 18,749 | 18,748 | 18,746 | 18,749 |
| Single-copy cis introns (bp) | 18,727 | 18,733 | 18,749 | 18,749 | 18,748 | 18,746 | 18,749 |
| rRNA genes (bp) | 12,065 | 12,065 | 12,065 | 12,065 | 5,389 | 8,727 | 12,065 |
| Single-copy rRNA genes (bp) | 5,389 | 5,389 | 5,389 | 5,389 | 5,389 | 5,389 | 5,389 |
| tRNA genes (bp) | 1,746 | 2,282 | 1,746 | 1,746 | 1,746 | 1,453 | 1,746 |
| Single-copy tRNA genes (bp) | 1,449 | 1,449 | 1,449 | 1,449 | 1,449 | 1,303 | 1,449 |
| Pseudogenes and pseudoexons (bp) | 1,180 | 1,177 | 1,180 | 1,180 | 1,180 | 524 | 1,180 |
| Single-copy pseudogenes and pseudoexons (bp) | 1,180 | 1,106 | 1,180 | 1,180 | 1,180 | 524 | 1,180 |
| Coding totals | |||||||
| Total known genes (bp) | 41,504 | 51,832 | 41,504 | 42,404 | 35,980 | 38,298 | 41,504 |
| Total single-copy genes (bp) | 34,531 | 35,677–35,980a | 34,531 | 34,531 | 35,683 | 34,810 | 34,531 |
| Total genes, % total genome | 11.25 | 10.35 | 11.37 | 11.52 | 9.51 | 11.22 | 10.77 |
| Single-copy genes, % complexity | 10.32 | 10.17–10.26a | 10.30 | 10.45 | 9.99 | 10.69 | 9.93 |
| ORFs | |||||||
| Total ORFs (bp) | 69,801 | 96,429 | 72,990 | 69,435 | 72,750 | 63,510 | 74,502 |
| Single-copy ORFs (bp) | 63,147 | 72,945 | 65,223 | 62,976 | 71,283 | 63,510 | 66,390 |
| ORFs % total genome | 18.93 | 19.25 | 20.00 | 18.87 | 19.22 | 18.61 | 19.34 |
| Single-copy ORFs, % complexity | 18.88 | 20.79 | 19.45 | 19.06 | 19.96 | 19.50 | 19.10 |
| Integrated plastid sequences | |||||||
| Total integrated sequences (bp) | 7,621 | 6,984 | 7,499 | 7,499 | 7,857 | 3,906 | 7,716 |
| Single-copy integrated sequences (bp) | 7,621 | 6,454 | 7,499 | 7,499 | 7,710 | 3,861 | 7,520 |
| % Total genome | 2.07 | 1.40 | 2.05 | 2.04 | 2.08 | 1.14 | 2.00 |
| % Complexity | 2.28 | 1.84 | 2.24 | 2.27 | 2.16 | 1.19 | 2.16 |
When considering short or long TK81-MS-cox2-exon2.
Integrated Plastid Sequences
The total length of plastid inserted sequences in mt genomes varied from 3,906 bp in CMS-G to 7,857 bp in CMS-E, representing from 1.19% to 2.28% of genome complexity (table 1). The size of inserted cp sequences varied from 30 bp in all genomes to 3,368 bp in mt genomes TK81-O, A, B, CMS-E, and macrocarpa (supplementary table S2, Supplementary Material online), with a median size per genome varying from 73.5 bp in CMS-G to 79 bp in TK81-O, A, and B.
Conserved Genes
Every one of the genomes we sequenced contained the same 29 protein coding genes (supplementary table S3, Supplementary Material online)—18 genes involved in ATP-generating electron transport: nine in Complex I (nad 1, 2, 3, 4, 4L, 5, 6, 7, and 9), one in Complex III (cob), three in Complex IV (cox 1, 2, and 3), and five in Complex V (atp 1, 4, 6, 8, and 9); three genes involved in biogenesis of cytochrome c (ccm B, FC, and FN); six genes coding for ribosomal proteins (rpl5, rps 3, 4, 7, 12, and 13); and one gene involved in independent membrane targeting and translocation system (tatC) and one maturase (mat-r).
Using TK81-O as reference, a total of 296 edited sites annotated in TK81-O protein coding genes, we found 294 conserved sites in A, B,CME-E, CMS-G, and B. macrocarpa annotated as edited. Thirty-three edited sites were predictably located in start or stop codons of protein coding genes. We found three genes where ACG was edited to become a start codon ATG (atp6, nad1, and nad4L) and two cases where editing generated a stop codon, CAA (atp6) or CGA (atp9) to TAA or TGA, respectively. The gene tatC did not contain a DNA-encoded AUG start codon and did not seem to be edited. We annotated its start codon as in TK81-MS (AUA). The stop codon was UAA for 16 genes (including edited atp genes): atp 1, 6, 8, cox 1, 2, nad 1, 2, 3, 4L, 5, 6, and 9—except for one copy of CMS-G—rpl5, rps 3, 4, and 7; UGA for 10 genes (atp9, ccm B,FC, FN, cob, cox2—for CMS-G and one copy of TK81-MS, cox3, nad4, rps 12, and 13); and UAG for four genes (mat-r, nad7, one copy of nad9 in CMS-G and TK81-MS, and tatC).
As in Kubo et al. (2000), 20 introns were found for seven protein coding genes, representing an average of 6% of the genome complexity. Six were trans-splicing introns for three genes (nad 1, 2, and 5) and 14 were cis-splicing introns for six genes (nad 1, 2, 4, 5, 7, and ccmFC). It must be noted that contrary to other plants, rps3 lacks introns in beet, even though a second copy of what is homologous to exon1 is found in the beet mt genomes (counted as pseudoexon in the present study). As previously described (Matsunaga et al. 2010), rps3 pseudoexon1 is contained in orf246 in TK81-O and it is not found in TK81-MS. We found this rps3 pseudoexon1 in the five sequenced genomes and at a distance of 190 bp from genuine rps3. In all these genomes (CMS and non-CMS), rps3 pseudoexon1 is contained in orf273 (homologous to TK81-O orf246).
The Beta genomes contained three RNA genes (rrn 5S, 8S, and 26S, supplementary table S3, Supplementary Material online). Although three copies of rrn26S were found in TK81-O, TK81-MS, A, B, and macrocarpa, only two copies were found in CMS-G and in only one copy in CMS-E. Copy deletion is most probably due to the remaining gaps of the two unfinished genomes.
Eighteen tRNAs were found as well as five potential pseudo-tRNAs (supplementary table S3, Supplementary Material online). Among the 18 tRNAs, 11 were native, six were cp-like, and one was of unknown origin: trnC2 (Kubo et al. 2000) (“native” = tRNA genes derived from the ancestral alpha-proteobacteria at the origin of mitochondria and “cp-like” = tRNA genes of plastidial origin inserted into the mt genome during evolution). The cp-like trnW was most likely present in CMS-G as well (in a nonintegrated contig of 5 kb, data not shown). Among the five pseudo-tRNAs as defined by Kubo et al. (2000), cp-like trnP and trnV were not found on unfinished mt genome CMS-G, whereas trnI was not found on CMS-E.
Polymorphic Protein Coding Genes
We found 19 protein coding genes that were polymorphic among the seven analyzed genomes (table 2). Overall, 60 mutations and one variable 5′ part of a gene (atp6) were found. Twenty-three mutations were specific to CMS-G, 14 to TK81-MS, six to TK81-O, three to CMS-E, and one to macrocarpa. Four variants were shared by TK81-MS and CMS-G, whereas the alternative allele was shared by TK81-O, A, B, CMS-E, and macrocarpa. Six variants were shared by TK81-MS and CMS-E, whereas the alternative allele was shared by TK81-O, A, B, CMS-E, and macrocarpa. One variant was shared by TK81-MS, CMS-G, and CMS-E, whereas the alternative allele was shared by TK81-O, A, B, and macrocarpa. On one site, one allele was unique to TK81-MS, one other allele was shared by A and B, and a third one by the remaining cytoplasms. On an other site, one allele was unique to TK81-MS, one other allele was unique to CMS-E, and a third one was shared by the remaining cytoplasms. Overall, there were 16 synonymous mutations and 44 nonsynonymous mutations. Among the 44 nonsynonymous mutations, when we focused on CMS-G totalizing 23 nonsynonymous mutations, one mutation generated a premature stop codon in exon2 of cox2, whereas one mutation modified the stop codon of nad9, resulting in a longer coding sequence of 42 bp, as previously described by our team (Ducos et al. 2001). The start codon was mutated in CMS-G-cox1, resulting in a shorter coding sequence of 87 bp or a longer one of 408 bp (with an alternative start codon found upstream, supplementary file S4, Supplementary Material online). Last, CMS-G-cox3 was also potentially modified with one nonsynonymous mutation. Three polymorphisms, two specific to TK81-O and one specific to TK81-MS (on nad2 exon4, rps3, and rps12), are expected to be edited resulting in a nonmodified protein sequence.
Table 2.
Substitutions and Indels in Beta mt Genes (Macro Corresponds to Beta macrocarpa)
| Genes | Position in TK81-O Genome | Genomes |
||||||
| TK81-O | TK81-MS | A | B | CMS-E | CMS-G | Macro | ||
| atp1 | 1386 | gaT → D | gaT → D | gaT → D | gaT → D | gaT → D | gaG → E | gaT → D |
| atp6 | First 1,146 bp specific to TK81-MS and CMS-E | First 1,152 bp specific to TK81-MS and CMS-E | ||||||
| 1–3 | Acg → M | Gcg → A | Acg → M | Acg → M | Gcg → A | Acg → M | Acg → M | |
| 4–6 | ATT → I | GGA → G | ATT → I | ATT → I | GGA → G | ATT → I | ATT → I | |
| 7–9 | Acg → T | CGg → R | Acg → T | ACg → T | CGg → R | ACg → T | ACg → T | |
| 10–12 | CCc → P | ATc → I | CCc → P | CCc → P | ATc → I | CCc → P | CCc → P | |
| 13–15 | AAc → N | CCc → P | AAc → N | AAc → N | CCc → P | AAc → N | AAc → N | |
| 19–22 | ccA → P | ccC → P | ccA → P | ccA → P | ccG → P | ccA → P | ccA → P | |
| 23–26 | ctT → L | ctA → L | ctT → L | ctT → L | ctA → L | ctT → L | ctT → L | |
| 264 | gtT → V | gtT → V | gtT → V | gtT → V | gtG → V | gtT → V | gtT → V | |
| 489 | ccT → P | ccC → P | ccT → P | ccT → P | ccT → P | ccT → P | ccT → P | |
| ccmFC exon1 | 306 | ttA → L | ttA → L | ttA → L | ttA → L | ttA → L | ttC → F | ttA → L |
| ccmFC exon2 | 288 | cCa → P | cCa → P | cCa → P | cCa → P | cCa → P | cAa → Q | cCa → P |
| cox1 | 1–3 | AtG → M | AtG → M | AtG → M | AtG → M | AtG → M | TtT → Fa | AtG → M |
| 14 | gTt → V | gTt → V | gTt → V | gTt → V | gTt → V | gAt → D | gTt → V | |
| 131 | cGa → R | cGa → R | cGa → R | cGa → R | cGa → R | cAa → Q | cGa → R | |
| 153 | ggT → G | ggC → G | ggT → G | ggT → G | ggT → G | ggT → G | ggT → G | |
| 966 | atC → I | atA → I | atC → I | atC → I | atC → I | atC → I | atC → I | |
| 1179 | gcA → A | gcG → A | gcA → A | gcA → A | gcA → A | gcA → A | gcA → A | |
| 1206 | atC → I | atA → I | atC → I | atC → I | atC → I | atC → I | atC → I | |
| 1207 | Ttt → F | Ttt → F | Ttt → F | Ttt → F | Ttt → F | Gtt → V | Ttt → F | |
| cox2 Exon2 | 376 | tTa → L | tTa → Lb | tTa → L | tTa → L | tTa → L | tGa → c | tTa → L |
| cox3 | 151 | Att → I | Att → I | Att → I | Att → I | Att → I | Ctt → L | Att → I |
| mat-r | 750 | aaT → N | aaT → N | aaT → N | aaT → N | aaG → K | aaT → N | aaT → N |
| 1086 | gtC → V | gtA → V | gtC → V | gtC → V | gtC → V | gtC → V | gtC → V | |
| 1215 | aaT → N | aaG → K | aaT → N | aaT → N | aaT → N | aaT → N | aaT → N | |
| nad1 Exon1 | 7 | Ata → T | Ata → T | Ata → T | Ata → T | Ata → T | Gta → V | Ata → T |
| nad1 Exon3 | 160–161 | CGt → R | GCt → A | GCt → A | GCt → A | GCt → A | GCt → A | GCt → A |
| nad2 Exon4 | 14 | ccC → P | ccT → P | ccT → P | ccT → P | ccT → P | ccT → P | ccT → P |
| 74 | atA → I | atA → I | atA → I | atA → I | atA → I | atC → I | atA → I | |
| nad4L | 17 | tAt → Y | tAt → Y | tAt → Y | tAt → Y | tAt → Y | tTt → F | tAt → Y |
| 19 | Ttt → F | Ttt → F | Ttt → F | Ttt → F | Ttt → F | Gtt → V | Ttt → F | |
| nad5 Exon1 | 13 | Atc → I | Atc → I | Atc → I | Atc → I | Ctc → L | Atc → I | Atc → I |
| nad5 Exon4 | 3 | Aat → N | Cat → H | Aat → N | Aat → N | Aat → N | Cat → H | Aat → N |
| nad7 Exon1 | 15 | atC → I | atC → I | atC → I | atC → I | atC → I | atG → M | atC → I |
| nad9 | 59 | aAa → K | aAa → K | aAa → K | aAa → K | aAa → K | aCa → T | aAa → K |
| 66 | atA → I | atC → I | atA → I | atA → I | atA → I | atA → I | atA → I | |
| 74 | tCa → S | tCa → S | tCa → S | tCa → S | tCa → S | tTa → L | tCa → S | |
| 118 | Caa → Q | Aaa → K | Caa → Q | Caa → Q | Caa → Q | Caa → Q | Caa → Q | |
| 261 | cgG → R | cgC → R | cgC → R | cgC → R | cgC → R | cgC → R | cgC → R | |
| 262 | Cta → L | Gta → V | Gta → V | Gta → V | Gta → V | Gta → V | Gta → V | |
| 318 | ccA → P | ccA → P | ccA → P | ccA → P | ccA → P | ccG → P | ccA → P | |
| 525 | ttT → F | ttT → F | ttT → F | ttT → F | ttT → F | ttG → L | ttT → F | |
| 559 | Cgt → R | Cgt → R | Cgt → R | Cgt → R | Cgt → R | Ggt → G | Cgt → R | |
| 557 | Taa → c | Taa → c | Taa → c | Taa → c | Taa → c | Gaa → Ed | Taa → c | |
| atp4 | 463 | Cac → H | Cac → H | Cac → H | Cac → H | Cac → H | Aac → N | Cac → H |
| rps3 | 85 | Agt → S | Ggt → G | Agt → S | Agt → S | Agt → S | Ggt → G | Agt → S |
| 106 | Ctc → L | Atc → I | Atc → I | Atc → I | Atc → I | Atc → I | Atc → I | |
| 755 | tCc → S | tTc → F | tTc → F | tTc → F | tTc → F | tTc → F | tTc → F | |
| 1106 | aTa → I | aTa → I | aTa → I | aTa → I | aTa → I | aGa → R | aTa → I | |
| 1232 | aTa → I | aGa → R | aTa → I | aTa → I | aTa → I | aGa → R | aTa → I | |
| 1240 | Gct → A | Cct → P | Gct → A | Gct → A | Gct → A | Cct → P | Gct → A | |
| rps4 | 103 | Aag → K | Gag → E | Aag → K | Aag → K | Aag → K | Aag → K | Aag → K |
| 527 | cTg → L | cGg → R | cTg → L | cTg → L | cTg → L | cTg → L | cTg → L | |
| 573 | cgC → R | cgC → R | cgC → R | cgC → R | cgC → R | cgA → R | cgC → R | |
| 745–746 | TAt → Y | GAt → D | TCt → S | TCt → S | TAt → Y | TAt → Y | TAt → Y | |
| rps7 | 198 | gtC → V | gtA → V | gtC → V | gtC → V | gtA → V | gtA → V | gtC → V |
| rps12 | 269 | tCg → S | tTg → L | tCg → S | tCg → S | tCg → S | tCg → S | tCg → S |
| 326 | gAt → D | gGt → G | gAt → D | gAt → D | gAt → D | gAt → D | gAt → D | |
| tatC | 323 | aGa → R | aGa → R | aGa → R | aGa → R | aGa → R | aGa → R | aTa → I |
| 567 | tcT → S | tcC → S | tcT → S | tcT → S | tcT → S | tcT → S | tcT → S | |
Undefined start codon (beginning 408 bp before or 87 bp after).
Two copies, one is identical to Nv and the other have the first 198 bp identicals and 506 bp unique.
Stop codon
Leading to supplementary 42 bp.
As described by Satoh et al. (2004), two copies of cox2 exon2 were found in TK81-MS: one copy was identical to the cox2 exon2 found in the other genomes and the other copy being identical in the 158 first nucleotides and then composed of a specific sequence of 506 bp (supplementary file S4, Supplementary Material online).
Finally, as described by Yamamoto et al. (2005), atp6 was longer in TK81-MS with an additional 1,146 bp upstream. CMS-E was found to exhibit an additional atp6 5′-leader sequence, which was 88% identical to TK81-MS with 1% of gap. This sequence specific to CMS-E was identical to the atp6 found in I-12CMS(3), a wild CMS found in Pakistan, suggesting the identity between CMS-E and I-12CMS(3) (Onodera et al. 1999) (supplementary file S4, Supplementary Material online).
Open Reading Frames
Out of a total of 235 ORFs (putative coding sequence with a minimum size of 300 bp), 34 were found to overlap genes, 20 to overlap inserted plastid sequences, and 13 were chimeric (with at least a 16 bp sequence similar to an mt gene) (supplementary table S5, Supplementary Material online). Certain ORFs were specific to a given CMS and were therefore potential candidate sterilizing genes: 38 ORFs were specific to TK81-MS, 21 to CMS-G, and 14 to CMS-E. Four ORFs were shared between TK81-MS and CMS-G, three between TK81-MS and CMS-E, one between CMS-E and CMS-G, and four were common to all three CMSs.
In reference to Satoh et al. (2004) where TK81-O and TK81-MS were compared, orf317 found in TK81-O but not in TK81-MS was also found in other genomes, TK81-MS having a smaller corresponding ORF (orf221). Orf518 found in TK81-O was also found in macrocarpa but was absent in all other genomes. Its homolog, orf496, found in TK81-MS was also found in B and CMS-E. Orf518 and orf496 are similar to ccmC (Mower and Palmer 2006). None of these orfs has been found in CMS-G, but the genome is not fully sequenced. Orf324 and orf119c were specific to TK81-MS, orf214 was only shared by TK81-O and TK81-MS, and orf129b and orf145 only found in TK81-O, but corresponding ORFs (orf122b and orf176b) described as specific to TK81-MS were found in all other genomes.
Among the ORFs specific to CMS-E, we found orf129, described by Yamamoto et al. (2008), as a candidate sterilizing gene in I-12CMS(3), suggesting once again that CMS-E is identical or at least genetically close to I-12CMS(3).
Concerning the ORFs that might be specific to or shared among non-CMSs, it must be noted that due to the incompleteness of CMS-E and CMS-G genomes, their specificity is not guaranteed. With this in mind, 26 ORFs were found to be specific to TK81-O, five were specific to macrocarpa, one was specific to A, 10 were shared by all genomes except TK81-MS, six were found in all genomes except TK81-MS and CMS-G, three were shared by TK81-O, A, and macrocarpa, 13 were found in all but CMS-G, one was shared by A and macrocarpa, and one was shared by A and B (supplementary table S5, Supplementary Material online).
Supplementary table S5 (Supplementary Material online) summarizes the chimeric ORFs found among the seven genomes. Only TK81-MS and TK81-O exhibited specific chimeric ORFs, two ORFs for each.
Repeated Sequences
Figure 1 gives a representation of duplicates found in beet mt genomes. Overall, 20 repeats longer than 0.5 kb were detected over the seven mt genomes (supplementary table S6, Supplementary Material online). The largest genome, TK81-MS, featured the two largest duplications, R87 and R51, respectively, 87 kbp and 51 kbp long. TK81-O, A, B, and macrocarpa shared a 23-kbp long (R23) duplication, which was a subfragment of R87. A duplication of 12.5 kbp (R12.5) only found in macrocarpa was also a subfragment of R87. All the other duplications were shorter than 10 kb and usually specific to a given genome even though they shared common sequences with duplications found in other genomes. It can be noted that three copies of duplicate R6.0 were found in A, B, and macrocarpa and that R6.0 was partially included in R6.2 of TK81-O, which also featured three copies. Interestingly, TK81-MS also exhibited a triplication with R7.6, which partially included R6.2 and R6.0. This could be the signature of an ancestral triplication with subsequent rearrangements in the Beta mt genomes.
Phylogeny and Molecular Evolution Rate of Beta mt Genomes
We constructed a concatenated sequence for each Beta mt genome composed of the genomic sequence shared by all seven Beta genomes: the backbone sequence. The consensus sequence length is 265.553 kbp (including gaps), and, for each genome, the length varied between 255.223 kbp (TK81-O) and 265.371 kbp (macrocarpa) (mean of 265.341 kbp and median of 265.362 kbp). This enabled us to measure pairwise substitution rates, pairwise indel rates (indels less than 16 bp), and mutation (substitution and indel) rates among genomes (supplementary table S7a, b, and c, Supplementary Material online). Pairwise substitution rates varied from 1.09 substitutions per 10,000 bp (between A and B) to 19.28 substitutions per 10,000 bp (between TK81-O and TK81-MS), with a median value of 15.75. When considering small indels, the pairwise indel rates vary from 0.23 (between A and B) to 16.68 indels (between TK81-O and TK81-MS) per 10,000 bp with a median value of 7.87. Globally, when considering substitutions and small indels, the pairwise mutation rates varied from 1.32 mutations per 10,000 bp (between A and B) to 35.97 mutations per 10,000 bp (between TK81-O and TK81-MS) with a median value of 20.72. Substitution and indel rates were highly correlated in Beta (r = 0.861, P = 0.001). It must be noted that T81-MS and CMS-G exhibited the highest rate with macrocarpa. Using backbone sequences, a phylogenetic tree rooted with B. macrocarpa was built with strong bootstrap values (fig. 2A). The same topology was found using NJ or ML methodologies. The same was true when considering only variable sites (data not shown). In addition, the four tests of phylogenetic signal saturation implemented in PhyloBayes were rejected by this data set (P > 0.51). These experiments suggest that signal saturation in our data is accurately anticipated under model assumptions and that it is not expected to significantly bias our estimates. The tree is composed of two clades, one formed by A, B, and TK81-O constituting a non-CMS clade and the other forming a CMS clade with CMS-E, CMS-G, and TK81-MS. CMS-G and TK81-MS form long branches. We assessed whether the evolutionary dynamics was different between CMS and non-CMS mt genomes in B. v. ssp. maritima. For each of the 29 protein coding genes (total size of 29,220 bp), we assessed the average synonymous and nonsynonymous nucleotide diversities (πs and πa) within CMS (CMS-E, CMS-G, and TK81-MS) or non-CMS genomes (A, B, and TK81-O) and their average synonymous and nonsynonymous nucleotide divergence (Ks and Ka) with macrocarpa, as well as individual Ks and Ka for each genome (table 3).
FIG. 2.—

Phylogenetic analyses of Beta genomes. Phylogenetic trees based on nucleotide distances, (A) with backbone sequences of mt genomes and (B) with chloroplastic sequences, were constructed using BIONJ and Tree-Puzzle. (C) Phylogenetic tree based on rearrangement distances built with BIONJ. The trees were rooted using Beta macrocarpa. Branch lengths are proportional to substitution or rearrangement rates. For (A and B), bootstrap values (upper values for distance and lower values for likelihood) are reported.
Table 3.
Synonymous and Nonsynonymous Nucleotide Diversity and Divergence with Beta macrocarpa of CMS and Non-CMS mt Genomes
| mt_Genes (29,220 bp) | πs | πa | πa/πs | Ks | Ka | Ka/Ks |
| CMS | 0.00160 | 0.00130 | 0.810 | 0.00109 | 0.00090 | 0.820 |
| E | 0.00081 | 0.00060 | 0.749 | |||
| G | 0.00061 | 0.00114 | 1.893 | |||
| TK81-MS | 0.00187 | 0.00094 | 0.503 | |||
| Non-CMS | 0.00010 | 0.00016 | 1.577 | 0.00005 | 0.00014 | 2.839 |
| A | 0 | 0.00010 | — | |||
| B | 0 | 0.00010 | — | |||
| TK81-O | 0.00015 | 0.00024 | 1.577 |
NOTE.—means cannot be calculated.
The average Ks along CMS branches was on average 6.6 higher than along non-CMS branches. The same phenomenon was observed at the diversity level (πs): mt genes were an average of five times more diverse in CMSs than in non-CMSs, suggesting a higher rate of synonymous substitution rate in the sterile lineage. To formally test this hypothesis, we used HyPhy to test for a variation of the synonymous rate (dS) across lineages. The null hypothesis of no variation was strongly rejected (P < 0.0001), suggesting that the CMS lineage experienced an increase in synonymous substitution rate. This effect was apparently strong, with an average dS of 0.0051 along non-CMS lineages but an average dS of 0.02916 along CMS lineages, that is, a 57-fold increase (supplementary table S8, Supplementary Material online).
The nonsynonymous divergence of mt genes with macrocarpa (Ka) was an average of six times higher in CMSs than in non-CMSs but demonstrated a similar Ka/Ks ratio. Nucleotide nonsynonymous diversities were about five times higher in CMSs than in non-CMSs, resulting in a similar πa/πs among the two groups. Among CMSs, CMS-G exhibits a high Ka and a subsequent Ka/Ks ratio higher than 1 suggesting that a positive selection occurred on this peculiar genome.
Mitochondrial and Chloroplastic Evolution
In order to determine whether longer branches associated with CMSs were specific to mt genomes, we sequenced 31 chloroplastic fragments totalizing a size of 17.740 kbp. These fragments were concatenated to build a phylogenetic tree (fig. 2B). Tree resolution was not optimal, and some nodes are not resolved, but it can be noted that branches of CMS chloroplastic genomes are not longer than non-CMS branches. Therefore, it seems that the long branches found on CMS mt genomes are the signature of fast-evolving mt genomes within the species.
Rearrangement Distances and Phylogeny
A rearrangement distance matrix was generated from SA sequence comparison (supplementary table S9, Supplementary Material online). Rearrangement distances vary from 4 (A to B) to 42 (TK81-MS to CMS-G) with median value of 27 and mean of 22. Consequently, at the structural level, A and B are the closest, whereas TK81-MS and CMS-G are the most distant. A phylogenetic tree was built from rearrangement distances (fig. 2C). This tree shows the same topology as the one based on backbone sequences with two clades composed of CMS and non-CMS mt genomes. Long branches found on CMS mt genomes with backbone sequences were also found in this rearrangement tree. In particular, we found a correlation between rearrangement and nucleotidic distances (Spearman coefficient of correlation r = 0.80, P = 0.02).
Discussion
In this study, we present the (nearly) whole sequences of five new mt genomes in Beta genus, four from B. vulgaris, and one from B. macrocarpa, a sister species belonging to the same Beta section. Pooling our results with two previously sequenced genomes of B. vulgaris, we were able to assess genome diversity at a species level for the first time in a eudicot species. The occurrence of male sterility is an important feature in the B. vulgaris breeding system (called gynodioecy) and is expected to affect mt gene/ORF content and diversity through the emergence and selection of CMS-specific mt genes (Charlesworth 2002; Touzet and Delph 2009). The phylogenetic analysis revealed that CMSs and non-CMSs formed separate clades. We therefore compared the two groups of mt genomes, CMSs versus non-CMSs, and found that the CMS lineage might have evolved rapidly in sequence and structure.
CMS in B. vulgaris
As no general nomenclature is available for B. vulgaris mt genomes, a secondary outcome of our study is the possibility to identify CMSs that have been characterized by independent studies. It seems that what we call CMS-E following Cuguen et al. (1994) corresponds to what Hokkaido's team called I-12CMS(3) (Onodera et al. 1999; Yamamoto et al. 2008). Indeed, CMS-E exhibits a 5′-leader atp6 sequence and orf129 that are only found in I-12CMS(3) (Onodera et al. 1999; Yamamoto et al. 2008). CMS-E is the most frequent source of CMS in wild beet populations on the European Coasts and thus seems to be found on an even larger geographical scale, as I-12CMS(3) is found in wild populations in Pakistan. Therefore, it seems that the number of CMSs in wild beet might be limited to four (out of a total of 20 different mt genomes—Desplanque et al. 2000) and that the occurrence of CMS is a rare event.
Previous studies have tentatively proposed candidate genes of male sterility in beet.
Regarding CMS Owen/TK81-MS, widely used in sugar beet breeding, Yamamoto et al. (2005) have shown that among the four specific ORFs detected when TK81-MS mt genome was compared with T81-O, only the ORF corresponding to a peculiar 5′-leader sequence of atp6 (preSatp6) was expressed at the protein level. It codes for a 35-kDa polypeptide that is specific to the Owen CMS. However, no effect of nuclear restoration was detected on the size or the amount of the preSATP6 polypeptide, and no transformation experiment has validated the sterilizing effect of preSATP6 (Yamamoto et al. 2008).
Regarding CMS-E/I-12CMS(3), Yamamoto et al. (2008) demonstrated that the E-specific orf129 was transcribed and coded for a specific 12-kDa polypeptide, which accumulated in the mitochondria of flower, root, and leaf. Transgenic expression in tobacco of orf129 fused with an mt targeting presequence led to male-sterile plants, demonstrating the sterilizing effect of ORF129. The question of the effect of restorer loci on this CMS remains because no effect on ORF129 abundance has been detected when plants were restored.
In a former study (Ducos et al. 2001), we had shown that CMS-G exhibited a modified genomic cox2 sequence that resulted in a truncated protein at the C-terminus. In addition, it was shown that the in vitro activity of cytochrome c oxidase was reduced by 50% in leaves, suggesting a possible effect of the observed mutations on the complex activity. However, we were unable to recover Complex IV by blue native electrophoresis in CMS-G plants, raising the question of the stability and/or the physicochemical characteristics of the CMS-G-Complex IV. In the present study, as expected, we found not only the modified cox2 sequence but also mutations on cox1 and cox3. For cox1, a mutation at the start codon, which is commonly found in other beet genomes, can potentially result in the translation of a longer protein with an extended N-terminus (660 vs. 524 amino acids (aa); estimated weight of 73.5 vs. 57.6 kDa) or a shorter one (495 aa; estimated weight 54.2 kDa). Note that a recent study of the same genome confirms that there is only one copy of cox1 (Kawanishi et al. 2010). We did not detect a long variant on previous sodium dodecyl sulfate (SDS)–PAGE from in organello S-labeled proteins in CMS-G, suggesting that the translated form of CMS-G-cox1 might be the shortest one, with a size variation that could not be detected in SDS–PAGE conditions (Ducos et al. 2001). However, in the truncated N-terminus, amino acids are expected to be involved in subunit I/III interface (S10), D-pathway (L19), or subunit I/VIIc interface (A25). In addition, two nonsynonymous polymorphisms were found: R180/Q and F403/V, two codons that are not associated to any known function (from the Pfam database—http://pfam.sanger.ac.uk/—refering to Tsukihara et al. 1996). For cox3, one nonsynonymous polymorphism was detected leading to an I/L variation on codon 51, with no known associated function. The polymorphism of Complex IV could be the signature of coadaptation or the result of a relax in selection, enabling the accumulation of nonsynonymous mutations, once the sterilizing mutations have been selected through disruption of COX activity. This polymorphism could imply compensatory mutations on COX nuclear genes in fertility restoration. More generally, it raises the question of the evolution of nuclear–mt interaction and coevolution of protein coding genes involved in the same respiratory complex (Moison et al. 2010).
Evolution Rates of mt Genomes
A phylogeny based on chloroplastic sequences constructed by our group (Fénart et al. 2006) suggested that the sterile cytoplasms emerged independently from a nonsterile cytoplasm. However, the resolution was very low due to the lack of polymorphism. In the present study, through pairwise substitution rates among the mt genomes, we were able to build phylogenetic trees with three different methods where two clusters appeared, one composed of the three CMSs and one of the three non-CMSs. We then observed that the two groups had distinct features not only in nucleotide diversity but also in rearrangement rates. We acknowledge that the grouping of the two most divergent lineages (in sequence and in structure), that is, TK81-MS and CMS-G, could be due to long-branch attraction. Nevertheless, trees obtained from different methods were congruent knowing that the two lineages shared 334 out of 837 variable sites. Even though the topology might be discussed, it remains that the two lineages are characterized by a high rate of evolution. Indeed, it appears that CMSs seem to exhibit not only higher synonymous divergence with B. macrocrapa but also a higher rate of structure rearrangement. A first explanation could be that the CMS genomes are older than expected due to balancing selection that may have favored their maintenance over larger timescales than non-CMS genomes (Charlesworth 2002; Touzet and Delph 2009). This would explain why CMS genomes are found on longer branches in the sequence and rearrangement trees. It would imply that the trees are therefore wrongly rooted with macrocarpa. However, when we compared chloroplastic sequences that should have followed the same pattern since they are cotransmitted with mt genomes, we found that cp chloroplastic sequences related to CMS mt genomes were no more divergent than non-CMS ones. Consequently, we concluded that the CMS lineages are probably not older but have a faster rate of evolution at the sequence level (probably through a higher mutation rate in these lineages) and also at the structure level. Interestingly, this correlation between molecular evolution rates and genome rearrangement rates has been found at the interspecific level in animal mt genomes (Xu et al. 2006). It was proposed that the variation of the accuracy of the replication process in mt genomes among species, due to deleterious mutations in the involved nuclear genes, could lead to the correlated variation of both rates. In plants, to our knowledge, no such study is available, but a high variation of mt substitution rate among species has been documented (Cho et al. 2004; Parkinson et al. 2005; Barr et al. 2007; Mower et al. 2007; Sloan et al. 2008, 2010). It was also suggested that this could be due not only to nuclear mutations causing error-prone replications or defective repair but also to any biological factors that could affect the mt mutation rate, such as the production or detoxification of oxygen free radicals that are believed to damage DNA (Cho et al. 2004). In this study, we show that a variation of substitution rate and thus possibly of mutation rate can even be seen at the intraspecies level. This result is reminiscent of a recent study in gynodioecious Silene vulgaris, where the variation of synonymous substitution rate was also described among lineages, suggesting the occurrence of fast-evolving mt genomes (Sloan et al. 2008). Interestingly, not only does there seem to be fast-evolving mt genomes in beet but also they seem to be restricted to CMSs. Could it be that an episode of increased mutation and rearrangement rates is at the origin of the emergence of male-sterilizing factors in beet, being either new ORFs through intragenomic recombination as generally assumed (Chase 2007) or the outputs of deleterious mutations on essential mt genes positively selected as they disrupt pollen production (Budar et al. 2003)? Finally, the fast evolution of CMS genomes in beet raises the question of a direct involvement of male sterility in the evolution of CMS mt genomes, when one considers that a dysfunction in mitochondria that disrupts pollen production might also generate the accumulation of mutagenous free radicals, as well as a relax of purifying selection.
Conclusion
The study of similar data in other species where CMS and non-CMS mt genomes are found, such as maize (Allen et al. 2007), is required in order to assess whether the trend found in beet, that is, fast-evolving CMS mt genomes, is also found in other gynodioecious species. More generally, it is clear that the increasing number of whole sequenced mt genomes in plants will facilitate the emergence of a better picture of the evolutionary forces that shape content, size, and structure of mt genomes (Lynch et al. 2006; Alverson et al. 2010). But as exemplified by the present study, the analysis of several representative genomes per species will also be a necessary step to draw a complete picture.
Funding
Agence Nationale de laRecherche (ANR-06-JCJC-0074); Région Nord Pas de Calais and the European Community (Arcir PLANT-TEQ6) to P.T.; PPF Bioinformatique of University of Lille1 to P.T., J.-S.V.; PhD fellowship from French Research Ministry to A.D.
Supplementary Material
Supplementary tables S1–S9 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals .org/).
Acknowledgments
We wish to thank Benjamin Brachi for the scripts in R (fig. 1), Samuel Blanquart for the PhyloBayes analyses, Shahinaz Gas in the process of EMBL submission and Licia Huffman-Touzet for English editing. This work was part of the Genoscope project entitled BeetMito (AP06 #65).
References
- Allen JO, et al. Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genetics. 2007;177:1173–1192. doi: 10.1534/genetics.107.073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverson AJ, Wei X, Rice DW, Stern DB, Barry K, Palmer JD. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae) Mol Biol Evol. 2010;27:1436–1448. doi: 10.1093/molbev/msq029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Nielsen R, Yang Z. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics. 2003;164:1229–1236. doi: 10.1093/genetics/164.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr CM, Keller SR, Ingvarsson PK, Sloan DB, Taylor DR. Variation in mutation rate and polymorphism among mitochondrial genes of Silene vulgaris. Mol Biol Evol. 2007;24(8):1783–1791. doi: 10.1093/molbev/msm106. [DOI] [PubMed] [Google Scholar]
- Budar F, Touzet P, De Paepa R. The nucleo-mitochondrial conflict in cytoplasmic male sterilities revisited. Genetica. 2003;117(1):3–16. doi: 10.1023/a:1022381016145. [DOI] [PubMed] [Google Scholar]
- Charlesworth D. What maintains male-sterility factors in plant populations. Heredity. 2002;89:408–409. doi: 10.1038/sj.hdy.6800193. [DOI] [PubMed] [Google Scholar]
- Chase CD. Cytoplasmic male sterility: a window to the world of plant mitochondrial-nuclear interactions. Trends Genet. 2007;23:81–90. doi: 10.1016/j.tig.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Cho W, Mower JP, Qiu Y-L, Palmer JD. Mitochondrial substitution rates are extraordinarily elevated and variable in a genus of flowering plants. Proc Natl Acad Sci U S A. 2004;101(51):17741–17746. doi: 10.1073/pnas.0408302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuguen J, Wattier R, Saumitou-Laprade P, Forcioli D, Mörchen M, Van Dijk H, Vernet P. Gynodioecy and mitochondrial DNA polymorphism in natural populations of Beta vulgaris ssp maritima. Genet Sel Evol. 1994;26:87–101. [Google Scholar]
- Darling AC, Mau B, Blatter FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darracq A, Varré J-S, Touzet P. A scenario of mitochondrial genome evolution in maize based on rearrangement events. BMC Genomics. 2010;11:233. doi: 10.1186/1471-2164-11-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplanque B, Viard F, Bernard J, Forcioli D, Saumitou-Laprade P, Cuguen J, Van Dijk H. The linkage disequilibrium between chloroplast DNA and mitochondrial DNA haplotypes in Beta vulgaris ssp. maritima (L.): the usefulness of both genomes for population genetic studies. Mol Ecol. 2000;9:141–154. doi: 10.1046/j.1365-294x.2000.00843.x. [DOI] [PubMed] [Google Scholar]
- Ducos E, Touzet P, Boutry M. The male sterile G cytoplasm of wild beet displays modified mitochondrial respiratory complexes. Plant J. 2001;26:171–180. doi: 10.1046/j.1365-313x.2001.01017.x. [DOI] [PubMed] [Google Scholar]
- Dufaÿ M, Cuguen J, Arnaud J-F, Touzet P. Sex ratio variation among gynodioecious populations of wild beet: can it be explained by negative frequency-dependent selection? Evolution. 2009;63:1483–1497. doi: 10.1111/j.1558-5646.2009.00653.x. [DOI] [PubMed] [Google Scholar]
- Dufaÿ M, Touzet P, Maurice S, Cuguen J. Modelling the maintenance of a male fertile cytoplasm in a gynodioecious population. Heredity. 2007;99:349–356. doi: 10.1038/sj.hdy.6801009. [DOI] [PubMed] [Google Scholar]
- Fénart S, Touzet P, Arnaud J-F, Cuguen J. Emergence of gynodioecy in wild beet (Beta vulgaris ssp. maritima L.): a genealogical approach using chloroplastic nucleotide sequences. Proc R Soc Lond B Biol Sci. 2006;273:1391–1398. doi: 10.1098/rspb.2005.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14:685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- Gray MW, Burger G, Lang BF. Mitochondrial Evolution. Science. 1999;283:1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23(2):254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- Kawanishi Y, Shinada H, Matsunaga M, Masaki Y, Mikami T, Kubo T. A new source of cytoplasmic male sterility found in wild beet and its relationship to other CMS types. Genome. 2010;53:251–256. doi: 10.1139/g10-003. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2004;21(5):676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Kubo T, Newton KJ. Angiosperm mitochondrial genomes and mutations. Mitochondrion. 2008;8:5–14. doi: 10.1016/j.mito.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Kubo T, Nishizawa S, Sugawara A, Itchoda N, Estiati A, Mikami T. The complete nucleotide sequence of the mitochondrial genome of sugar beet (Beta vulgaris L.) reveals a novel gene for tRNACys(GCA) Nucleic Acids Res. 2000;28:2571–2576. doi: 10.1093/nar/28.13.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. PhyloBayes 3. A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25(17):2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Koskella B, Schaak S. Mutation pressure and the evolution of organelle genomic architecture. Science. 2006;311:1727–1730. doi: 10.1126/science.1118884. [DOI] [PubMed] [Google Scholar]
- Matsunaga M, Nagano H, Mikami T, Kubo T. Large 3' UTR of sugar beet rps3 is truncated in cytoplasmic male-sterile mitochondria. Plant Cell Rep. 2010;30:231–238. doi: 10.1007/s00299-010-0912-y. [DOI] [PubMed] [Google Scholar]
- Moison M, Roux F, Quadrado M, Duval R, Ekovich M, Le D-H, Verzaux M, Budar F. Cytoplasmic phylogeny and evidence of cyto-nuclear co-adaptation in Arabidopsis thaliana. Plant J. 2010;63:728–738. doi: 10.1111/j.1365-313X.2010.04275.x. [DOI] [PubMed] [Google Scholar]
- Mower JP, Palmer JD. Patterns of partial RNA editing in mitochondrial genes of Beta vulgaris. Mol Genet Genomics. 2006;276:285–293. doi: 10.1007/s00438-006-0139-3. [DOI] [PubMed] [Google Scholar]
- Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD. Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol Biol. 2007;7:135. doi: 10.1186/1471-2148-7-135. doi:10.1186/1471-2148-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa S, Mikami T, Kubo T. Mitochondrial DNA phylogeny of cultivated and wild beets: relationships among cytoplasmic male-sterility inducing and nonsterilizing cytoplasms. Genetics. 2007;177:1703–1712. doi: 10.1534/genetics.107.076380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noe L, Kucherov G. YASS: enhancing the sensitivity of DNA similarity search. Nucleic Acids Res. 2005;33(2):W540–W543. doi: 10.1093/nar/gki478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera Y, Yamamoto MP, Kubo T, Mikami T. Heterogeneity of the atp6 presequences in normal and different sources of male-sterile cytoplasms of sugar beet. J Plant Physiol. 1999;155:656–660. [Google Scholar]
- Palmer JD, Herbon LA. Plant mitochondrial DNA evolved rapidly in structure, but slowly in sequence. J Mol Evol. 1988;28(1):87–97. doi: 10.1007/BF02143500. [DOI] [PubMed] [Google Scholar]
- Parkinson CL, Mower JP, Qiu Y-L, Shirk AJ, Song K, Young ND, dePamphilis CW, Palmer JD. Multiple major increases and decreases in mitochondrial substitution rates in the plant family Geraniaceae. BMC Evol Biol. 2005;5:73. doi: 10.1186/1471-2148-5-73. doi:10.1186/1471-2148-5-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Sanshez-Delbarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Satoh M, Kubo T, Nishizawa S, Estiati A, Itchoda N, Mikami T. The cytoplasmic male-sterile type and normal type mitochondrial genomes of sugar beet share the same complement of genes of known function but differ in the content of expressed ORFs. Mol Genet Genomics. 2004;272:247–256. doi: 10.1007/s00438-004-1058-9. [DOI] [PubMed] [Google Scholar]
- Schmidt HA. Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Scotti N, Cardi T, Maréchal-Drouard L. Mitochondrial DNA and RNA isolation from small amounts of potato tissue. Plant Mol Biol Rep. 2001;19:1–8. [Google Scholar]
- Sloan DB, Barr CM, Olson MS, Keller SR, Taylor DR. Evolutionary rate variation at multiple levels of biological organization in plant mitochondrial DNA. Mol Biol Evol. 2008;25(2):243–246. doi: 10.1093/molbev/msm266. [DOI] [PubMed] [Google Scholar]
- Sloan DB, Oxelman B, Rautenberg A, Taylor DR. Phylogenetic analysis of mitochondrial substitution rate variation in the angiosperm tribe Sileneae. BMC Evol Biol. 2010;10:12. doi: 10.1186/1471-2148-9-260. doi:10.1186/1471-2148-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzet P, Delph LF. The effect of breeding system on polymorphism in mitochondrial genes of Silene. Genetics. 2009;181:631–644. doi: 10.1534/genetics.108.092411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- Vallenet D, et al. Comparative analysis of acinetobacters: three genomes for three lifestyles. PLoS ONE. 2008;3(3):e1805. doi: 10.1371/journal.pone.0001805. doi:10.1371/journal.pone.0001805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Jameson D, Tang B, Higgs PG. The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J Mol Evol. 2006;63:375–392. doi: 10.1007/s00239-005-0246-5. [DOI] [PubMed] [Google Scholar]
- Yamamoto MP, Kubo T, Mikami T. The 5’-leader sequence of sugar beet mitochondrial atp6 encodes a novel polypeptide that is characteristic of Owen cytoplasmic male sterility. Mol Genet Genomics. 2005;273:342–349. doi: 10.1007/s00438-005-1140-y. [DOI] [PubMed] [Google Scholar]
- Yamamoto MP, Shinada H, Onodera Y, Komaki C, Mikami T, Kubo T. A male sterility-associated mitochondrial protein in wild beets causes pollen disruption in transgenic plants. Plant J. 2008;54(6):1027–1036. doi: 10.1111/j.1365-313X.2008.03473.x. [DOI] [PubMed] [Google Scholar]
- Yang Z, Sankoff D. Natural parameter values for generalized gene adjacency. Lect Notes Comput Sci. 2009;5817:13–23. doi: 10.1089/cmb.2010.0099. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Adam Z, Choi V, Sankoff D. Generalized gene adjacencies, graph bandwidth, and clusters in yeast evolution. IEEE/ACM Trans Comput Biol Bioinform. 2009;6(2):213–220. doi: 10.1109/TCBB.2008.121. [DOI] [PubMed] [Google Scholar]