

Abstract
We have shown that Wnt signals are necessary and sufficient for neural crest cells to adopt pigment cell fates. nacre, a zebrafish homolog of MITF, is required for pigment cell differentiation. We isolated a promoter region of nacre that contains Tcf/Lef binding sites, which can mediate Wnt responsiveness. This promoter binds to zebrafish Lef1 protein in vitro, and a nacre reporter construct is strongly repressed by dominant-negative Tcf in melanoma cells. Mutation of Tcf/Lef sites abolishes Lef1 binding and reporter function in vivo. Wnt signaling therefore directly activates nacre, which in turn leads to pigment cell differentiation.
Keywords: Wnt, β-catenin, nacre, neural crest, MITF
Although the Wnt/β-catenin signaling pathway functions throughout development (Cadigan and Nusse 1997), few target genes have been identified that mediate the roles of Wnt signaling in cell fate determination. Previous Wnt/β-catenin target genes have been identified by promoter analysis, in which canonical Tcf/Lef binding sites have been identified (Riese et al. 1997; Eastman and Grosschedl 1999). Although vertebrate targets have included genes responsible for cell cycle progression (He et al. 1998; Tetsu and McCormick 1999) and dorsal–ventral axis specification (Brannon et al. 1997), none are directly associated with specifying a particular cell fate.
Our recent experiments have shown that Wnt signals are necessary and sufficient to promote the specification of neural crest as pigment cells (Dorsky et al. 1998). Wnt-1 and Wnt-3a are expressed at the correct time and place to influence fate decisions made by premigratory neural crest cells. In the zebrafish, neural crest cells adjacent to domains of Wnt expression primarily produce pigment, whereas cells more distant from the Wnt signals primarily produce neurons and glia. By misexpressing mRNA in individual cells, we were able to demonstrate that β-catenin promotes pigment cell formation, whereas dominant-negative forms of Tcf and Wnt inhibit the pigment fate. We were therefore interested in whether a Wnt target gene exists in neural crest cells, providing a direct link between extracellular signals and cell fate specification.
One possible mechanism by which pigment cell specification could take place is the direct control of genes necessary and sufficient for adopting the pigment cell fate. A candidate Wnt target gene that fulfills this role in zebrafish is nacre, which encodes a helix-loop–helix leucine zipper transcription factor homologous to MITF (microphthalmia-associated transcription factor) (Lister et al. 1999). nacre is expressed in differentiating neural crest-derived melanophores, as well as the retinal pigmented epithelium (Lister et al. 1999), and the product of its mammalian homolog MITF directly binds the promoters of pigment cell-specific genes such as TRP-1 (Yasumoto et al. 1997; Bertolotto et al. 1998b). Misexpression of nacre or MITF can promote pigment cell phenotypes in nonpigment cells (Tachibana et al. 1996; Lister et al. 1999), demonstrating that it is a regulatory gene sufficient to specify pigment cell fate. Moreover, loss of nacre or MITF function results in a failure of neural crest-derived melanocytes to differentiate (Opdecamp et al. 1997; Lister et al. 1999), demonstrating that it is required for this cell fate. These properties led us to hypothesize that nacre may be directly regulated by Wnt signaling in the premigratory neural crest.
In this paper we show that an 838-bp nacre promoter/enhancer, which is sufficient to drive tissue-specific expression in vivo, contains three Tcf/Lef binding sites. These sites are necessary for specific interaction with zebrafish Lef1 in vitro and are necessary for regulation of the promoter/enhancer by Wnt signaling in a melanoma cell line. Finally, we show that the Tcf/Lef binding sites are necessary for proper expression in zebrafish embryos. Taken together, our results support a model in which the nacre gene is a direct target of Wnt signaling and establish a molecular pathway from extracellular signal to specification of cell fate.
Results
We observed that the human melanocyte-specific MITF promoter (Fuse et al. 1996) contains several putative Tcf/Lef binding sites, hallmarks of Wnt/β-catenin-mediated transcriptional control (Riese et al. 1997; Eastman and Grosschedl 1999). To test the hypothesis that nacre is a target of the Wnt/β-catenin pathway in zebrafish, we isolated a promoter/enhancer region for this gene from a genomic phage library. This genomic fragment contains the translation initiation site for nacre and an 836-bp upstream region with a potential transcription start site, a cAMP response element (CRE), and several consensus Tcf/Lef binding sites (Fig. 1A). These CRE and Tcf/Lef sites are not absolutely conserved between the human and zebrafish promoters in either their sequences or their positions from the transcription start site (data not shown). To examine whether the putative nacre promoter/enhancer can direct proper spatial control of gene expression, we injected one -cell zebrafish embryos with the 836-bp DNA fragment driving a green fluorescent protein (GFP) reporter gene (nac>GFP, Fig. 1B). Mosaic gene expression is typical for transient transgenics in zebrafish, and because the neural crest is derived from very few precursor cells, reporter efficiency is decreased further. Despite these obstacles, we were able to analyze the fate of the few reporter-expressing cells in injected animals. In this transient expression assay, 18% of injected embryos showed GFP expression in one or more pigment cells (Fig. 2A,B); and strikingly, no expression was observed in other crest derivatives or outside the neural crest.
Figure 1.

(A) Schematic diagram of the 836-bp nacre promoter/enhancer. Three Tcf/Lef binding sites, a cAMP response element (CRE), and the transcription start site are indicated. The sequences of the Tcf/Lef consensus and sites S1, S2, and S3 are listed, and the bases mutated to inactivate them are listed below. Arrows indicate an additional consensus base (C or G) that appears in these and all other published binding sites. The non-consensus base in site 3 is boxed. (B) Constructs used in this study are referenced in the text by the names indicated (left). To rescue function in the construct with three mutant Tcf/Lef sites, five replacement binding sites were inserted in nacMS+Lef>nac at the indicated position.
Figure 2.
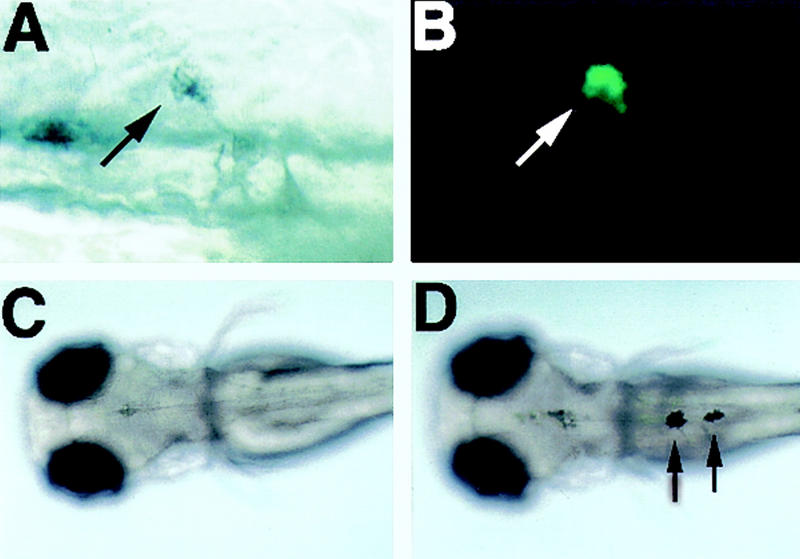
Analysis of nacre promoter/enhancer function in vivo. (A) (Arrow) A melanophore in a wild-type embryo injected at the one-cell stage with the nac>GFP construct; (B) the same cell is labeled with GFP; (C) 48-hr nacre−/− embryo lacks melanophores (dorsal view); (D) a nacre−/− embryo injected at the one-cell stage with the nac>nac construct contains melanophores (arrows).
To determine whether control elements are sufficient for the correct initiation of gene expression, we used the promoter/enhancer to drive expression of wild-type nacre cDNA in nacre−/− embryos, again by DNA injection at the one-cell stage. For cDNA expression to rescue the nacre−/− phenotype, it must be expressed in premigratory neural crest cells, at the same time as the endogenous gene. In 34% of nacre−/− embryos injected with the nac>nac construct (Fig. 1B), we observed rescue of melanophores, which are normally absent (Fig. 2C,D; Table 1). In addition, all of the melanin-containing cells that we observed had normal melanophore shape and were found in correct embryonic locations, in contrast to the pigment cells derived after nacre was misexpressed by mRNA injection or heat shock induction (Lister et al. 1999). We conclude that this 836-bp fragment contains both a functional promoter and all of the elements sufficient to drive normal nacre expression at the correct time and place to influence pigment cell formation.
Table 1.
Rescue of nacre−/− embryos
| Promoter construct
|
Embryos injected (n)
|
Embryos with melanophores (n)
|
|---|---|---|
| nac > nac | 123 | 42 |
| nacMS > nac | 165 | 0 |
| nacMS + Lef > nac | 107 | 31 |
If the nacre promoter/enhancer is directly controlled by Wnt/β-catenin signaling, it should bind to Tcf/Lef proteins in vitro, consistent with known signaling mechanisms (Bienz 1998; Eastman and Grosschedl 1999). We initially identified two consensus binding sites (S1, S2; Fig. 1A) in the 836-bp fragment, based on existing Tcf/Lef binding sequences (Waterman et al. 1991; Brannon et al. 1997; Riese et al. 1997). In EMSAS, a fragment of the promoter/enhancer containing these sites bound recombinant zebrafish Lef1 protein (Dorsky et al. 1999; Fig. 3A, lane 1). However, mutation of both sites did not abolish Lef1 binding (Fig. 3B, lane 2), indicating that at least one cryptic binding site exists in this fragment. DNase I footprinting (not shown) identified an additional binding site in this region (S3; Fig. 1A), and an oligonucleotide containing this site competes for Lef1 binding of the entire fragment (Fig. 3A, lanes 2,3). In contrast, a version of this oligonucleotide with mutated Tcf/Lef binding nucleotides is unable to compete for Lef1 binding (Fig. 3A, lanes 4,5). Although S3 differs from the consensus Tcf/Lef binding sequence by a single base (box; Fig. 1A), it contains an additional conserved base identified previously in an optimal binding sequence (van de Wetering et al. 1997). We found that a C or G nucleotide at this position is present in all published Tcf/Lef sites and the three sites in the nacre promoter/enhancer (arrows; Fig. 1A). Mutation of all three Tcf/Lef sites confirmed that they are necessary to promote binding to Lef1 protein in vitro (Fig. 3B, lane 4). We conclude that the nacre promoter/enhancer region contains three legitimate Tcf/Lef binding sites.
Figure 3.

Binding of Lef1 to the nacre promoter/enhancer. (A) The 836-bp fragment binds to in vitro-translated zebrafish Lef1 (Dorsky et al. 1999) (lane 1). Excess cold S3 probe is able to compete for Lef1 binding (lanes 2,3). In contrast, excess mutant S3 probe (see Fig. 1A for sequence) is unable to compete for Lef1 binding (lanes 4,5). (B) When S1 and S2 are mutated (MS 1,2), the promoter/enhancer still binds Lef1 (lanes 1,2). Mutation of all three sites (MS 1,2,3) eliminates Lef1 binding (lanes 3,4).
Before testing the importance of the Tcf/Lef sites in vivo, we first needed to confirm the prediction that β-catenin and Tcf/Lef could regulate these sites in a well-established cell culture assay. Either activation by β-catenin or inhibition by dominant-negative Tcf would establish that nacre was a target of the Wnt/β-catenin pathway. Because the nacre promoter/enhancer, like the human MITF promoter (Fuse et al. 1996), acts in a tissue-specific fashion, we chose the mouse B16 melanoma cell line that had been used previously to study the regulation of MITF (Bertolotto et al. 1996). The 836-bp promoter/enhancer was inserted in front of the luciferase (luc) gene in the pGL3 reporter vector (nac>luc; Fig. 1B) and transfected into B16 cells. nac>luc showed high basal luciferase expression in these cells, which could not be increased by cotransfection with β-catenin (not shown). Because reporter activity was already very high in these cells, we tested its response to zebrafish Δtcf, similar to a Xenopus construct that blocks activation of Wnt/β-catenin target genes (Molenaar et al. 1996). We used Tcf rather than Lef in this assay because Lef is unable to repress siamois transcription in Xenopus (Brannon et al. 1999). Cotransfection of Δtcf efficiently repressed the activity of nac>luc in B16 cells (Fig. 4). We then cotransfected nac>luc with full-length zebrafish tcf3, in the presence or absence of excess β-catenin. Although overexpression of full-length tcf3 was able to repress activity of nac>luc, just as shown previously with the Xenopus siamois promoter (Brannon et al. 1997), addition of β-catenin was able to restore reporter activity to basal levels (Fig. 4). Strikingly, the repression of nac>luc by Δtcf was largely eliminated when all three Tcf/Lef sites were mutated in the construct nacMS>luc (Fig. 1B), even though the basal activity was unaffected (Fig. 4). The high basal activity of nacMS>luc suggests that additional factors in B16 melanoma cells may be able to activate this construct in a Wnt-independent manner. Taken together, these data demonstrate that the Tcf/Lef binding sites are required for regulation of nacre expression by the Wnt pathway.
Figure 4.

The nacre promoter/enhancer is regulated by the Wnt pathway. A wild-type promoter/enhancer-luciferase reporter construct (nac>luc, Fig. 1B) is repressed 6.8-fold by cotransfection with Δtcf in B16 melanoma cells. This reporter construct is also repressed by full-length tcf3, but this repression can be relieved by excess β-catenin. In contrast, a construct with all three Tcf/Lef sites mutated (nacMS>luc, Fig. 1B) is not significantly repressed by Δtcf. Luciferase activity is represented in arbitrary units, mean of four experiments normalized to cotransfection with β-galactosidase. Error bars, s.e.m.
Based on these results we hypothesized that nacre is a direct target of the Wnt signaling pathway via β-catenin and Tcf/Lef-mediated transcription in vivo. To test this hypothesis, we used the mutant promoter/enhancer to drive nacre cDNA expression and attempted to rescue the nacre−/− phenotype by DNA injections. As mentioned previously, the nac>nac construct is able to promote melanophore development in nacre−/− embryos (Table 1). In contrast, mutation of the three Tcf/Lef binding sites (nacMS>nac; Fig. 1B) completely eliminated the ability of the promoter/enhancer to rescue the melanophore fate (Table 1). To assess whether replacing Tcf/Lef binding sites in the mutant construct was sufficient to restore normal expression, we reintroduced five synthetic sites into the mutant promoter/enhancer between S3 and the CRE (nacMS+Lef>nac; Fig. 1B). The replacement Tcf/Lef sites promoted melanophore formation in 29% of injected embryos (Table 1), indicating that the absolute positions of Tcf/Lef sites are not critical to their ability to rescue promoter function. These experiments show that transcriptional control of nacre expression via Tcf/Lef sites is necessary for the normal function of this gene in the developing embryo. Moreover, these data further support the hypothesis that Wnt signaling normally serves to activate nacre transcription in melanophore precursors.
Discussion
Our data indicate that Tcf/Lef sites in the nacre promoter/enhancer are directly modulated by Wnt and β-catenin. We found that β-catenin overexpression relieves repression by Tcf in B16 cells, consistent with other Wnt-responsive genes (Bienz 1998; Eastman and Grosschedl 1999). By mutation of Tcf/Lef binding sites, it is possible to distinguish whether target gene expression is modulated by direct activation or by relief of repression. For example, mutation of the siamois promoter results in a derepression of reporter expression in the Xenopus embryo (Brannon et al. 1997). We propose that Tcf/Lef sites in the nacre promoter/enhancer are required for the activation of transcription, because mutations result in the loss of expression in vivo.
A seemingly contradictory result is the difference in reporter activity in B16 melanoma cells, where our mutant construct was highly active. These data could lead to the conclusion that Tcf/Lef sites are required for activation in one case and repression in the other. However, we believe that these experiments illustrate the fundamental differences between a committed melanocyte lineage and unspecified neural crest cells. Because B16 cells are derived from tissue that has become committed to produce melanocytes, they already express high levels of MITF (Bertolotto et al. 1996; Mahalingam et al. 1997). In contrast, unspecified neural crest cells in vivo are still multipotent and are not likely to express tissue-specific factors found in melanoma lines. It is therefore probable that initial activation of nacre transcription in the embryo requires Wnt/β-catenin signaling as well as other factors, whereas this activation has already taken place in B16 cells. Furthermore, in both systems we can show that the nacre promoter/enhancer responds to Wnt signals in a manner dependent on Tcf/Lef binding sites.
Although Wnt signaling is present in many places during embryonic development, the nacre gene is activated specifically in melanophores and retinal pigmented epithelium, suggesting that other tissue-specific elements regulate its expression. Consistent with this idea, we found that the nac>luc construct is inactive in other, nonmelanocyte cell lines, even when cotransfected with β-catenin and/or Lef1 (not shown). Similar integration of signal and tissue specificity was observed with the human MITF promoter, through response to cAMP (Bertolotto et al. 1998a) and transactivation by PAX3 (Watanabe et al. 1998). Our data suggest that integration of multiple signals such as cAMP, pax3, and Wnt could serve to limit nacre expression to a subset of neural crest in zebrafish.
Taken with our previous work demonstrating that Wnt signaling is necessary and sufficient for the pigment cell fate (Dorsky et al. 1998), the present study extends our knowledge of cell fate specification by identifying a key regulatory gene that is directly activated by localized Wnt signals. The position of nacre/MITF at the top of a hierarchy of pigment cell specification genes (Tachibana et al. 1996) and the function of Nacre/MITF protein as a transcription factor (Yasumoto et al. 1994) allow a direct link to be drawn between cell–cell signaling and fate determination. Interestingly, although Wnt signaling promotes multiple pigment cell fates, including xanthophores (Dorsky et al. 1998), nacre function is not required for xanthophore development (Lister et al. 1999). Additional target genes besides nacre may therefore be downstream of Wnt signaling, resulting in the promotion of other pigment cell types. As a general mechanism, the regulation of nacre by Wnt signaling may serve as a model demonstrating how environmental signals promote cell type diversity during development.
Materials and methods
Cloning of nacre promoter/enhancer
A probe comprising the first 271 nucleotides of the nacre cDNA was used to screen a zebrafish genomic phage library (Strategene) using standard methods. Positive clones were purified and characterized by Southern blot analysis. A 2.1-kb fragment was subcloned and sequenced and found to contain the 5′ end of the nacre cDNA. A 836-bp fragment including the first 10 amino acids of nacre was fused to GFP, and fragments ending in the nacre 5′-untranslated region were fused to luciferase (pGL3, Promega) and nacre cDNAs. The sequence for the nacre promoter/enhancer has been submitted to GenBank under accession no. AF211890.
DNA microinjections and EMSAS
Embryos were obtained from wild-type lines and a viable stock homozygous for a null mutation in nacre (Lister et al. 1999). DNA constructs were diluted to 100 ng/μl and were injected into one-cell zebrafish embryos using standard methods. Approximately 0.25–1 nl of DNA was injected into each embryo.
DNA probes were gel purified and end-labeled by standard procedures using Klenow DNA polymerase. Zebrafish Lef1 protein (Dorsky et al. 1999) was produced using the TNT SP6 reticulocyte lysate kit (Promega). Mobility shift assays and competition analyses were performed as described previously (Brannon et al. 1997).
Mutagenesis and Tcf/Lef site replacement
nacre promoter/enhancer constructs were mutated using the GeneEditor mutagenesis kit (Promega). Mutagenic oligonucleotides were 28–32 bases in length and contained the mismatched bases shown in Figure 1. All mutations introduced new restriction sites. Tcf/Lef site replacement oligonucleotides were designed using the sequence of TOPflash (van de Wetering et al. 1997) and included ends compatible with a unique SpeI site in the nacre promoter/enhancer. Oligonucleotides were annealed, kinased, and ligated into a mutant construct digested with SpeI and dephosphorylated. This ligation destroyed the SpeI site and created a new DraIII site. All clones were confirmed by sequencing.
B16 cell transfections and luciferase assays
B16 cells (gift from Dr. Vincent Hearing, NCI, Bethesda, MD) were grown in DMEM + 10% FBS. Cells were plated to a density of 1 × 105/well and transfected with Lipofectamine Plus (GIBCO) following the manufacturer's instructions. After 24 hr, cells were lysed and assayed for luciferase activity with a Berthold Luminomiter. β-Galactosidase activity was measured using Galactolite (Tropix).
Acknowledgments
We thank Vincent Hearing for providing B16 cells, James Lister for providing the nacre cDNA and embryos, Marian Waterman for providing Lef1 protein and antibodies, Jeffrey Miller for assistance with Western blots, members of the Nathanson and McKnight laboratories for assistance with luciferase assays, and Julie Kiefer and David Kimelman for comments on the manuscript. R.I.D. acknowledges support as an Associate, and R.T.M. as an Investigator of the HHMI. This research was funded by the HHMI (R.I.D and R.T.M) and the NIH (D.W.R.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rtmoon@u.washington.edu; FAX (206) 543-0858.
References
- Bertolotto C, Bille K, Ortonne JP, Ballotti R. Regulation of tyrosinase gene expression by cAMP in B16 melanoma cells involves two CATGTG motifs surrounding the TATA box: Implication of the microphthalmia gene product. J Cell Biol. 1996;134:747–755. doi: 10.1083/jcb.134.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotto C, Abbe P, Hemesath TJ, Bille K, Fisher DE, Ortonne JP, Ballotti R. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J Cell Biol. 1998a;142:827–835. doi: 10.1083/jcb.142.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotto C, Busca R, Abbe P, Bille K, Aberdam E, Ortonne JP, Ballotti R. Different cis-acting elements are involved in the regulation of TRP1 and TRP2 promoter activities by cyclic AMP: Pivotal role of M boxes (GTCATGTGCT) and of microphthalmia. Mol Cell Biol. 1998b;18:694–702. doi: 10.1128/mcb.18.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz M. TCF: Transcriptional activator or repressor? Curr Opin Cell Biol. 1998;10:366–372. doi: 10.1016/s0955-0674(98)80013-6. [DOI] [PubMed] [Google Scholar]
- Brannon M, Gomperts M, Sumoy L, Moon RT, Kimelman D. A β-catenin/XTcf-3 complex binds to the siamois promoter to regulate dorsal axis specification in Xenopus. Genes & Dev. 1997;11:2359–2370. doi: 10.1101/gad.11.18.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannon M, Brown JD, Bates R, Kimelman D, Moon RT. XCtBP is a XTcf-3 co-repressor with roles throughout Xenopus development. Development. 1999;126:3159–3170. doi: 10.1242/dev.126.14.3159. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R. Wnt signaling: A common theme in animal development. Genes & Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Moon RT, Raible DW. Control of neural crest cell fate by the Wnt signalling pathway. Nature. 1998;396:370–373. doi: 10.1038/24620. [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Snyder A, Cretekos CJ, Grunwald DJ, Geisler R, Haffter P, Moon RT, Raible DW. Maternal and embryonic expression of zebrafish Lef1. Mech Dev. 1999;86:147–150. doi: 10.1016/s0925-4773(99)00101-x. [DOI] [PubMed] [Google Scholar]
- Eastman Q, Grosschedl R. Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr Opin Cell Biol. 1999;11:233–240. doi: 10.1016/s0955-0674(99)80031-3. [DOI] [PubMed] [Google Scholar]
- Fuse N, Yasumoto K, Suzuki H, Takahashi K, Shibahara S. Identification of a melanocyte-type promoter of the microphthalmia-associated transcription factor gene. Biochem Biophys Res Commun. 1996;219:702–707. doi: 10.1006/bbrc.1996.0298. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Lister JA, Robertson CP, Lepage T, Johnson SL, Raible DW. nacre encodes a zebrafish microphthalmia-related protein that regulates neural-crest-derived pigment cell fate. Development. 1999;126:3757–3767. doi: 10.1242/dev.126.17.3757. [DOI] [PubMed] [Google Scholar]
- Mahalingam H, Watanabe A, Tachibana M, Niles RM. Characterization of density-dependent regulation of the tyrosinase gene promoter: role of protein kinase C. Exp Cell Res. 1997;237:83–92. doi: 10.1006/excr.1997.3773. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson Maduro J, Godsave S, Korinek V, Roose J, Destr'ee O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Opdecamp K, Nakayama A, Nguyen MT, Hodgkinson CA, Pavan WJ, Arnheiter H. Melanocyte development in vivo and in neural crest cell cultures: crucial dependence on the Mitf basic-helix-loop-helix-zipper transcription factor. Development. 1997;124:2377–2386. doi: 10.1242/dev.124.12.2377. [DOI] [PubMed] [Google Scholar]
- Riese J, Yu X, Munnerlyn A, Eresh S, Hsu SC, Grosschedl R, Bienz M. LEF-1, a nuclear factor coordinating signaling inputs from wingless and decapentaplegic. Cell. 1997;88:777–787. doi: 10.1016/s0092-8674(00)81924-8. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Takeda K, Nobukuni Y, Urabe K, Long JE, Meyers KA, Aaronson SA, Miki T. Ectopic expression of MITF, a gene for Waardenburg syndrome type 2, converts fibroblasts to cells with melanocyte characteristics. Nat Genet. 1996;14:50–54. doi: 10.1038/ng0996-50. [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Cavallo R, Dooijes D, van Beest M, van Es J, Loureiro J, Ypma A, Hursh D, Jones T, Bejsovec A, et al. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. 1997;88:789–799. doi: 10.1016/s0092-8674(00)81925-x. [DOI] [PubMed] [Google Scholar]
- Watanabe A, Takeda K, Ploplis B, Tachibana M. Epistatic relationship between Waardenburg syndrome genes MITF and PAX3. Nat Genet. 1998;18:283–286. doi: 10.1038/ng0398-283. [DOI] [PubMed] [Google Scholar]
- Waterman ML, Fischer WH, Jones KA. A thymus-specific member of the HMG protein family regulates the human T cell receptor C α enhancer. Genes & Dev. 1991;5:656–669. doi: 10.1101/gad.5.4.656. [DOI] [PubMed] [Google Scholar]
- Yasumoto K, Yokoyama K, Shibata K, Tomita Y, Shibahara S. Microphthalmia-associated transcription factor as a regulator for melanocyte-specific transcription of the human tyrosinase gene. Mol Cell Biol. 1994;14:8058–8070. doi: 10.1128/mcb.14.12.8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasumoto K, Yokoyama K, Takahashi K, Tomita Y, Shibahara S. Functional analysis of microphthalmia-associated transcription factor in pigment cell-specific transcription of the human tyrosinase family genes. J Biol Chem. 1997;272:503–509. doi: 10.1074/jbc.272.1.503. [DOI] [PubMed] [Google Scholar]