

Abstract
A number of pathogenic and proinflammatory stimuli, and the transforming growth factor-β (TGF-β) exert opposing activities in cellular and immune responses. Here we show that the RelA subunit of nuclear factor κB (NF-κB/RelA) is necessary for the inhibition of TGF-β-induced phosphorylation, nuclear translocation, and DNA binding of SMAD signaling complexes by tumor necrosis factor-α (TNF-α). The antagonism is mediated through up-regulation of Smad7 synthesis and induction of stable associations between ligand-activated TGF-β receptors and inhibitory Smad7. Down-regulation of endogenous Smad7 by expression of antisense mRNA releases TGF-β/SMAD-induced transcriptional responses from suppression by cytokine-activated NF-κB/RelA. Following stimulation with bacterial lipopolysaccharide (LPS), or the proinflammatory cytokines TNF-α and interleukin-1β (IL-1β, NF-κB/RelA induces Smad7 synthesis through activation of Smad7 gene transcription. These results suggest a mechanism of suppression of TGF-β/SMAD signaling by opposing stimuli mediated through the activation of inhibitory Smad7 by NF-κB/RelA.
Keywords: NF-κB, SMADs, TGF-β, signal transduction, cytokines
The survival of organisms in complex environments depends on their capability to adapt effectively to a diverse spectrum of injury and infection. The IκB/NF-κB family of proteins constitutes an evolutionarily conserved and ubiquitous signaling pathway that functions as a coordinating element in the response of an organism to situations of stress, infection, and inflammation (Ghosh et al. 1998). Following this paradigm, inducers of NF-κB include a wide variety of pathogenic signals and proinflammatory cytokines (Baeuerle and Henkel 1994). Genes regulated by the inducible transcriptional activator NF-κB include cell adhesion molecules, cytokines and hematopoietic growth factors, acute phase proteins, transcription factors, and viral genes. The most abundant form of NF-κB is a heterodimer of p50 and p65/RelA subunits, in which the p65/RelA subunit contains the transcriptional activator domain. In the absence of inducers of NF-κB, NF-κB heterodimers remain in the cytoplasm bound to inhibitory proteins known as inhibitors of κB (IκBs). Exposure of cells to stimuli of NF-κB activates signaling pathways leading to the phosphorylation, ubiquitination, and degradation of IκB proteins. The released NF-κB translocates to the nucleus and induces gene expression of NF-κB-responsive genes (Baldwin, Jr. 1996).
Transforming growth factor-β (TGF-β) is the prototype of a cytokine superfamily that controls cell fates, including cell cycle arrest, differentiation, and apoptosis. TGF-β initiates signaling through the ligand-dependent activation of a complex of heteromeric transmembrane serine/threonine kinases, consisting of type I (RI) and type II (RII) receptors (Wrana et al. 1994; Derynck and Feng 1997). Activated RI phosphorylate and thus activate Smad2 and/or Smad3, two signaling mediators of the SMAD protein family (Heldin et al. 1997; Hoodless and Wrana 1998; Kretzschmar and Massagué 1998; Nakao et al. 1997b). Following phosphorylation at a carboxy-terminal SSXS motif (Macias-Silva et al. 1996; Liu et al. 1997), Smad2 and Smad3 associate with the shared partner Smad4 and translocate to the nucleus in which SMAD protein complexes participate in transcriptional activation of target genes (Derynck et al. 1998).
The TGF-β/SMAD signaling system is notable for an autoinhibitory feedback loop, which involves Smad7, a structurally and functionally divergent SMAD protein of the subfamily of inhibitory SMADs (Nakao et al. 1997a). In contrast to the so-called substrate SMADs, which are generally regulated by post-translational modifications, the expression of the Smad7 gene is induced by TGF-β itself (Nakao et al. 1997), and by fluid shear stress acting on endothelial cells (Topper et al. 1997). Smad7 binds to the ligand-activated RI and interferes with the phosphorylation of substrate SMADs (Hayashi et al. 1997; Nakao et al. 1997a).
A number of observations document unique and essential roles for TGF-β in regulating inflammatory and adaptive immune responses, which on balance, suggest an antiinflammatory and immunosuppressive role for TGF-β (for review, see Letterio and Roberts 1997). In particular, TGF-β antagonizes the activation of important target genes of proinflammatory stimuli of NF-κB in macrophages and lymphocytes, such as inducible nitric oxide synthetase (iNOS) and major histocompatibility complex (MHC) class I and class II antigens (Geiser et al. 1993; Vodovotz et al. 1996). Conversely, several stimuli of NF-κB inhibit activities of TGF-β in matrix synthesis, inflammation, apoptosis, and hematopoiesis (Oberhammer et al. 1992; Snoeck et al. 1996; Inagaki et al. 1995). It is therefore likely that the interplay of opposing NF-κB and TGF-β signaling pathways is key to a coordinated cellular response dependent on physiological context. However, the molecular mechanisms of signaling cross talk between these two pathways remain obscure.
In this report, we demonstrate that transmodulation between opposing pathways and the TGF-β/SMAD signaling pathway is mediated through NF-κB-dependent activation of the inhibitory Smad7. We show that the NF-κB subunit p65/RelA is required for transcriptional activation of Smad7 by bacterial lipopolysaccharides (LPS), and the proinflammatory cytokines interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α). TNF-α/NF-κB induced Smad7 suppresses TGF-β/SMAD signaling through its direct interaction with the RI upon TGF-β ligand-receptor binding. The increased occupancy of activated TGF-β receptor complexes with Smad7 correlates with suppressed phosphorylation, nuclear translocation, and DNA binding of substrate SMAD-signaling complexes. Finally, we show that down-modulation of endogenous Smad7 with antisense mRNA releases the suppression of TGF-β/SMAD-mediated transcriptional responses by TNF-α activated NF-κB/RelA. These results suggest a mechanism for subversion of opposing activities of TGF-β by diverse stimuli of the NF-κB/RelA pathway.
Results
Activation of NF-κB/RelA inhibits TGF-β/SMAD-dependent signaling
To examine whether activation of the ubiquitous NF-κB/RelA signaling pathway inhibits TGF-β/SMAD-mediated signaling and transcriptional regulation, we used a genetically defined system of NF-κB/RelA-deficient (RelA−/−) and wild-type (RelA+/+) mouse fibroblast cell lines derived from RelA-deficient mice and wild-type littermates (Beg et al. 1995). We confirmed that RelA−/− fibroblasts used in our studies were deficient in both RelA mRNA and protein by Northern blot and immunofluorescence analysis (data not shown). The proinflammatory cytokine TNF-α is a well-characterized activator of NF-κB (Ghosh et al. 1998) with opposite effects to TGF-β on a number of target genes (Baldwin 1996). We sought to determine whether NF-κB/RelA, when activated by TNF-α, was able to interfere with TGF-β/SMAD signaling by preventing nuclear translocation of endogenous Smad2. Nuclear translocation is a hallmark of activation of Smad2 and/or Smad3 in the TGF-β/SMAD signaling cascade (Heldin et al. 1997). Most untreated RelA+/+ and RelA−/− fibroblasts showed cytoplasmic and nuclear staining for Smad2, and only few cells (14% and 26%, respectively) showed exclusively nuclear staining (Fig. 1A, Control). After 30 min of exposure to TGF-β1 alone, exclusively nuclear staining of Smad2 was detectable in 79% and 96% of cells, respectively (Fig. 1A, TGF-β). In contrast, incubation with TNF-α alone had no significant effect on subcellular localization of Smad2 (Fig. 1A, TNF-α). Addition of TNF-α 30 min prior to TGF-β suppressed the TGF-β-induced nuclear translocation of Smad2 in RelA+/+ cells, but not in RelA−/− cells (Fig. 1A, TNF-α TGF-β; 28% and 92% of cells showed exclusively nuclear staining, respectively).
Figure 1.
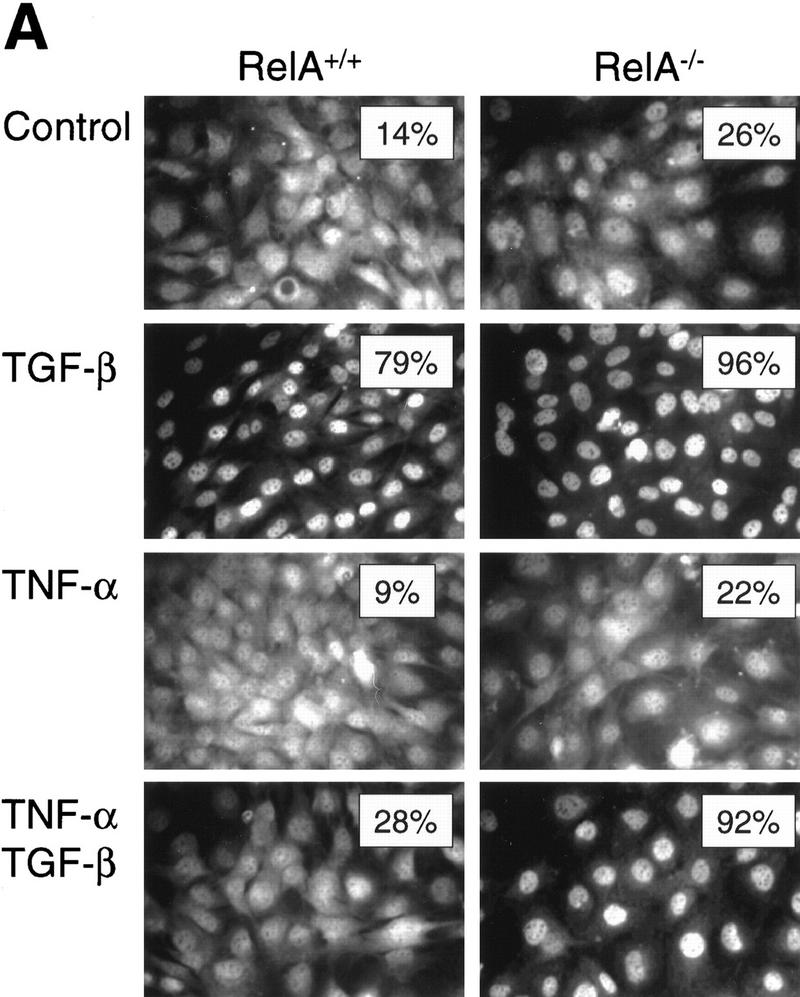


The RelA subunit of NF-κB is required for inhibition of TGF-β/SMAD signaling by TNF-α. (A) Smad2 immunofluorescence in RelA+/+ and RelA−/− fibroblasts left untreated (Control), or treated with either 0.1 ng/ml TGF-β, 0.2 ng/ml TNF-α, or both (TNF-α and TGF-β). (Insets) The percentage of cells with exclusively nuclear staining for Smad2. (B) EMSA with a radiolabeled oligonucleotide probe containing a consensus SMAD protein binding site (Zawel et al. 1998). Nuclear protein extracts derived from both RelA+/+ (lane 2) and RelA−/− (lane 9) fibroblasts, treated with TGF-β, contained four distinct DNA-binding protein complexes marked as a, b, c, and d. Pretreatment of cells with TNF-α prior to TGF-β-treatment prevents formation of DNA-binding protein complexes in RelA+/+ (lane 4), but not in RelA−/− fibroblasts (lane 11). Antibody interference analysis showing the effect of preincubation of nuclear extracts from TGF-β-treated RelA+/+ fibroblasts with nonimmune IgG (lane 5), anti-Smad4 (lane 6), anti-Smad3 (lane 7), and anti-Smad2 (lane 8). (Lane 7) a*, b* denotes supershifted complex(es). (C) Transient transcriptional response assays with the TGF-β-responsive reporter constructs 3TPLux (histogram a), or SBE4-Luc (histogram b) (Zawel et al. 1998). RelA+/+ (a and b, left) and RelA−/− fibroblasts (a and b, right) were treated as indicated 20 hr after transfection. Luciferase activities were expressed as ratios (Fold-induction) of normalized luciferase activities in treated cells and untreated cells. Histograms represent mean ±S.D. of at least three experiments.
Next, we examined whether TNF-α suppressed the TGF-β-induced DNA binding of SMAD-transcriptional activator complexes. Complementary oligonucleotides containing a palindromic GTCTAGAC sequence motif, were reported previously to bind recombinant Smad3 and Smad4 protein complexes (Zawel et al. 1998). By use of this probe, EMSAs revealed four distinct and specific DNA-binding protein complexes, labeled a, b, c, and d, in nuclear protein extracts prepared from RelA+/+ fibroblasts after treatment with TGF-β for 90 min (Fig. 1B, lane 2). These nuclear DNA-binding protein complexes were not detected in nuclear extracts prepared from untreated cells (Fig. 1B, lane 1), or from cells incubated with TNF-α alone (Fig. 1B, lane 3), or from cells treated with TNF-α for 30 min prior to the addition of TGF-β (Fig. 1B, lane 4). To verify whether SMAD proteins were present in complexes a, b, c, or d, we incubated nuclear protein extracts derived from TGF-β-treated RelA+/+ fibroblasts with nonimmune control IgG, anti-Smad4, anti-Smad3, or anti-Smad2 antibodies prior to the addition of radiolabeled oligonucleotide probe. Control IgG had no effect on DNA–protein complex formation (Fig. 1B, lane 5). Preincubation with anti-Smad4 antibody interfered with complexes a, b, and d (Fig. 1B, lane 6), whereas the anti-Smad3 antibody interfered with binding of complexes a and b, and resulted in the appearance of a higher molecular weight complex(es), labeled a*, b* (Fig. 1B, lane 7). The anti-Smad2 antibody interfered with DNA binding of complex a. The identity of nuclear proteins contained in complex c remains to be determined. All three antibodies specifically detected their cognate Smad antigen in Western blot analysis (data not shown). Together, our antibody interference experiments in RelA+/+ nuclear protein extracts suggest that DNA-binding protein complexes containing either Smad2 and/or Smad2 plus Smad4 (complex a), Smad3 and Smad4 (complex b), or Smad4 and an unidentified protein(s) (complex d) specifically assemble on a SMAD consensus binding element following treatment of cells with TGF-β. However, the presence of TNF-α in the culture medium inhibits completely the DNA-binding activity of SMAD proteins induced by TGF-β. Whereas Zawel et al. (1998) reported binding of recombinant Smad3 and/or Smad4, but not Smad2 to the SBE-containing probe (Zawel et al. 1998), our studies demonstrated binding of Smad2-containing complexes (Fig. 1B). This difference is most likely attributable to the use of whole nuclear protein extracts in our gel-shift experiments, as opposed to recombinant, purified SMAD proteins in the studies by Zawel et al. (1998). In fact, it has been well documented that Smad2 requires the presence of cofactors such as Fast-1 for efficient DNA binding (Chen et al. 1996).
To determine whether the inhibitory activity of TNF-α on activation of SMAD-DNA binding in response to TGF-β is mediated by NF-κB/RelA, we conducted similar experiments as described in the previous paragraph, but using nuclear extracts prepared from RelA−/− fibroblasts. TGF-β-induced formation of DNA-binding protein complexes a, b, c, and d in RelA−/− cells, although complex a appeared weaker than in RelA+/+ fibroblasts. However, TNF-α pretreatment in RelA−/− cells did not suppress the DNA-binding activities of SMAD protein complexes induced by TGF-β (Fig. 1B, cf. lanes 9 and 11). Our results demonstrate that activation of NF-κB/RelA by TNF-α inhibits the DNA binding of SMAD proteins normally induced by TGF-β in these murine fibroblasts.
Next, we sought to determine whether the NF-κB-dependent inhibition of nuclear translocation and DNA binding of TGF-β-activated SMAD protein complexes altered transcriptional responses normally induced by TGF-β/SMAD. TGF-β treatment significantly (P<0.05) increased the luciferase activity of the TGF-β-responsive reporter 3TPLux by 3.0-fold and 3.2-fold in RelA+/+ and RelA−/− fibroblasts, respectively (Fig. 1C; histogram a). However, addition of TNF-α 45 min prior to TGF-β significantly (P<0.05, compared with TGF-β alone) suppressed the induction of 3TPLux activity by TGF-β only in RelA+/+ (1.4-fold induction), but not in RelA−/− (2.5-fold induction) fibroblasts (Fig. 1C). Similar results were obtained in analogous experiments with the TGF-β-responsive transcriptional reporter SBE4-Luc (Zawel et al. 1998; Fig. 1C, histogram b). Taken together, these results suggest that TNF-α antagonizes TGF-β/SMAD transcriptional responses at an extranuclear step in the TGF-β/SMAD signaling cascade, and that this transmodulation requires activation of NF-κB/RelA.
NF-κB/RelA activates the transcription of inhibitory Smad7
Smad7 was first identified as a gene that is activated by fluid shear stress in endothelial cells (Topper et al. 1997), and by TGF-β itself (Nakao et al. 1997a). It has been proposed that Smad7 interacts stably with the ligand-activated RI, thereby preventing binding and phosphorylation/activation of substrate Smad2 and/or Smad3 by the receptor (Hayashi et al. 1997; Nakao et al. 1997a). We reasoned that Smad7 may play a role in the NF-κB/RelA-dependent suppression of TGF-β/SMAD signaling by TNF-α. Treatment with TNF-α increased steady-state mRNA levels of Smad7 in RelA+/+ fibroblasts, but not in RelA−/− fibroblasts (Fig. 2A). Induction was seen as early as 30 min and peaked between 30 and 180 min of exposure. In contrast, TGF-β treatment increased Smad7 mRNA levels irrespective of the RelA genotype (Fig. 2A). These results suggest that NF-κB/RelA is required for up-regulation of Smad7 steady-state mRNA levels by TNF-α, but not by TGF-β. The induction of Smad7 mRNA expression by TNF-α was not restricted to RelA+/+ fibroblasts, but also detected in NIH-3T3 fibroblasts, mink lung epithelial cells (Mv1Lu), and rat hepatic stellate cells (HSC) (Fig. 2B).
Figure 2.


RelA is required for transcriptional activation of Smad7 by TNF-α. (A) Northern blot analysis of Smad7 transcript levels in RelA+/+ and RelA−/− fibroblasts treated with TNF-α (10 ng/ml) or TGF-β (1 ng/ml) for the indicated time periods. The same blots were probed with GAPDH to control for RNA loading. (B) Smad7 mRNA in murine fibroblasts (NIH-3T3), Mv1Lu, and HSC. Cells were exposed to TNF-α for 1 hr. 18S rRNA was probed to control for loading. (C) RelA+/+ and RelA−/− fibroblasts were incubated with TNF-α (10 ng/ml), IL-1β (1 ng/ml), LPS (10 μg/ml), and IFN-γ (250 U/ml) for 1 hr, respectively. (D) RelA+/+ fibroblasts were incubated with TNF-α (10 ng/ml) for 1 hr in the absence or presence of various compounds including cycloheximide (10 μg/ml), SB203580 (10 μm), U0126 (10 μm), actinomycin D (10 μg/ml), or DMSO alone as control (28S rRNA as loading control).
To examine whether the essential role for NF-κB/RelA in the induction of Smad7 is restricted to stimulation by TNF-α, or whether NF-κB-dependent up-regulation of Smad7 was inducible by a variety of pathogenic and proinflammatory stimuli, we treated RelA+/+ and RelA−/− fibroblasts with the well-characterized activators of NF-κB/RelA, IL-1β and LPS derived from gram-negative Escherichia coli (Muller et al. 1993). In addition, we used interferon-γ, an activator of the Jak/Stat pathway that has recently been shown to induce Smad7 steady-state mRNA expression (Ulloa et al. 1999). TNF-α, IL-1β, and LPS all increased Smad7 mRNA expression in RelA+/+ fibroblasts (Fig. 2C, lanes 2–4). In contrast, TNF-α and LPS had no effect on Smad7 mRNA expression in RelA−/− fibroblasts, and the induction by IL-1β was significantly reduced when compared with RelA+/+ fibroblasts (Fig. 2C, lanes 7–9). However, IFN-γ had no significant effect on Smad7 mRNA levels irrespective of the RelA genotype in these IFN-γ-responsive fibroblasts (Fig. 2C, lanes 5 and 10, respectively). These data demonstrate that a variety of pathogenic (LPS) and proinflammatory (TNF-α and IL-1β) stimuli up-regulate Smad7 mRNA expression through activation of NF-κB/RelA. Immunofluorescence labeling studies with an anti-p65/RelA antibody showed that treatment of RelA+/+ cells with either TNF-α, IL-1β, or LPS resulted in nuclear translocation of p65/RelA, confirming activation of NF-κB/RelA by these stimuli (data not shown).
Next, we treated RelA+/+ fibroblasts with TNF-α in the presence of either cycloheximide, the p38 inhibitor SB203580, the MEK1 inhibitor U0126, or the RNA-synthesis inhibitor actinomycin D. Preincubation with cycloheximide for 30 min prior to addition of TNF-α did not prevent the up-regulation of Smad7 mRNA expression when compared with cells exposed to TNF-α alone (Fig. 2D, lanes 2,3), indicating that up-regulation of Smad7 expression by TNF-α did not require de novo protein synthesis. The solvent DMSO alone had no effect on Smad7 expression (Fig. 2D, lane 4). Similarly, preincubation of RelA+/+ cells with the p38 inhibitor SB203580, or the MEK1 inhibitor U0126 had no effect on the induction of Smad7 mRNA expression by TNF-α (Fig. 2D, lanes 6 and 7, respectively). In contrast, the TNF-α effect was completely blocked by pretreatment with actinomycin D prior to TNF-α. Together, these results suggest that TNF-α up-regulates transcription of the Smad7 gene through activation of NF-κB/RelA. In addition, we have recently identified a functional NF-κB/RelA responsive element located upstream from the transcription initiation site in the 5′-flanking region of the human Smad7 gene, providing further evidence that Smad7 is a novel target gene of NF-κB/RelA (G. von Gersdorff and E. Böttinger, unpubl.).
NF-κB/RelA induces Smad7 to interfere with TGF-β type I receptor signaling
It has been proposed that Smad7 inhibits TGF-β signaling through its stable and direct interaction with the RI upon TGF-β ligand-receptor binding, thereby preventing the association of this essential signaling receptor with substrate SMAD proteins (Hayashi et al. 1997; Liu et al. 1997). To determine whether Smad7 binding to TGF-β receptor complexes is enhanced by TNF-α, we used [125I]TGF-β1 to label and activate the TGF-β receptor complexes. The amounts of receptors bound to Smad7 were assessed by coimmunoprecipitation with anti-Smad7 antibodies. An increased number of multimeric TGF-β ligand-receptor complexes, consisting of RI, RII, and betaglycan (RIII), were bound to Smad7 in RelA+/+ fibroblasts pretreated with TNF-α, when compared with untreated cells (Fig. 3A, lanes 1,2). In contrast, TNF-α treatment did not increase the amount of Smad7-associated TGF-β receptor complexes in RelA−/− fibroblasts (Fig. 3B, lanes 1,2). Using gel electrophoresis of whole cell lysates from these experiments, we determined that TNF-α pretreatment did not significantly affect the expression and [125I]TGF-β1 ligand binding of cell surface RI, RII, and RIII in either RelA+/+, or RelA−/− fibroblasts (Fig. 3A and B, lanes 3 and 4, respectively). Western blotting with an anti-Smad7 antibody revealed that TNF-α treatment increased the level of endogenous Smad7 in RelA+/+ fibroblasts, but not in RelA−/− fibroblasts (Fig. 3C).
Figure 3.


TNF-α induces the association of inhibitory Smad7 and TGF-β receptor complexes, and inhibits TGF-β-induced phosphorylation of substrate SMADs. (A) Coimmunoprecipitation of affinity-labeled TGF-β receptors with anti-Smad7 antibody from whole-cell lysates of untreated (lane 1), or TNF-α-treated RelA+/+ fibroblasts (lane 2). Abundance of affinity-labeled TGF-β receptor complexes in the same lysates run on SDS–polyacrylamide gels (7.5%) prior to immunoprecipitation (lanes 3,4). RI, RII, and RIII denote TGF-β type I receptors, type II receptors, and TGF-β-binding protein betaglycan affinity labeled with [125I]TGF-β1, respectively. (B) Same experiment as described in A was performed with RelA−/− fibroblasts and samples were run on 10% SDS–polyacrylamide gels. (C) Western blot analysis of aliquots of affinity-labeled cell lysates showing Smad7 protein levels in untreated and TNF-α-treated RelA+/+ or RelA−/− fibroblasts. The same blot was probed for GDP dissociation inhibitor (GDI) to control for equal protein loading. (D) Immunoprecipitation of endogenous phospho-Smad2 [32P] Smad2 in metabolically labeled RelA+/+ fibroblasts following cytokine treatments as indicated. Western blotting of total endogenous Smad2 protein in cell lysates prior to immunoprecipitation.
To test whether the enhanced association of Smad7 and TGF-β receptors that we observed following TNF-α/NF-κB stimulation was correlated with decreased substrate SMAD phosphorylation, we labeled RelA+/+ fibroblasts with [32P]orthophosphate and subjected cell lysates to immunoprecipitation with anti-Smad2 antibody. Low levels of phosphorylated Smad2 were detected in untreated and TNF-α-treated cells (Fig. 3D, lanes 1,2). TGF-β alone strongly induced the phosphorylation of Smad2 (Fig. 3D, lane 3). This effect was almost completely reversed in the presence of TNF-α (Fig. 3D, lane 4). Whole cell lysates were immunoblotted to demonstrate comparable Smad2 protein content in all samples (Fig. 3D). TGF-β-induced phosphorylation of Smad3 was also suppressed by pretreatment of RelA+/+ fibroblasts with TNF-α (data not shown).
Our results suggest that TNF-α raises the intracellular levels of Smad7 protein through NF-κB/RelA, and enhances the binding of Smad7 to TGF-β receptor complexes upon addition of TGF-β1. The enhanced interaction of Smad7 and TGF-β receptor complexes induced by TNF-α/NF-κB decreases phosphorylation of substrate Smad2 and/or Smad3, probably by interfering with the association of substrate SMADs and activated RI (Hayashi et al. 1997; Nakao et al. 1997a).
Smad7 mediates suppression of TGF-β/SMAD signaling by TNF-α
We reasoned that down-modulation of Smad7 expression would result in loss of inhibition of TGF-β/SMAD signaling by TNF-α. Because cells with genetic deletions of Smad7 are currently not available, we used an antisense mRNA approach. A full-length Smad7 cDNA was ligated in the reverse orientation into the pcDNA3 expression vector to generate the plasmid pSmad7AS, which expressed a full-length Smad7 antisense mRNA after transfection into cells (data not shown). We performed a series of cotransfection and immunofluorescence labeling experiments in RelA+/+ fibroblasts to verify that the Smad7 antisense mRNA specifically reduced expression of Smad7 protein, but not Smad2 or Smad3 expression (Fig. 4A,B,C). Endogenous Smad7 staining was absent in cells cotransfected with pSmad7AS, and pEGFP (enhanced green fluorescence protein) vector as a marker for transfected cells (Fig. 4A, a, b, c, and d). In contrast, all untransfected cells stained for Smad7. Smad7 antisense mRNA did not significantly reduce staining for Smad2 (Fig. 4A, e and f), and did not affect expression of Flag–Smad3 in RelA+/+ fibroblasts cotransfected with pFSmad3 and pSmad7AS (Fig. 4B). These data indicated that the Smad7 antisense mRNA specifically inhibits Smad7 protein synthesis. Furthermore, after transfection of COS cells with pF-Smad7 expression vector, we detected abundant Flag-tagged Smad7 protein by anti-Flag immunoblotting (Fig. 4C, lane 1), and coexpression of increasing amounts of Smad7 antisense vector pSmad7AS inhibited the synthesis of Flag–Smad7 protein (Fig. 4C, lanes 2,3).
Figure 4.
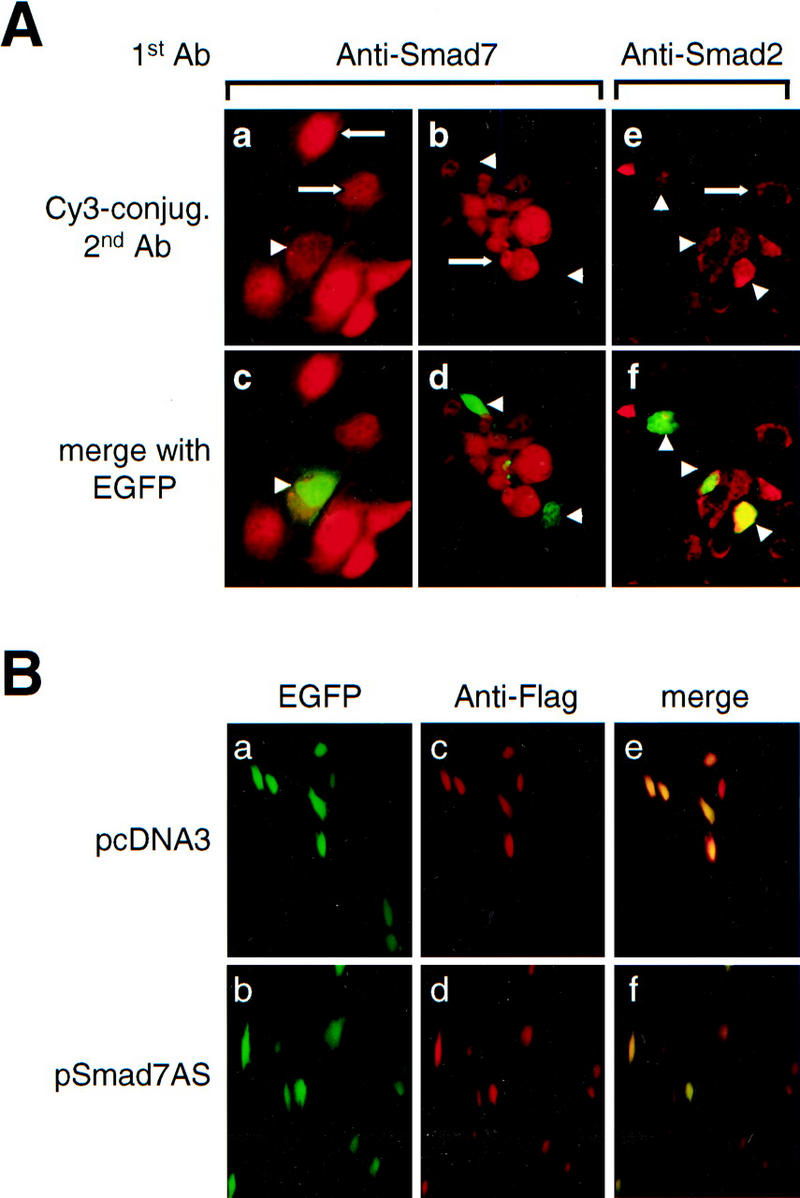

Down-modulation of Smad7 releases TGF-β/SMAD signaling from suppression by TNF-α. (A) Indirect immunofluorescence labeling with anti-Smad7 antibody (a,b,c,d) and anti-Smad2 antibody (e,f) in RelA+/+ fibroblasts cotransfected with constructs expressing Smad7 antisense mRNA (pSmad7AS) and pEGFP. (a,b,e) Red fluorescence signal for Cy3-conjugated secondary anti-rabbit (a,b) and anti-mouse IgG (e) only; (c,d,f) identical fields with red and green (GFP) fluorescence merged to identify transfected cells. (Arrows) Staining for endogenous Smad7 (a,b) and Smad2 (c) in untransfected cells. (Arrowheads) Transfected cells that are identified by green fluorescence (EGFP) and reveal decreased expression of Smad7 (a,b), but normal expression of Smad2 (e), respectively. (B) EGFP fluorescence (a,b), indirect immunofluorescence staining for anti-Flag (c,d), and merged fluorescence image (e,f) in RelA+/+ fibroblasts cotransfected either with Flag-tagged Smad3 (pFSmad3) and empty vector (pcDNA3) (a,c,e), or with Flag-tagged Smad3 (pFSmad3) and Smad7 antisense mRNA expression vector (pSmad7AS) (b,d,f). (C) Anti-Flag immunoblotting of Flag Smad7 in COS cells cotransfected with Flag-Smad7 plasmid (pF-Smad7) and empty vector DNA (pcDNA3) (lane 1), or with increasing amounts of Smad7 antisense plasmid (pSmad7AS) (lanes 2,3). (D) RelA+/+ fibroblasts were cotransfected with either 3TPLux reporter vector (histogram a), or SBE4 Luc reporter vector (histogram b), together with either empty control vector pcDNA3, or the same vector expressing Smad7 antisense mRNA (pSmad7AS) as indicated. Cells were incubated with or without TNF-α for 45 min prior to treatment with or without TGF-β1 for an additional 4 hr. Results are expressed as ratios (Fold-induction) of normalized luciferase activities in treated and untreated cells. Bars, mean ± s.d. of three independent experiments, respectively.
Next, we cotransfected RelA+/+ fibroblasts with the TGF-β responsive reporter constructs 3TPLux (histogram a), or SBE4-Luc (histogram b), together with either empty control vector pcDNA3, or the Smad7 antisense expression vector pSmad7AS (Fig. 4D). The transfected cell cultures were treated with TNF-α and/or TGF-β as indicated 24 hr after transfection. TGF-β1 significantly (P<0.05) induced 3TPLux and SBE4-Luc reporter activity by 2.1-fold and 1.9-fold, respectively, in cells cotransfected with the reporter and empty control vectors (Fig. 4D, a and b). When transfected cells were incubated with TNF-α for 45 min prior to treatment with TGF-β, the induction of 3TPLux (1.2-fold vs. 2.1-fold, respectively, P<0.05) and SBE4-Luc (1.1-fold vs. 1.9-fold, respectively, P<0.05) reporter constructs by TGF-β was significantly reduced (Fig. 4D, a and b). In contrast, when RelA+/+ cells were cotransfected with the reporter vectors and pSmad7AS, TNF-α pretreatment did not significantly inhibit the TGF-β-mediated induction of 3TPLux (2.3-fold vs. 2.8-fold, respectively) and SBE4-Luc (1.8-fold vs. 2.1-fold, respectively) reporter constructs (Fig. 4D, a and b). Our results suggest that the suppression of TGF-β/SMAD signaling by TNF-α requires Smad7 synthesis.
Discussion
We report a mechanism of suppression of TGF-β signaling by NF-κB/RelA-dependent pathways. Our results suggest that activation of NF-κB/RelA by a variety of pathogenic and proinflammatory stimuli increases transcription of the Smad7 gene and elevates intracellular levels of Smad7 protein. This effect promotes the association of inhibitory Smad7 and RI upon TGF-β stimulation, at the expense of receptor accessibility for substrate SMADs. The intracellular blockade of TGF-β receptor complexes by Smad7 inhibits phosphorylation, nuclear translocation, and DNA binding of SMAD transcriptional activator complexes, resulting in down-modulation of target gene activation by TGF-β. We propose that control of Smad7 transcription may provide a general mechanism that determines counteractive signaling thresholds for TGF-β, dependent on physiological requirements.
Repression of TGF-β receptor signaling by NF-κB/RelA
Antagonistic transmodulation between proinflammatory and immunostimulatory signals and TGF-β has been well documented, in particular in the context of tissue injury and tissue repair, apoptosis and immune homeostasis (Letterio and Roberts 1998). NF-κB is a pivotal mediator of inflammation that induces the acute phase response (Grilli et al. 1993), activates leukocytes, and enhances extravasation and migration of neutrophils and monocytes (Ghosh et al. 1998). It also triggers the local production of inflammatory cytokines and the up-regulation of MHC class I and II antigens to boost adaptive immune responses (Brown et al. 1994; Kishimoto et al. 1994; Ouaaz et al. 1999). Although context-dependent proinflammatory activities of TGF-β have been described (Wahl 1994), TGF-β generally exerts anti-inflammatory and immunosuppressive activities such as suppression of leukocyte extravasation (Lefer et al. 1993) and repression of inducible nitric oxide synthetase (Vodovotz et al. 1996), or repression of MHC class I and II expression (Geiser et al. 1993).
A complex pattern of transmodulation between TGF-β and NF-κB has been described in the control of programmed cell death, extracellular matrix synthesis, and hematopoiesis. NF-κB is required for cellular protection against apoptosis in the liver (Beg et al. 1995; Arsura et al. 1997), and suppresses TGF-β-induced apoptosis in hepatocytes (Oberhammer et al. 1992). Conversely, TGF-β can induce apoptosis in immature B-cells by suppressing NF-κB activity through up-regulation of its cytoplasmic inhibitor, IκB (Arsura et al. 1996). Interestingly, transcriptional activation of the human immunodeficiency virus 1 enhancer by TGF-β in keratinocytes and pre-B cells is mediated through activation of NF-κB in an IκB-independent pathway (Li et al. 1998). However, we show here that NF-κB is not required for the transcriptional activation of Smad7 by TGF-β. The induction of collagen type I synthesis by TGF-β is inhibited by TNF-α through a NF-κB-dependent mechanism (Inagaki et al. 1995; Chung et al. 1996), indicating opposing activities for these pathways in tissue remodeling. In experimental models of early hematopoiesis, TNF-α abrogates the inhibitory effect of ambient TGF-β to allow primitive stem cells to proliferate (Snoeck et al. 1996). Here, we report a mechanism by which activators of NF-κB may subvert TGF-β activities.
We show that activated NF-κB inhibits TGF-β signaling at the level of TGF-β type I receptor function through induction of synthesis and receptor binding of the inhibitory Smad7 protein. The following lines of evidence support our conclusions. First, exposure to a variety of pathogenic and proinflammatory activators of NF-κB/RelA increased Smad7 mRNA expression in several cell types including wild-type fibroblasts, but not in RelA-deficient fibroblasts. Interestingly, neither exposure to TNF-α, nor TGF-β altered Smad7 mRNA expression in endothelial cells (Topper et al. 1997), indicating that the regulation of Smad7 expression may be subject to cell-type-dependent constraints. Second, Smad7 protein levels are increased, and Smad7-bound TGF-β receptor complexes form in TNF-α-induced RelA wild-type, but not RelA-deficient cells. The inhibitory function of Smad7 in TGF-β signaling has been linked to its stable interaction with ligand-activated type I receptor, thus blocking the association and phosphorylation of its substrates, Smad2 and/or Smad3 (Hayashi et al. 1997; Nakao et al. 1997a). Complexes of Smad7 and activated type I receptors are stable, whereas interactions of substrate SMADs and activated type I receptor are transient (Hayashi et al. 1997a). Thus, increases in cellular Smad7 levels induced by NF-κB may result in effective suppression of type I receptor function in the event of increased cellular exposure to TGF-β ligand. In addition, it is intriguing to speculate that NF-κB-dependent signals may enhance the affinity of Smad7 for type I receptor binding and/or promote accumulation of Smad7 at clusters of activated TGF-β receptors (M. Bitzer and E. Böttinger, unpubl.). Third, we provide evidence that blockade of activated type I receptor with increased Smad7 is a critical event that results in inhibition of all subsequent steps in the TGF-β/SMAD signaling cascade. Thus, TNF-α activated NF-κB/RelA significantly suppressed the effects of TGF-β on phosphorylation and nuclear translocation of Smad2, on binding of Smad2/Smad4 and Smad3/Smad4 hetero-oligomeric nuclear complexes to a consensus Smad-binding sequence element, and on transcriptional activation of two TGF-β/SMAD-responsive luciferase reporter genes in wild-type fibroblasts. Our observations that the inhibitory activity of TNF-α on any of these steps of TGF-β/SMAD signal transduction is absent in RelA-deficient fibroblasts further support a central role for NF-κB/RelA. Fourth, down-modulation of Smad7 protein synthesis with antisense mRNA released the suppression of TGF-β/SMAD transcriptional responses by TNF-α, demonstrating that Smad7 is a downstream effector of the TNF-α/NF-κB pathway in this system.
A general role for Smad7 as molecular switch to suppress TGF-β activity
Smad7 was originally identified as shear stress-inducible gene in endothelial cells (Topper et al. 1997). Subsequently, it has been shown that Smad7 is expressed in many mammalian cells and tissues, and that its expression is induced by TGF-β itself, perhaps as mediator of an autoinhibitory feedback loop (Nakao et al. 1997a). A recent report demonstrates that inhibition of TGF-β/SMAD signaling by IFN-γ is mediated through increased binding of inhibitory Smad7 and TGF-β receptors, following induction of Smad7 expression by the Jak1/Stat1 pathway (Ulloa et al. 1999). Together, these observations, and the data presented in this report, suggest that Smad7 may function as a general negative regulator of TGF-β receptor signaling that is capable of mediating both autoinhibitory feedback signaling and down-modulation of TGF-β signaling strength by major opposing pathways including the Jak/Stat pathway and the NF-κB pathway. Experimental systems with a conditional targeted deletion of Smad7 and/or inducible expression of Smad7 will enable experimental validation of this model.
Interestingly, a fluid shear stress response element has been identified in the promoters of several shear stress-induced genes and consists of functional NF-κB and AP-1 transcription factor binding sites (Ballermann et al. 1998). Because the induction of Smad7 expression by cytokine-activated NF-κB/RelA (this work), and TGF-β and IFN-γ is mediated through transcriptional activation (G. von Gersdorff and E. Böttinger, unpubl.), it will be important to identify the corresponding cis-acting elements and trans-activating factors of the Smad7 gene. The 5′-flanking region of the Smad7 gene contains nonoverlapping consensus sequences for a putative Smad-binding element and multiple potential NF-κB and Stat1-binding sites. We have recently identified distinct transcriptional control elements that mediate activation of Smad7 gene transcription by TGF-β and NF-κB, respectively (G. von Gersdorff and E. Böttinger, unpubl.). Together, these observations support a model in which the Smad7 gene is activated through nonoverlapping signaling pathways and diverse transcriptional activator complexes.
Suppression of TGF-β receptor signaling function by inhibitory Smad7 may represent an effective and general mechanism to alter the balance between signaling pathways with opposing effects on a wide variety of target genes and complex cellular responses such as immune cell function and inflammation, and cell proliferation and programmed cell death. Thus, stimuli of the NF-κB pathway may subvert opposing TGF-β activities through this mechanism.
Materials and methods
Cell culture and materials
Fibroblast cell lines established from RelA-deficient (RelA−/−) and wild-type (RelA+/+) control mice were described previously (Beg et al. 1995; Ouaaz et al. 1999). Mv1Lu, COS, and NIH-3T3 cells were obtained from the American Type Culture Collection. HSC were described previously (Garcia-Trevijano et al. 1999). All cells were cultured in DMEM with 10% FBS and antibiotics. Cytokines and inhibitors were obtained from the following suppliers: recombinant human TGF-β1 and recombinant mouse IL-1β (R & D Systems); recombinant mouse TNF-α (Boehringer Mannheim); recombinant mouse IFN-γ (Genzyme, Cambridge, MA); actinomycin D, cycloheximide, and lipopolysaccharide E. coli 026:B6 (all from Sigma); p38 inhibitor SB203580 (Calbiochem, San Diego, CA); and MEK1 inhibitor U0126 (Promega, Madison, WI).
RNA analysis
RNA was isolated from cells with Trizol Reagent (GIBCO BRL, Gaithersburg, MD) following the manufacturer's protocol. For Northern blot analysis, RNA was electrophoresed on 1% agarose gels and transferred to a filter. Filters were then hybridized in QuickHyb solution (Stratagene, La Jolla, CA) with 32P-labeled probes for murine Smad7 cDNA generated from pF-Smad7, and analyzed by phosphorimagery. Glyceraldehyde-3-phosphate dehydrogenase, 18S rRNA, and 28S rRNA probes were used for normalization as indicated.
Immunofluorescence
Cells were treated with TGF-β1 and/or TNF-α as indicated and processed for immunofluorescence as described (Mundel et al. 1997). Endogenous Smad2 or the Flag epitope were visualized with monoclonal anti-Smad2 antibody (Transduction Laboratories, Lexington, Kentucky) or monoclonal anti-Flag M5 (Sigma, St. Louis, MO), and Cy3-conjugated anti-mouse immunoglobulin (Jackson Immuno Research, West Grove, PA). Slides were counterstained with DAPI to visualize cell nuclei. The percentage of cells with exclusively nuclear staining was evaluated counting at least 100 stained cells for each treatment group. Smad7 was detected with affinity-purified rabbit anti-Smad7 antibody (gift from Peter ten Dijke, Ludwig Cancer Research Institute, Uppsala, Sweden), and Cy3-conjugated anti-rabbit immunoglobulin (Jackson Immuno Research, West Grove, PA). To identify immunofluorescence signals in transiently transfected cells, we cotransfected various expression plasmids as indicated together with a plasmid that constitutively expresses pEGFP (Promega, Madison, WI).
Nuclear lysates
Nuclear lysates were prepared from 100-mm dishes. Cells were washed twice in cold PBS and lysed in 1 ml of ice-cold hypotonic lysis buffer (10 mm HEPES at pH 7.9, 10 mm KCl, 0.1 mm EDTA at pH 8.0, 0.1 mm EGTA, 1 mm DTT, 0.6% NP-40 containing AEBSF, leupeptin, aprotinin, pepstatinA, antipain, sodium vanadate, sodium fluoride, and okadaic acid at concentrations recommended by the manufacturers). The cells were allowed to swell for 15 min, then scraped, collected, and washed with hypotonic lysis buffer without detergent. Nuclei were pelleted by centrifugation at 13,000 rpms for 20 sec in a microcentrifuge and resuspended in 20μl of nuclear extraction buffer (lysis buffer with 20mm HEPES at pH 7.9 and 420mm NaCl). Lysates were incubated for 20 min on a shaker and cleared of debris by centrifugation.
EMSAs
EMSAs were performed with nuclear extracts prepared from either untreated cells, or cells treated with TNF-α for 120 min, TGF-β1 for 90 min, or both (TNF-α was added 30 min prior to TGF-β). Complementary oligonucleotides containing a Smad-binding consensus sequence (Zawel et al. 1998) were synthesized, 32P-end labeled with T4-DNA polynucleotide kinase and annealed. Oligonucleotide probes (50,000 cpm) were incubated with 10 μg of nuclear extracts in binding buffer (16% glycerol, 20 mm HEPES at pH 7.9, 0.1 mm EDTA, 30 mm KCl, 3 μg of polydI:dC, 0.8 mm NaPi at pH 7.8, 4 mm spermidine, 4 mm MgCl2) with or without preincubation for 10 min with 100-fold molar excess of cold competitor at 4°C for 30 min. For antibody interference studies (supershift analysis) of SMADs, nuclear extracts were incubated overnight at 4°C with 2 μg of anti-Smad2 (S-20-X), anti-Smad3 (I-20-X), or anti-Smad4 (C-20-X) antibodies (Santa Cruz Biotech, Santa Cruz, CA) prior to addition of radiolabeled oligonucleotide probe. DNA-binding protein complexes were separated by native 5% PAGE at 150 V at 4°C and visualized by autoradiography.
Affinity labeling and immunoprecipitation
Receptor affinity labeling and immunoprecipitation were performed essentially as described previously (Böttinger et al. 1997). In brief, porcine TGF-β1 was iodinated and used for labeling of cell surface binding proteins on cells in 100-mm culture dishes (Geiser et al. 1992). Aliquots of cell lysates containing equal amounts of protein were subjected to SDS-PAGE and complexes of [125I]TGF-β1 ligand and bound receptors demonstrated by autoradiography. Additional aliquots of these lysates, containing equal amounts of protein, were incubated for 1 hr at 4°C with 1.4 μg of N-19 polyclonal anti-Smad7 antibody (Santa Cruz Biotech, Santa Cruz, CA). Antigen-antibody complexes were precipitated with protein A/G-Sepharose and eluted by boiling in 2× SDS sample loading buffer containing 5% β-mercaptoethanol prior to SDS-PAGE and autoradiography.
Western blotting
Aliquots (100 μg of protein) from various cell lysates were loaded on an SDS-polyacrylamide gel (7.5% or 10% acrylamide). After transfer to nitrocellulose, membranes were probed with antibodies against Smad7 and Smad2, and GDP dissociation inhibitor (GDI, kind gift from Dr. Philipp Scherer, Albert Einstein College of Medicine, Bronx, NY) to control for protein loading, as indicated. Bound primary antibodies were detected with horseradish peroxidase-labeled anti-mouse or anti-rabbit secondary antibodies, respectively, and developed with enhanced chemiluminescence reagents purchased from Pierce, Rockford, IL.
Transfections and transcriptional reporter assays
The 3TPLux and SBE4-Luc reporter constructs were gifts from Joan Massagué, Memorial Sloan Kettering Cancer Center, and Bert Vogelstein, Johns Hopkins University, respectively (Wrana et al. 1992; Zawel et al. 1998). For transcriptional reporter assays, cells (2.5–4 × 104/well) were seeded in 24-well dishes and transfected with the luciferase reporter constructs and pRSV-Gal (Promega Corp., Madison, WI) with or without either pcDNA3 control vector DNA or pSmad7AS vector as indicated, with the Superfect Reagent (Qiagen) following the manufacturers' protocol. After transfection, cells were incubated in 0.2% FBS starvation medium for 20 hr and then left either untreated (controls), or treated with either TNF-α (0.2 ng/ml) for 4 hr 45 min, TGF-β1 (0.1 ng/ml) for 4 hr, or TNF-α followed by TGF-β for the indicated time periods. Cells were lysed and processed by a luciferase assay system (Promega Corp., Madison, WI), and luciferase activity was assayed with an AutoLumat LB953 (E G & G Berthold) luminometer. Luciferase activities were normalized for galactosidase activity assayed by a galactosidase assay system (Promega Corp., Madison, WI). Results were expressed as ratio (Fold-induction) of normalized luciferase activities in cytokine treated cells and untreated cells of each experiment, respectively.
Acknowledgments
We thank Joan Massagué for the 3TPLux plasmid, Bert Vogelstein and Kenneth Kinzler for the SBE4-Luc plasmid, Peter ten Dijke for the pFlag-Smad7 plasmid and the anti-Smad7 antibody, and Dr. Philipp Scherer for the anti-GDI antibody. We thank Mark de Caestecker, Robert Lechleider, Antonio Iavarone, Victor Schuster, and Peter Mundel for their critical reading of the manuscript. This research was supported in part by a Young Investigator award of the National Kidney Foundation of New York/New Jersey and by the City of New York Council Speaker's Fund for Biomedical Research: Towards the Science of Patient Care to E.P.B., and National Institutes of Health grants DK-56077-01 to E.P.B., AA-09231 and AA-10541 to M.R. M.B. is the recipient of a research fellowship from the Deutsche Forschungsgemeinschaft (DFG). G.v.G. is the recipient of the National Kidney Foundation/Kevin and Gloria Keily Fellow research fellowship award.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL bottinge@aecom.yu.edu; FAX (718) 430-8963.
References
- Arsura M, Wu M, Sonenshein GE. TGF beta 1 inhibits NF-kappa B/Rel activity inducing apoptosis of B cells: Transcriptional activation of I kappa B alpha. Immunity. 1996;5:31–40. doi: 10.1016/s1074-7613(00)80307-6. [DOI] [PubMed] [Google Scholar]
- Arsura M, FitzGerald MJ, Fausto N, Sonenshein GE. Nuclear factor-kappaB/Rel blocks transforming growth factor beta1-induced apoptosis of murine hepatocyte cell lines. Cell Growth Differ. 1997;8:1049–1059. [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Baldwin AS., Jr The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Ballermann BJ, Dardik A, Eng E, Liu A. Shear stress and the endothelium. Kidney Int Suppl. 1998;67:S100–S108. doi: 10.1046/j.1523-1755.1998.06720.x. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Böttinger EP, Jakubczak JL, Roberts IS, Mumy M, Hemmati P, Bagnall K, Merlino G, Wakefield LM. Expression of a dominant-negative mutant TGF-beta type II receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. EMBO J. 1997;16:2621–2633. doi: 10.1093/emboj/16.10.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Linhoff MW, Stein B, Wright KL, Baldwin AS, Jr, Basta PV, Ting JP. Function of NF-kappa B/Rel binding sites in the major histocompatibility complex class II invariant chain promoter is dependent on cell-specific binding of different NF-kappa B/Rel subunits. Mol Cell Biol. 1994;14:2926–2935. doi: 10.1128/mcb.14.5.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Rubock MJ, Whitman M. A transcriptional partner for MAD proteins in TGF-beta signalling. Nature. 1997;383:691–696. doi: 10.1038/383691a0. [DOI] [PubMed] [Google Scholar]
- Chung KY, Agarwal A, Uitto J, Mauviel A. An AP-1 binding sequence is essential for regulation of the human alpha2(I) collagen (COL1A2) promoter activity by transforming growth factor-beta. J Biol Chem. 1996;271:3272–3278. doi: 10.1074/jbc.271.6.3272. [DOI] [PubMed] [Google Scholar]
- Derynck R, Feng XH. TGF-beta receptor signaling. Biochim Biophys Acta. 1997;1333:F105–F150. doi: 10.1016/s0304-419x(97)00017-6. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang Y, Feng XH. Smads: Transcriptional activators of TGF-beta responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- Garcia-Trevijano ER, Iraburu MJ, Fontana L, Dominguez-Rosales JA, Auster A, Covarrubias-Pinedo A, Rojkind M. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology. 1999;29:960–970. doi: 10.1002/hep.510290346. [DOI] [PubMed] [Google Scholar]
- Geiser AG, Burmester JK, Webbink R, Roberts AB, Sporn MB. Inhibition of growth by transforming growth factor-beta following fusion of two nonresponsive human carcinoma cell lines. Implication of the type II receptor in growth inhibitory responses. J Biol Chem. 1992;267:2588–2593. [PubMed] [Google Scholar]
- Geiser AG, Letterio JJ, Kulkarni AB, Karlsson S, Roberts AB, Sporn MB. Transforming growth factor beta 1 (TGF-beta 1) controls expression of major histocompatibility genes in the postnatal mouse: Aberrant histocompatibility antigen expression in the pathogenesis of the TGF-beta 1 null mouse phenotype. Proc Natl Acad Sci. 1993;90:9944–9948. doi: 10.1073/pnas.90.21.9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Grilli M, Chiu JJ, Lenardo MJ. NF-kappa B and Rel: Participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MAJ, Wrana JL, Falb D. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Hoodless PA, Wrana JL. Mechanism and function of signaling by the TGF beta superfamily. Curr Top Microbiol Immunol. 1998;228:235–272. doi: 10.1007/978-3-642-80481-6_10. [DOI] [PubMed] [Google Scholar]
- Inagaki Y, Truter S, Tanaka S, Di Liberto M, Ramirez F. Overlapping pathways mediate the opposing actions of tumor necrosis factor-alpha and transforming growth factor-beta on alpha 2(I) collagen gene transcription. J Biol Chem. 1995;270:3353–3358. doi: 10.1074/jbc.270.7.3353. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Taga T, Akira S. Cytokine signal transduction. Cell. 1994;76:253–262. doi: 10.1016/0092-8674(94)90333-6. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Massagué J. SMADs: Mediators and regulators of TGF-beta signaling. Curr Opin Genet Dev. 1998;8:103–111. doi: 10.1016/s0959-437x(98)80069-5. [DOI] [PubMed] [Google Scholar]
- Lefer AM, Ma XL, Weyrich AS, Scalia R. Mechanism of the cardioprotective effect of transforming growth factor beta 1 in feline myocardial ischemia and reperfusion. Proc Natl Acad Sci. 1993;90:1018–1022. doi: 10.1073/pnas.90.3.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letterio JJ, Roberts AB. TGF-beta: a critical modulator of immune cell function. Clin Immunol Immunopathol. 1997;84:244–250. doi: 10.1006/clin.1997.4409. [DOI] [PubMed] [Google Scholar]
- ————— Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Li JM, Shen X, Hu PP, Wang XF. Transforming growth factor beta stimulates the human immunodeficiency virus 1 enhancer and requires NF-kappaB activity. Mol Cell Biol. 1998;18:110–121. doi: 10.1128/mcb.18.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sun Y, Constantinescu SN, Karam E, Weinberg RA, Lodish HF. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc Natl Acad Sci. 1997;94:10669–10674. doi: 10.1073/pnas.94.20.10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias-Silva M, Abdollah S, Hoodless PA, Pirone R, Attisano L, Wrana JL. MADR2 is a substrate of the TGFbeta receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell. 1996;87:1215–1224. doi: 10.1016/s0092-8674(00)81817-6. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Muller JM, Ziegler-Heitbrock HW, Baeuerle PA. Nuclear factor kappa B, a mediator of lipopolysaccharide effects. Immunobiology. 1993;187:233–256. doi: 10.1016/S0171-2985(11)80342-6. [DOI] [PubMed] [Google Scholar]
- Mundel P, Reiser J, Borja AZ, Pavenstadt H, Davidson GR, Kriz W, Zeller R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997a;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- Nakao A, Roijer E, Imamura T, Souchelnytskyi S, Stenman G, Heldin CH, ten Dijke P. Identification of Smad2, a human Mad-related protein in the transforming growth factor beta signaling pathway. J Biol Chem. 1997b;272:2896–2900. doi: 10.1074/jbc.272.5.2896. [DOI] [PubMed] [Google Scholar]
- Oberhammer FA, Pavelka M, Sharma S, Tiefenbacher R, Bursch W, Schulte-Hermann R. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc Natl Acad Sci. 1992;89:5408–5412. doi: 10.1073/pnas.89.12.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouaaz F, Li M, Beg AA. A critical role for the RelA subunit of nuclear factor kappaB in regulation of multiple immune-response genes and in Fas-induced cell death. J Exp Med. 1999;189:999–1004. doi: 10.1084/jem.189.6.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeck HW, Weekx S, Moulijn A, Lardon F, Lenjou M, Nys G, Van Ranst PC, Van Bockstaele DR, Berneman ZN. Tumor necrosis factor alpha is a potent synergistic factor for the proliferation of primitive human hematopoietic progenitor cells and induces resistance to transforming growth factor beta but not to interferon gamma. J Exp Med. 1996;183:705–710. doi: 10.1084/jem.183.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper JN, Cai J, Qiu Y, Anderson KR, Xu YY, Deeds JD, Feeley R, Gimeno CJ, Woolf EA, Tayber O, et al. Vascular MADs: Two novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc Natl Acad Sci. 1997;94:9314–9319. doi: 10.1073/pnas.94.17.9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397:710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- Vodovotz Y, Geiser AG, Chesler L, Letterio JJ, Campbell AJ, Lucia MS, Sporn MB, Roberts AB. Spontaneously increased production of nitric oxide and aberrant expression of the inducible nitric oxide synthase in vivo in the transforming growth factor beta 1 null mouse. J Exp Med. 1996;183:2337–2342. doi: 10.1084/jem.183.5.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SM. Transforming growth factor beta: The good, the bad, and the ugly. J Exp Med. 1994;180:1587–1590. doi: 10.1084/jem.180.5.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massagué J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]