

Abstract
Bcl-3 is an atypical member of the IκB family that has the potential to positively or negatively modulate nuclear NF-κB activity in a context-dependent manner. Bcl-3's biologic impact is complex and includes roles in tumorigenesis and diverse immune responses, including innate immunity. Bcl-3 may mediate LPS tolerance, suppressing cytokine production, but it also appears to contribute to defense against select systemic bacterial challenges. However, the potential role of Bcl-3 in organ-specific host defense against bacteria has not yet been addressed. Here we investigate the relevance of Bcl-3 in a lung challenge with the Gram-negative pathogen Klebsiella pneumoniae. In contrast to wild-type mice, Bcl-3 deficient mice exhibited significantly increased susceptibility towards K. pneumoniae pneumonia. The mutant mice showed increased lung damage marked by neutrophilic alveolar consolidation and they failed to clear bacteria in lungs, which correlated with increased bacteremic dissemination. Loss of Bcl-3 incurred a dramatic cytokine imbalance in the lungs characterized by higher levels of IL-10 and a near total absence of IFNγ. Moreover, Bcl-3 deficient mice displayed increased lung production of the neutrophil-attracting chemokines CXCL-1 and CXCL-2. Alveolar macrophages and neutrophils are important to antibacterial lung defense. In vitro stimulation of Bcl-3-deficient alveolar macrophages with LPS or heat-killed K. pneumoniae recapitulated the increase in IL-10 production and Bcl-3 deficient neutrophils were impaired in intracellular bacterial killing. These findings suggest that Bcl-3 is critically involved in lung defense against Gram-negative bacteria, modulating functions of several cells to facilitate efficient clearing of bacteria.
Keywords: Bcl-3, NF-κB, pneumonia, Klebsiella, IL-10
Introduction
Host defense against pathogen invasion and infection relies on a potent inflammatory response aimed at clearing the pathogen and restoring the function of the infected organ. This response requires a balanced and coordinated production of pro-inflammatory mediators to fight pathogens, but also anti-inflammatory mediators to avoid excessive immunopathological damage at the focus of infection or in distant organs. Upon infection, recognition of the pathogen-associated molecular patterns by pattern-recognition receptors such as Toll-like receptors triggers innate defenses via activation of intracellular mediators such as NF-κB transcription factors. NF-κB factors transcriptionally activate a large array of genes in various cells, including those encoding proinflammatory cytokines, chemokines and anti-pathogenic agents to initiate the immune response and defend the host against the invading pathogen.
NF-κB transcription factors are homo- or heterodimers that are comprised of various combinations of five different subunits, namely RelA (p65), RelB, c-Rel, p50 and p52. In contrast to other NF-κB subunits, p50/NF-κB1 and p52/NF-κB2 lack transactivation domains. Instead of activating transcription, p52 homodimers and the much more abundant p50 homodimers largely function as inhibitors of gene transcription, in part by association with histone deacetylases, but also by competing with transactivating dimers for κB binding sites (1, 2). NF-κB activity is regulated by members of the IκB family, including the classical inhibitors IκBα, IκBβ and IκBε, as well as the atypical regulators Bcl-3, IκBξ and IκBNS. In contrast to the classical IκB proteins, Bcl-3 does not interact with the typical transactivating NF-κB dimers and thus does not affect signal-induced liberation of NF-κB complexes; instead, Bcl-3 modulates nuclear NF-κB activity by interacting exclusively with p50 and p52 homodimers on DNA (3, 4). In this way Bcl-3 may function as a positive regulator of NF-κB activity, owing to its transactivation domains, or it may function as a negative regulator, depending on the specific promoter/enhancer context (5-8) and likely also depending on its phosphorylation and ubiquitination status (1, 9).
The potential of Bcl-3 to variably modulate NF-κB activity, depending on context, may explain the variety of attributed biologic roles. This modulator acts in stromal cells to help develop a proper splenic architecture and, as a consequence, germinal centers and antibody responses (10, 11). It also acts in thymic epithelial cells to help insure negative selection of autoreactive thymocytes (12). Furthermore, Bcl-3 is critical for mounting an effective Th1 response against Toxoplasma gondii infection, possibly acting indirectly although mechanisms remain to be determined (10), and it has been suggested to directly modulate functions of mature T cells (13-15). Finally, Bcl-3 has been implicated in tumorigenesis. In particular, various B cell malignancies have been shown to express high levels of Bcl-3 on account of recurrent t(14;19) translocations (16-19). As a consequence, upregulated Bcl-3 may induce expression of cyclin D1 and thus promote cell cycle progression (20, 21).
Several lines of evidence point to a role for Bcl-3 in innate immune responses. Bcl-3 has been implicated in the regulation of LPS–induced cytokine production by macrophages and dendritic cells (22-24), as well as anti-bacterial agents in epithelial cells (25), i.e.cells on the frontline of pathogen defense. Bcl-3 may dampen LPS-induced expression of pro-inflammatory cytokines such as TNF-α, IL-1β and IL-23p19 (22-24) and it may specifically mediate tolerance to LPS by stabilizing the repressive p50 homodimers on target genes such as TNFα (26, 27) (7). While these activities portend an anti-inflammatory role, Bcl-3 also ostensibly serves a pro-inflammatory role, as it inhibits LPS-induced expression of the anti-inflammatory cytokine IL-10 (7, 24). Because IL-10 can induce Bcl-3, this atypical IκB protein mediates a negative auto-regulatory loop limiting production of IL-10 (22-24, 28).
Also consistent with a role of Bcl-3 in innate immunity to pathogens, prior studies have revealed a high susceptibility of Bcl-3 deficient mice to systemic infections with Gram-positive Streptococcus pneumoniae and Listeria monocytogenes (11, 24), though not Gram-negative Escherichia coli (11). Of note, these infection models all relied on intra-peritoneal administration of the pathogen, a route of infection whose pathophysiology does not involve organ-specific defense against trans-epithelial bacterial invasion, the physiologic route of infection. To address the role of Bcl-3 in innate immune responses in more physiologic organ-specific bacterial infections, we challenged Bcl-3 deficient and sufficient mice with Gram-negative pneumonia induced by intratracheal instillation of Klebsiella pneumoniae.
Materials and Methods
Mice
Bcl-3 deficient mice were generated as previously described (10) and backcrossed to C57BL/6 (Taconic Farms, Derwood, MD) for 11 or more generations. C57BL/6 (Taconic Farms) wild-type (WT) controls were co-housed at all times. Mice were bred and housed in National Institute of Allergy and Infectious Diseases (NIAID) facilities, and all experiments were done with the approval of the NIAID Animal Care and Use Committee and in accordance with all relevant institutional guidelines.
Induction of pulmonary infection
Klebsiella pneumoniae (strain 43816, serotype 2; American type Culture collection) was grown overnight in Luria-Bertani (LB) broth at 37°C in a rotating shaker. The culture was 100-fold diluted in fresh LB broth and grown for 2 h, allowing the culture to reach early log phase. Bacteria were then washed, diluted in PBS, and the desired concentration was adjusted by spectrophotometry (absorbance at 600 nm) according to a reference curve. Bacterial concentration was systematically verified by quantitative culture of the inoculum onto LB agar plates. Pulmonary infection was initiated by intratracheal instillation of bacteria. For this purpose, short-duration anesthesia was induced by isoflurane inhalation and a gavage canula was inserted into the trachea. The intratracheal position was verified by respiratory oscillations of a droplet inside a 1-mL syringe. The desired quantity of bacteria was instilled in a volume of 50 μL. The survival status of animals was observed twice daily up to 14 days after induction of pneumonia.
Bronchoalveolar lavage
Mice were anesthesized by i.p. injection of ketamine/xylazine. The trachea was exposed and a bronchoalveolar lavage (BAL) was performed with 1 mL sterile PBS. BAL fluid (BALF) was subjected to 10-fold serial dilutions and cultured for 24 hours on LB agar plates to quantify the number of colony-forming units (CFU). Erythrocytes were lysed with ACK lysis buffer and the total number of viable cells per BAL was quantified by an automated hemocytometer. Neutrophils were identified as CD11b and Gr-1 double-positive cells by flow cytometry. To determine myeloperoxydase (MPO) activity, BAL fluid was incubated with o-dianisidine dihydrochloride in Hank's medium supplemented with 0.004% H2O2. The reaction was stopped by adding 1% NaN3 and MPO activity was determined by spectrophotometry.
Lung homogenates
The left lung was finely minced with scissors and homogenized with an electric mortar in 200μl PBS under sterile conditions. Lung homogenates were serially diluted and plated on LB agar at 37°C for 24h for the purpose of bacterial CFU quantification. The right lung was homogenized using the same technique in 200μl PBS containing protease inhibitor cocktail I (Millipore) and 8μg/ml AEBSF (Sigma). Samples were centrifuged at 4°C to pellet debris and the supernatant was snap frozen for the purpose of quantification of chemokines CXCL-1/KC and CXCL-2/MIP-2α.
Assessment of bacteremic dissemination
K. pneumoniae bacteremia was assessed through quantitative blood cultures on LB agar plates. Blood was collected by cardiac puncture. Bacteremic dissemination was also assessed through quantitative spleen cultures. Spleen was removed and mechanically homogenized under sterile conditions. Spleen homogenates were subjected to 10-fold serial dilutions and cultured for 24 hours on LB agar plates.
Histological analysis
Lungs were immersion-fixed in 10% paraformaldehyde, embedded in paraffin blocks and cut into 3-μm sections (Histoserv, Germantown, MD). Sections were stained with hematoxylin and eosine (H&E) and examined by light microscopy.
Lung digestion
Right lungs were perfused with 1 mL Collagenase/Dispase (1 mg/mL) and DNase I (0.5 mg/mL) (Roche) and incubated at room temperature for 45 min on a rotating shaker. Digested lungs were then smashed and passed through a 40-μm cell strainer. Erythrocytes were lysed with ACK lysis buffer. The total number of viable cells per lung was quantified by an automated cell counter and the proportion of neutrophils (CD11b+ Gr-1+) was determined by flow cytometry.
Flow cytometry analysis
FITC-conjugated Gr-1 and PE-conjugated CD11b and Fc-blocker antibodies were purchased from Becton Dickinson (San Diego, CA). Cell suspensions from BAL fluid and total lung were incubated with Fc-blocker antibody for 5 minutes, then stained with fluorescent antibodies for 30 min at 4°C. Acquisition was performed on a FACSCanto II flow cytometer (Becton Dickinson) and analysis was performed using the FlowJo software. Neutrophils were identified as CD11b+Gr-1+ cells.
Isolation and stimulation of alveolar macrophages
Alveolar macrophages were collected through successive BAL collections of 1 mL up to a total of 10 mL. BAL fluids from 6 to 10 mice were pooled, and red blood cells were lysed with ACK lysis buffer. Cells were seeded at a concentration of 105 cells per well onto flat-bottom 96-well plates in RPMI 1640 medium supplemented with 10% fetal calf serum. Following overnight plastic adherence, non-adherent cells were removed by medium replacement. Adherent cells were stimulated with either LPS (E. coli 0111:B4, Sigma) or heat-killed (95°C for 30 min) K. pneumoniae at a multiplicity of infection ratio of 1:100, based on the bacterial concentration before heat inactivation.
RNA isolation and real-time PCR
Lungs were frozen and ground on dry ice, and powder tissue was used for RNA isolation. 1 mL of TRIzol (Invitrogen, Carlsbad, CA) was added to 100 mg of powder tissue and homogenized with a needle syringe. For cell cultures of alveolar macrophages, adherent cells were lysed using RLT lysis buffer (Qiagen, Valencia, CA).
RNA cleanup/DNase digestion was carried out using the RNeasy mini kit (Qiagen) following the manufacturer's instructions. cDNA was synthesized using oligo(dT) and Superscript III reverse transcriptase (Invitrogen). Expressions of IL-10, TNF-α, IFN-γ, IL-12p40, IL-17, IL23p19, CXCL-1/KC, CXCL-2/MIP-2α and β-actin were quantified by real-time PCR using the TaqMan Fast Universal PCR Master Mix and gene-specific primers on a StepOnePlus thermocycler (Applied Biosystems, Foster City, CA). Pulmonary gene expression was expressed as 2-ΔCt where Δ cycle threshold (Ct) = (Cttarget − Ctβ-actin) for each individual sample. Gene induction results in alveolar macrophages were expressed as 2-ΔΔCt, where -ΔΔCt = (Cttarget – Ctβ-actin) for LPS or heat-killed bacteria stimulation - (Cttarget − Ctβ-actin) for untreated cells.
Quantification of cytokine and chemokine levels
IL-10, TNF-α, IFN-γ and IL-17 concentrations were quantified in BALF, and CXCL-1/KC and CXCL-2/MIP-2α were quantified in supernatants of lung homogenates using ELISA assays according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
Phagocytosis and killing assay
Alveolar macrophages were infected with K. pneumoniae at a multiplicity of infection ratio of 1:10 in flat-bottom 96-well plates. The plates were centrifuged at 1500 rpm for 10 min at 4°C to initiate the contact between cells and bacteria, and were incubated at 37°C for 30 min. Extracellular bacteria were then removed by washing with PBS and killed with 100 μg/mL Gentamycin (Mediatech, Inc, VA). Phagocytosis was assessed after 30 min (t=0). Cells were washed twice with PBS and were lysed with reporter lysis buffer (Promega, WI). The number of intracellular bacteria was determined by serial dilutions of lysates plated onto LB agar plates. The intracellular killing of the bacteria was assessed by the same method after 2h.
Neutrophils were purified from the bone marrow of WT and Bcl-3 deficient mice using Ly6G beads (Miltenyi Biotec) and were separated using an AutoMACS (Miltenyi Biotec). Cells were then resuspended in Krebs Ringer bicarbonate buffer (Sigma Aldrich) and incubated at 37°C for 1 h with K. pneumoniae at a multiplicity of infection ratio of 1:100. Cells were then washed with Krebs Ringer buffer containing 100μg/mL Gentamicin to remove extracellular bacteria. Samples were taken for t=0 and additional samples were incubated for a further 30 and 60 min to measure bacterial killing. At time of collection cells were lysed in 200μl PBS containing 0.1% Triton 100 and 20μl were plated on LB agar and grown overnight at 37°C. Colonies were counted the next day. Killing was calculated from the percentage of colonies present at t=30 or t=60 min compared to t=0 (i.e. 100 - (number of CFUt=30 / number of CFUt=0).
Statistical analysis
Survival curves were analyzed using the Kaplan-Meier method and compared using the log-rank test. Continuous variables were expressed either as scatterplots, or median and interquartile range using the boxplot representation and compared using the Mann-Whitney test, or expressed as mean ± SD and compared using the Student t-test. P values lower than 0.05 indicated statistically significant differences.
Results
Bcl-3 deficient mice display increased susceptibility towards K. pneumoniae pneumonia
To determine whether Bcl-3 has a role in the host response to Gram negative bacterial pneumonia, we subjected WT and Bcl-3 deficient mice to an experimental pulmonary infection with the Gram-negative bacillus K. pneumoniae. Following intratracheal (i.t.) instillation of 104 bacteria, approximately 60% of WT mice died within the 14-day follow-up. In contrast, all but one of the Bcl-3 deficient mice died within 6 days, 40% of them within 3 days (Fig. 1A). Similar results were obtained when using a five-fold lower bacterial inoculum (2×103 CFU) (Fig. 1B). While the survival rate of WT mice was slightly higher (approximately 60% were still alive after 14 days), Bcl-3 deficient mice remained significantly more susceptible to the bacterial challenge. These results suggest that Bcl-3 makes important contributions to the host response against Gram negative pulmonary infection. In order to further explore the role of Bcl-3 in lung defense, we first assessed the time course of Bcl-3 expression in lungs of WT mice subjected to K. pneumoniae pulmonary infection. As compared to non-infected mice (time point 0h), pulmonary infection led to an early (6 hours after instillation) and sustained (at least 72 hours) increase in Bcl-3 expression (Fig. 2).
Figure 1. Bcl-3 deficient mice display increased susceptibility towards K. pneumoniae pneumonia.
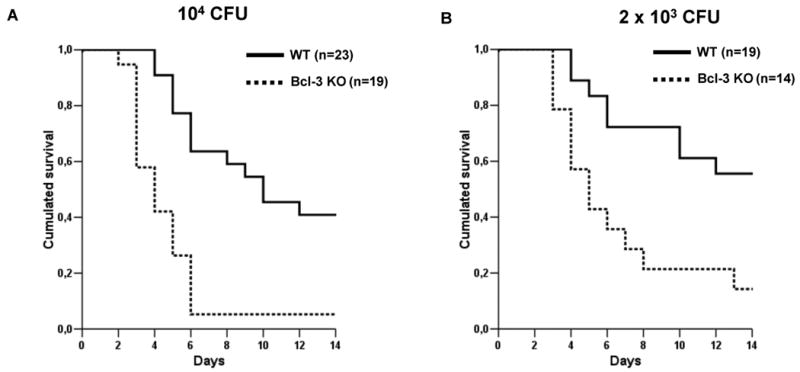
Survival of wild-type (WT, continuous line) and Bcl-3 deficient (Bcl-3 KO, dotted line) mice subjected to intratracheal instillation of 104 (A) and 2×103 (B) CFU of K. pneumoniae. Survival was monitored for 14 days. Survival curves summarize four distinct experiments. P< 0.001 between WT and Bcl-3 KO mice for both bacterial inoculums.
Figure 2. K. pneumoniae pulmonary infection upregulates Bcl-3 lung expression.

Wild-type mice were subjected to intratracheal instillation of 104 CFU of K. pneumoniae. Bcl-3 gene expression was assessed in the whole lung by quantitative RT-PCR at the indicated time points from induction of pulmonary infection, and expressed as 2-ΔCt. Data are represented as boxplots (n=10-12 animals per time point from three different experiments). * P < 0.01 as compared to non-infected (0h) mice.
Bcl-3 controls lung bacterial clearance during K. pneumoniae pneumonia
To test whether Bcl-3 deficiency affects bacterial clearance in the lung, we performed quantitative bacterial cultures of the BAL fluid (Fig. 3A) and of lung homogenates (Fig. 3B). No significant differences were observed 24h after inoculation; however, Bcl-3 deficient mice displayed significantly higher pulmonary bacterial loads than their WT counterparts at 48h and 72h. Local impairment in pathogen control is usually associated with increased bacteremic dissemination. Accordingly, bloodstream bacterial concentrations were much higher in Bcl-3 deficient mice than in WT mice 72h after inoculation (Fig. 3C). Since bacteremia is transient and may not reliably assess the circulating bacterial burden, we also concurrently performed quantitative bacterial cultures of spleen homogenates. Consistent with the blood results, the amount of bacteria in spleen was higher in Bcl-3 deficient mice (Fig. 3D). Altogether these results suggest that Bcl-3 contributes to efficient lung clearance and the prevention of bacteremic dissemination.
Figure 3. Bcl-3 deficient mice display impaired bacterial lung clearance and increased bacteremia.

Wild-type and Bcl-3 deficient mice were subjected to intratracheal instillation of 104 CFU of K. pneumoniae (WT, open triangles; Bcl-3 deficient, solid triangles). Lung bacterial load was assessed at the indicated time point by quantitative cultures of BAL fluid (BALF) (A) and of supernatants from lung homogenates (B). Bacteremia was quantified at the indicated time point by quantitative cultures of whole blood (C) and of spleen homogenates (D). Data are expressed as scatterplots from three different experiments. * P < 0.05, ** P < 0.01 between WT and Bcl-3 deficient mice.
Exacerbated lung damage in K. pneumoniae-infected Bcl-3 deficient mice
48h and 72h after induction of pneumonia, WT mice exhibited moderate neutrophil infiltration, predominantly in the peribronchial areas, but the overall structure of the lung was largely preserved. In contrast, the lungs of Bcl-3 mice were characterized by extensive foci of consolidation related to massive alveolar infiltration by neutrophils (Fig. 4A). Neutrophils account for the large majority of cells recovered in BALF in the setting of bacterial pneumonia. Neutrophil counts in BAL fluid (Fig. 4B) as well as MPO activity, a marker of neutrophil activation (Fig. 4C), did not markedly differ between WT and Bcl-3 deficient mice. However, consistent with neutrophil accumulation in consolidated areas of the lung in infected Bcl-3 deficient mice, the total number of neutrophils obtained from digested lung of these mutant mice was notably higher than in WT mice at 72h (Fig. 4D). Of note, the lifespan of neutrophils is measured in a few hours and is even shorter following infection. It is thus likely that many dead cells also contributed to the large infiltrates seen in histologic sections, but such dead cells were not enumerated in the analyses of BALFs and digested lung tissue, which only focused live cells.
Figure 4. Bcl-3 deficient mice exhibit major lung damage following K. pneumoniae pulmonary infection.
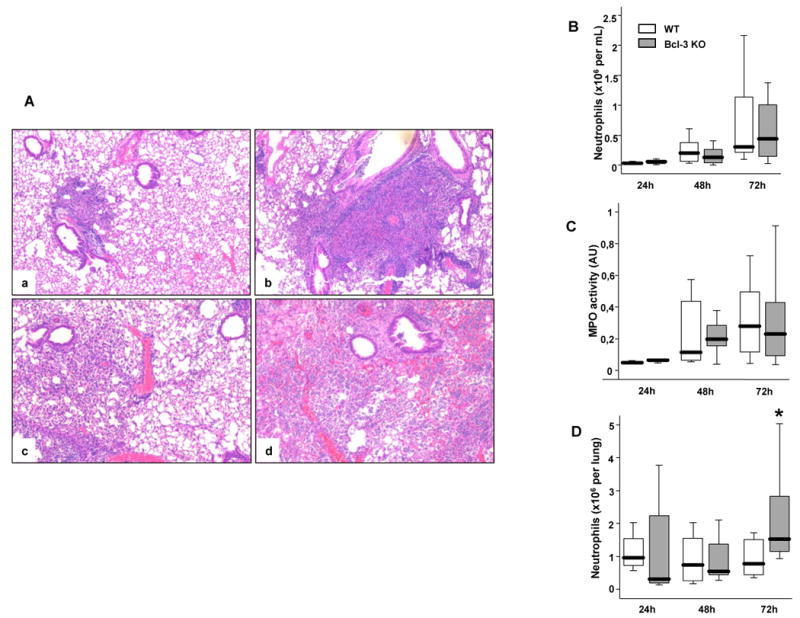
Wild-type (WT) and Bcl-3 deficient (Bcl-3 KO) mice were subjected to intratracheal instillation of 104 CFU of K. pneumoniae. A. H&E-stained lung sections from WT (a, c) and Bcl-3 KO mice (b, d) mice 48-hours (a, b) and 72-hours (c, d) after intra-tracheal inoculation ((40× magnification). The pictures shown are representative of three animals per group and per condition. B. BALF were collected at the indicated time points following inoculation. The absolute number of neutrophils was determined by the relative percentage of live cells expressing both Gr-1 and CD11b. C. The BAL fluid was tested for myeloperoxydase (MPO) activity (expressed as arbitrary units= absorbance at 450 nm). D. Neutrophils were quantified in the lungs of infected mice using flow cytometry. The absolute number of neutrophils was determined by the relative percentage of live cells expressing both Gr-1 and CD11b. Data are expressed as boxplots (12 animals per group from three different experiments). * P < 0.05 between WT (open rectangles) and Bcl-3 KO mice (filled rectangles).
Bcl-3 deficiency induces imbalanced cytokine and chemokine production during K. pneumoniae pneumonia
Pro-inflammatory and anti-inflammatory cytokines are major regulators of the anti-bacterial response. We investigated the cytokine production profile in WT and Bcl-3 deficient mice with quantitative RT-PCR of whole lung (Fig. 5A). While IL-10 levels remained minimal in WT mice throughout the first 72 hours of infection, Bcl-3 mice displayed increased levels of IL-10 after 48h and 72h. In contrast, IFN-γ levels were dramatically lower in Bcl-3 deficient when compared to WT mice at all post-infection times queried, consistent and concomitant with lower IL-12p40 levels. Of note, IL-12p35 transcript levels were very low in most samples (data not shown). We also observed some differences in IL-23p19 and IL-17; there were lesser amounts of IL-17 in Bcl-3 deficient mice after 24h and there was a similar trend after 72h (p= 0.08). Consistent with this, IL-23p19 was reduced in Bcl-3 deficient mice, at least at 72h after infection. We also observed somewhat lower levels of TNF-α mRNA in Bcl-3 deficient mice at 72h. The dramatic IL-10/IFN-γ mRNA imbalance of Bcl-3 deficient as compared to WT mice was confirmed by the quantification of these cytokine proteins in the BAL fluid (Fig. 5B). In addition, protein analyses also showed a trend towards lower production of IL-17, but failed to show significant differences in the production of TNFα (Fig. 5B).
Figure 5. Bcl-3 deficient mice display imbalanced pulmonary production of cytokines during K. pneumoniae pneumonia.


Wild-type (WT) and Bcl-3 deficient (Bcl-3 KO) mice were subjected to intratracheal instillation of 104 CFU of K. pneumoniae. A. The amounts of IL-10, TNF-α, IFN-γ, IL-12p40, IL-17 and IL-23p19 mRNA were determined in the whole lung by quantitative RT-PCR at the times indicated following the challenge. Baseline levels of mRNA expression were very low or even undetectable, WT and Bcl-3 KO being indistinguishable. B. The concentrations of the cytokines IL-10, TNF-α, IFN-γ and IL-17 were determined by ELISA in the BAL fluid. Data are expressed as boxplots (12 animals per group from three different experiments). * P < 0.05, ** P < 0.01 between WT (open rectangles) and Bcl-3 KO mice (filled rectangles).
Since the recruitment of neutrophils at the site of infection is highly dependent on chemokines, we assessed the lung production of two major neutrophil-attracting chemokines, CXCL-1/KC and CXCL-2/MIP-2α via quantitative RT-PCR (Fig. 6A) and protein quantification (Fig. 6B) in whole lung homogenates. Interestingly, Bcl-3 deficient mice produced higher levels of mRNA and protein of these chemokines at 48h after infection. These results suggest a potential role for Bcl-3 in the control of neutrophil influx within the infected organ.
Figure 6. Bcl-3 deficient mice display increased pulmonary production of chemokines during K. pneumoniae pneumonia.

Wild-type (WT) and Bcl-3 deficient (Bcl-3 KO) mice were subjected to intratracheal instillation of 104 CFU of K. pneumoniae. A. The amounts of CXCL-1/KC and CXCL-2/MIP-2α mRNA were determined in the whole lung by quantitative RT-PCR at the times indicated following the challenge. Baseline levels of mRNA expression were very low or even undetectable, WT and Bcl-3 KO being indistinguishable. (12 animals per group from three different experiments) B. The concentrations of the chemokines CXCL-1/KC and CXCL-2/MIP-2α were determined by ELISA in supernatants of lung homogenates. Data are expressed as boxplots (10-12 animals per group from three different experiments). * P < 0.05, ** P < 0.01 between WT (open rectangles) and Bcl-3 KO mice (filled rectangles).
Loss of Bcl-3 results in increased production of IL-10 as well as IFN-γ by alveolar macrophages
Alveolar macrophages represent the first line of lung defense against invading pathogens through phagocytosis and production of inflammatory mediators. We assessed the antibacterial capacities of alveolar macrophages towards live K. pneumoniae. We found no differences in ex vivo bacterial uptake or bacterial killing between wild-type and Bcl-3 deficient alveolar macrophages (Fig. 7A). Similar results were observed with bone marrow-derived macrophages (data not shown). Upon exposure to pathogens, alveolar macrophages also produce various cytokines. Since we observed imbalanced cytokine production in Bcl-3 deficient mice subjected to Gram negative pneumonia, we assessed the ex vivo cytokine response of alveolar macrophages after stimulation with LPS or heat-killed K. pneumoniae via quantitative RT-PCR analyses (Fig. 7B). While TNF-α production was similar in WT and Bcl-3 deficient alveolar macrophages, production of IL-10 was notably higher in Bcl-3 deficient mice, consistent with higher levels observed in vivo. Surprisingly, LPS or bacterial stimulation also led to an increased production of IFN-γ in Bcl-3 deficient alveolar macrophages, contrasting with the dramatically lower levels of this cytokine in lungs of infected mutant mice. However, the protein levels of IFNγ and IL-10 in the supernatants were too low to be reliably assessed under our experimental conditions. Therefore, Bcl-3 deficient alveolar macrophages may contribute to the increased production of IL-10 seen in vivo, but are not likely to be a major source of IFN-γ release in the setting of pulmonary infection in vivo.
Figure 7. Bcl-3 deficiency does not affect antibacterial functions and increases both IL-10 and IFN-γ production in alveolar macrophages.

Alveolar macrophages were collected by BAL from non-infected wild-type (WT) or Bcl3 deficient (Bcl-3 KO) mice and pooled (10 mice per group).
A. Cells were incubated at 37°C for 30 min with K. pneumoniae at a multiplicity of infection ratio of 1:10. Extracellular bacteria were then removed by washing with Gentamycin. Cells were lysed with reporter lysis buffer for determination of bacterial uptake (t=0h) and intracellular killing (t=2h). The number of intracellular bacteria was determined by serial dilutions of lysates plated onto LB agar plates. The experiment shown is representative of two independent experiments performed in duplicates.
B & C. Cells were stimulated for 2, 4 or 6 hours with either LPS (1μg/mL) (B) or heat-killed K. pneumoniae (pre-killed multiplicity of infection 100) (C). Unstimulated cells served as reference. Production of the cytokines IL-10, IFN-γ and TNF-α was determined by quantitative RT-PCR. Data are reported as 2-ΔΔCt. The experiment shown is representative of three independent experiments.
Bcl-3 deficient neutrophils are impaired in killing bacteria
K. pneumoniae pneumonia in Bcl-3 deficient mice is characterized by impaired bacterial clearance despite an increase in the local recruitment of neutrophils, the major phagocytic cell in this context. This suggests that Bcl-3 deficient neutrophils might be defective in bacterial uptake and/or killing. We assessed phagocytosis and bacterial killing of K. pneumoniae with bone-marrow derived neutrophils in vitro. The absence of Bcl-3 did not appear to affect the bacterial uptake by these neutrophils (Fig. 8A), but it did significantly impair intracellular killing (Fig. 8B). While about half of bacteria were killed within 60 min in WT neutrophils, the number of intracellular bacteria did not significantly decrease in Bcl-3 deficient cells. Of note, we did not observe any differences in apoptotic rates between WT and Bcl-3 deficient K. pneumoniae-infected neutrophils (data not shown). These data suggest that an apparent defect in the ability of Bcl-3 deficient neutrophils to kill bacteria may contribute to the impaired lung clearance of K. pneumoniae observed in Bcl-3 deficient mice.
Figure 8. Bcl-3 deficiency impairs bacterial killing in neutrophils.

Neutrophils were collected from the bone marrow of non-infected wild-type (WT) or Bcl3 deficient (Bcl-3 KO) mice and incubated at 37°C for 1 h with K. pneumoniae. Cells were then washed with buffer containing Gentamicin to remove extracellular bacteria, and were lysed at various time points after washing (t=0 for reference, t=30 min and t=60 min). Live intracellular K. pneumoniae was quantified by culture. Bacterial uptake was assessed at the time point t=0 (A). Killing was calculated from the percentage of colonies present at t=30 min or t=60 min compared to t=0 (i.e. 100-(number of CFUt=30 / number of CFUt=0) (B). The result shown here is a combination of two independent experiments in duplicates with a total of 7 WT mice and 10 Bcl-3 KO mice. * P < 0.01 between WT (open rectangles) and Bcl-3 KO mice (filled rectangles).
Discussion
The present study employs a physiologically relevant lung infection model to reveal a critical role for the atypical IκB protein Bcl-3 in the innate immune defense against Gram negative K. pneumoniae. Bcl-3 was required to locally contain bacteria. Unlike wild-type mice, Bcl-3 deficient mice were unable to prevent the rapid rise of bacterial counts in the lung within 48h, and arguably in consequence, these mutant mice exhibited significantly higher systemic infection by 72h, resulting in the demise of the infected animals between 3-6 days post infection. Since the outcome of the infection is decided within 96h, well before adaptive immune response can be established, our findings highlight the importance of Bcl-3 in mounting an effective innate immune response against this bacterial infection. Mice lacking Bcl-3 also exhibited lung damage marked by extensive areas of neutrophilic consolidation that was consistent with increased pulmonary production of neutrophil-attracting chemokines. Furthermore, the balance between IL-10 and IFNγ was skewed towards an anti-inflammatory pattern in Bcl-3 deficient lungs. As discussed below these changes could account for the inability to control bacterial counts. In addition to the in vivo findings, BCL-3 deficiency was associated with some functional defects in two important innate cell types. Bcl-3 deficient neutrophils appeared to be defective in killing bacteria and Bcl-3 deficient alveolar macrophages displayed altered cytokine levels upon stimulation.
Gram-negative bacteria are frequently involved in severe community-acquired and healthcare-associated pneumonia. Mechanisms of lung defense against K. pneumoniae have been addressed and require the integrity and early activation of Il-12p70 / IFNγ and IL-23 / IL-17 pathways in order to eradicate the pathogen (29, 30). In particular, some studies have highlighted the critical role of the IL-10 / IFNγ balance in this setting. Thus inhibition of IL-10 improved bacterial clearance and survival of mice subjected to a K. pneumoniae pneumonia, while IFNγ deficient mice were conversely more susceptible (31, 32). In the present study increased susceptibility of Bcl-3 deficient mice to K. pneumoniae pneumonia was associated with both an increased production of IL-10 and a concomitant decreased production of IFNγ, and to a lesser degree, IL-17. Skewing of these critical cytokines towards such an anti-inflammatory profile locally in lungs could therefore be responsible for the impaired bacterial lung clearance in Bcl-3 deficient mice, overwhelming local barrier functions and resulting in uncontrolled bacteremic dissemination during the first 48 - 72 h post infection. In addition, systemic defenses may have been compromised as well and failure to control K. pneumoniae systemically may have contributed to the worse outcome in Bcl-3 deficient mice.
Impaired production of IFNγ and IL-12 has also been noted in the serum of Bcl-3 deficient mice challenged systemically with Gram positive Listeria monocytogenes (24); these mutant mice exhibited defective bacterial clearance from spleens as well as increased mortality (11). Unlike the present study, however, these previous reports involved intra-peritoneal injections of the pathogen, which completely bypassed the organ-specific defense against trans-epithelial bacterial invasion. It was also reported that Bcl-3 deficient mice challenged with an intra-peritoneal injection of LPS exhibited a serum cytokine imbalance similar to what we observed in lungs, namely high levels of IL-10 and reduced levels of IFNγ and, to a lesser extent, of IL-12 (24). Furthermore, it was proposed that high levels of IL-10 were responsible for the suppression of IL-12, and to a lesser extent, of IFNγ. In our studies we also observed a rise in IL-10 locally in lungs of Bcl-3 deficient mice, which was presumably prevented in wild-type mice by the rise in Bcl-3 expression in response to Gram-negative pulmonary infection with K. pneumoniae. However, it does not necessarily follow that IL-10 was responsible for suppression of IFNγ in this organ-specific immune defense model: lung levels of IFNγ were already reduced in Bcl-3 deficient mice within 24h post infection, while the rise in IL-10 levels was not observed until later. A lung-specific regulatory network may control expression of these cytokines; in this setting Bcl-3 may be able to promote IFNγ expression in lungs of challenged mice by means other than by delimiting IL-10 production. In accordance with this hypothesis, administration of neutralizing antibodies against IL-10 was not able to prevent mortality in Bcl-3 deficient mice (unpublished observations). Apart from IL-10's anti-inflammatory effect, which is presumed to negatively impact pathogen clearance, IL-10 is also involved in the prevention of immunopathological damage within infected tissues. Thus, antibodies to IL-10 may also exacerbate harmful effects of the inflammatory responses.
In addition to cytokines, chemokines are also critical to the host's anti-bacterial response, as they direct the migration of circulating polymorphonuclear and mononuclear leukocytes to sites of infection. In accord with the increased recruitment of neutrophils within infected lungs of Bcl-3 deficient mice, we observed a rise in the neutrophil-attracting chemokines CXCL-1 / KC and CXCL-2 / MIP-2α, which may have contributed to the neutrophilic consolidation and extensive lung damage observed in this setting. However we cannot exclude that the increased lung production of CXCL-1 and CXCL-2 was related to the higher burden of bacteria. Interestingly, bone-marrow derived macrophages from Bcl-3 deficient mice exhibited increased LPS-induced expression of various chemokines, including CXCL-1 and CXCL-2 (7). In addition, splenocytes and CD4+ lymphocytes from Bcl-3 deficient mice were reported to produce elevated levels of chemokines as well (33). Altogether these findings suggest that loss of Bcl-3 in the context of a bacterial pulmonary infection evoked an abnormal chemokine response, potentially from several cellular sources, which in turn contributed to the observed pathophysiology.
It is worth noting that the response of mice to the systemic presence of LPS or Gram-negative bacteria in Bcl-3 deficient mice is not well understood. Counter to the notion that Bcl-3 promotes inflammation by delimiting expression of IL-10, Bcl-3 was also reported to induce tolerance to LPS-induced endotoxic shock by delimiting expression of pro-inflammatory cytokines (34). Furthermore, despite higher IL-10 and reduced IL-12/IFNγ levels in Bcl-3 deficient mice challenged with E. coli-derived LPS, these mutant mice were reportedly able to defend against systemic infections with E. coli (11). In contrast, the present study shows that Bcl-3 deficient mice are impaired in their lung response towards Gram negative K. pneumoniae pulmonary infection and thereby fail to delimit bacteremic dissemination. Thus, systemic responses may be distinct from organ-specific host defense, which in the present case relies on lung-specific containment of trans-epithelial invasion by bacteria.
Alveolar macrophages represent the first line of defense against bacterial invasion in lungs and play a key role in recognition of the pathogen and initiation of immune responses, including production of cytokines in the early phase of infection. Although no differences in phagocytosis or bacterial killing were observed between wild-type and Bcl-3 deficient alveolar macrophages in vitro, the mutant macrophages produced significantly higher levels of IL-10 mRNA in response to stimulation with either LPS or heat-killed K. pneumoniae, in line with the in vivo data. This finding is also consistent with previously reported observations in peritoneal or bone-marrow derived macrophages from Bcl-3 deficient mice (22, 24, 27). Unexpectedly, mutant alveolar macrophages also produced higher amounts of IFNγ mRNA in vitro when compared to their wild-type counterparts, despite significantly reduced levels of this cytokine in lungs of K. pneumoniae challenged mutant mice. Thus, the behavior of alveolar macrophages in vitro may not recapitulate all in vivo functions that are likely influenced by paracrine signals. Moreover, several cell types are involved in host defense against bacterial pulmonary infections, including epithelial cells and various other hematopoietic immune cells. IFNγ may be differently regulated in these latter cell types and alveolar macrophages most likely are not a primary source of IFNγ in this setting. Instead, innate T lymphocytes or NK cells are likely to significantly contribute to IFNγ release during pulmonary bacterial infections.
Neutrophils are essential to the host's ability to clear bacteria and are they represent the majority of cells recruited at the site of bacterial infection; they also directly contribute to lung damage through the release of reactive oxygen species and proteases. Limitation and termination of this inflammatory process requires neutrophil apoptosis. We investigated for possible defects of neutrophils through in vitro challenge with K. pneumoniae to measure antibacterial functions of neutrophils. While Bcl-3 deficiency did not impact the bacterial uptake, it did impair the intracellular bacterial killing functions of neutrophils. Of note, we did not observe any differences in apoptosis between WT and mutant cells (data not shown). Nevertheless, the in vitro functions of neutrophils might not reliably recapitulate their behavior in a particular in vivo environment. The generation of conditional Bcl-3 deficient mice may allow us to address the respective contributions of different cell types in lung defense against K. pneumoniae infections in the future.
In conclusion, our findings demonstrate that Bcl-3 is critically involved in innate host defense in lungs in a model of Gram-negative K. pneumoniae pneumonia. Bcl-3 appears to control this response by modulation of cytokine production, especially by promoting IFNγ expression and suppressing IL-10. In addition, Bcl-3 may also be involved in the regulation of neutrophil-attracting chemokines. Finally, Bcl-3 appears to contribute to bacterial killing by neutrophils. Together these findings implicate Bcl-3 in the control of a coordinated response to a bacterial challenge by way of modulating NF-κB controlled gene expression in multiple cell types. Whether the role of Bcl-3 in pneumonia is restricted to Gram-negative bacteria or also extends to non-Gram-negative bacteria devoid of LPS remains to be established.
Acknowledgments
We greatly appreciated the constructive inputs provided by all members of the laboratory. We are most grateful to Dr. Anthony S. Fauci for continued support.
This research was supported by the Intramural Research Program of NIAID, NIH.
References
- 1.Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- 2.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Q, Didonato JA, Karin M, McKeithan TW. BCL3 encodes a nuclear protein which can alter the subcellular location of NF-kappa B proteins. Mol Cell Biol. 1994;14:3915–3926. doi: 10.1128/mcb.14.6.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siebenlist U, Brown K, Claudio E. Control of lymphocyte development by nuclear factor-kappaB. Nat Rev Immunol. 2005;5:435–445. doi: 10.1038/nri1629. [DOI] [PubMed] [Google Scholar]
- 5.Franzoso G, Bours V, Park S, Tomita-Yamaguchi M, Kelly K, Siebenlist U. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-kappa B-mediated inhibition. Nature. 1992;359:339–342. doi: 10.1038/359339a0. [DOI] [PubMed] [Google Scholar]
- 6.Franzoso G, Bours V, Azarenko V, Park S, Tomita-Yamaguchi M, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 can facilitate NF-kappa B-mediated transactivation by removing inhibiting p50 homodimers from select kappa B sites. EMBO J. 1993;12:3893–3901. doi: 10.1002/j.1460-2075.1993.tb06067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science. 2007;317:675–678. doi: 10.1126/science.1142953. [DOI] [PubMed] [Google Scholar]
- 8.Palmer S, Chen YH. Bcl-3, a multifaceted modulator of NF-kappaB-mediated gene transcription. Immunol Res. 2008;42:210–218. doi: 10.1007/s12026-008-8075-4. [DOI] [PubMed] [Google Scholar]
- 9.Massoumi R, Paus R. Cylindromatosis and the CYLD gene: new lessons on the molecular principles of epithelial growth control. Bioessays. 2007;29:1203–1214. doi: 10.1002/bies.20677. [DOI] [PubMed] [Google Scholar]
- 10.Franzoso G, Carlson L, Scharton-Kersten T, Shores EW, Epstein S, Grinberg A, Tran T, Shacter E, Leonardi A, Anver M, Love P, Sher A, Siebenlist U. Critical roles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity. 1997;6:479–490. doi: 10.1016/s1074-7613(00)80291-5. [DOI] [PubMed] [Google Scholar]
- 11.Schwarz EM, Krimpenfort P, Berns A, Verma IM. Immunological defects in mice with a targeted disruption in Bcl-3. Genes Dev. 1997;11:187–197. doi: 10.1101/gad.11.2.187. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Wang H, Claudio E, Brown K, Siebenlist U. A role for the IkappaB family member Bcl-3 in the control of central immunologic tolerance. Immunity. 2007;27:438–452. doi: 10.1016/j.immuni.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell TC, Hildeman D, Kedl RM, Teague TK, Schaefer BC, White J, Zhu Y, Kappler J, Marrack P. Immunological adjuvants promote activated T cell survival via induction of Bcl-3. Nat Immunol. 2001;2:397–402. doi: 10.1038/87692. [DOI] [PubMed] [Google Scholar]
- 14.Valenzuela JO, Hammerbeck CD, Mescher MF. Cutting edge: Bcl-3 up-regulation by signal 3 cytokine (IL-12) prolongs survival of antigen-activated CD8 T cells. J Immunol. 2005;174:600–604. doi: 10.4049/jimmunol.174.2.600. [DOI] [PubMed] [Google Scholar]
- 15.Corn RA, Hunter C, Liou HC, Siebenlist U, Boothby MR. Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J Immunol. 2005;175:2102–2110. doi: 10.4049/jimmunol.175.4.2102. [DOI] [PubMed] [Google Scholar]
- 16.McKeithan TW, Takimoto GS, Ohno H, Bjorling VS, Morgan R, Hecht BK, Dube I, Sandberg AA, Rowley JD. BCL3 rearrangements and t(14;19) in chronic lymphocytic leukemia and other B-cell malignancies: a molecular and cytogenetic study. Genes Chromosomes Cancer. 1997;20:64–72. [PubMed] [Google Scholar]
- 17.Martin-Subero JI, Ibbotson R, Klapper W, Michaux L, Callet-Bauchu E, Berger F, Calasanz MJ, De Wolf-Peeters C, Dyer MJ, Felman P, Gardiner A, Gascoyne RD, Gesk S, Harder L, Horsman DE, Kneba M, Kuppers R, Majid A, Parry-Jones N, Ritgen M, Salido M, Sole F, Thiel G, Wacker HH, Oscier D, Wlodarska I, Siebert R. A comprehensive genetic and histopathologic analysis identifies two subgroups of B-cell malignancies carrying a t(14;19)(q32;q13) or variant BCL3-translocation. Leukemia. 2007;21:1532–1544. doi: 10.1038/sj.leu.2404695. [DOI] [PubMed] [Google Scholar]
- 18.Chapiro E, Radford-Weiss I, Bastard C, Luquet I, Lefebvre C, Callet-Bauchu E, Leroux D, Talmant P, Mozziconacci MJ, Mugneret F, Struski S, Raynaud S, Andrieux J, Barin C, Jotterand M, Mossafa H, Ramond S, Terre C, Lippert E, Berger F, Felman P, Merle-Beral H, Bernard OA, Davi F, Berger R, Nguyen-Khac F. The most frequent t(14;19)(q32;q13)-positive B-cell malignancy corresponds to an aggressive subgroup of atypical chronic lymphocytic leukemia. Leukemia. 2008;22:2123–2127. doi: 10.1038/leu.2008.102. [DOI] [PubMed] [Google Scholar]
- 19.Szymanowska N, Klapper W, Gesk S, Kuppers R, Martin-Subero JI, Siebert R. BCL2 and BCL3 are recurrent translocation partners of the IGH locus. Cancer Genet Cytogenet. 2008;186:110–114. doi: 10.1016/j.cancergencyto.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Westerheide SD, Mayo MW, Anest V, Hanson JL, Baldwin AS., Jr The putative oncoprotein Bcl-3 induces cyclin D1 to stimulate G(1) transition. Mol Cell Biol. 2001;21:8428–8436. doi: 10.1128/MCB.21.24.8428-8436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rocha S, Martin AM, Meek DW, Perkins ND. p53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-kappaB subunit with histone deacetylase 1. Mol Cell Biol. 2003;23:4713–4727. doi: 10.1128/MCB.23.13.4713-4727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuwata H, Watanabe Y, Miyoshi H, Yamamoto M, Kaisho T, Takeda K, Akira S. IL-10-inducible Bcl-3 negatively regulates LPS-induced TNF-alpha production in macrophages. Blood. 2003;102:4123–4129. doi: 10.1182/blood-2003-04-1228. [DOI] [PubMed] [Google Scholar]
- 23.Muhlbauer M, Chilton PM, Mitchell TC, Jobin C. Impaired Bcl3 up-regulation leads to enhanced lipopolysaccharide-induced interleukin (IL)-23P19 gene expression in IL-10(-/-) mice. J Biol Chem. 2008;283:14182–14189. doi: 10.1074/jbc.M709029200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riemann M, Endres R, Liptay S, Pfeffer K, Schmid RM. The IkappaB protein Bcl-3 negatively regulates transcription of the IL-10 gene in macrophages. J Immunol. 2005;175:3560–3568. doi: 10.4049/jimmunol.175.6.3560. [DOI] [PubMed] [Google Scholar]
- 25.Buchau AS, MacLeod DT, Morizane S, Kotol PF, Hata T, Gallo RL. Bcl-3 acts as an innate immune modulator by controlling antimicrobial responses in keratinocytes. J Invest Dermatol. 2009;129:2148–2155. doi: 10.1038/jid.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohuslav J, Kravchenko VV, Parry GC, Erlich JH, Gerondakis S, Mackman N, Ulevitch RJ. Regulation of an essential innate immune response by the p50 subunit of NF-kappaB. J Clin Invest. 1998;102:1645–1652. doi: 10.1172/JCI3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wessells J, Baer M, Young HA, Claudio E, Brown K, Siebenlist U, Johnson PF. BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J Biol Chem. 2004;279:49995–50003. doi: 10.1074/jbc.M404246200. [DOI] [PubMed] [Google Scholar]
- 28.Fujita S, Seino K, Sato K, Sato Y, Eizumi K, Yamashita N, Taniguchi M. Regulatory dendritic cells act as regulators of acute lethal systemic inflammatory response. Blood. 2006;107:3656–3664. doi: 10.1182/blood-2005-10-4190. [DOI] [PubMed] [Google Scholar]
- 29.Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, Odden AR, Shellito JE, Bagby GJ, Nelson S, Kolls JK. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–769. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Happel KI, Zheng M, Young E, Quinton LJ, Lockhart E, Ramsay AJ, Shellito JE, Schurr JR, Bagby GJ, Nelson S, Kolls JK. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol. 2003;170:4432–4436. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenberger MJ, Strieter RM, Kunkel SL, Danforth JM, Goodman RE, Standiford TJ. Neutralization of IL-10 increases survival in a murine model of Klebsiella pneumonia. J Immunol. 1995;155:722–729. [PubMed] [Google Scholar]
- 32.Moore TA, Perry ML, Getsoian AG, Newstead MW, Standiford TJ. Divergent role of gamma interferon in a murine model of pulmonary versus systemic Klebsiella pneumoniae infection. Infect Immun. 2002;70:6310–6318. doi: 10.1128/IAI.70.11.6310-6318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruan Q, Zheng SJ, Palmer S, Carmody RJ, Chen YH. Roles of Bcl-3 in the pathogenesis of murine type 1 diabetes. Diabetes. 2010;59:2549–2557. doi: 10.2337/db10-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carmody RJ, Chen YH. Nuclear factor-kappaB: activation and regulation during toll-like receptor signaling. Cell Mol Immunol. 2007;4:31–41. [PubMed] [Google Scholar]