

Abstract
Stat5 is activated by multiple receptors of hematopoietic cytokines. To study its role during hematopoiesis, we have generated primary chicken myeloblasts expressing different dominant-negative (dn) alleles of Stat5. This caused a striking inability to generate mature cells, due to massive apoptosis during differentiation. Bcl-2 was able to rescue differentiating cells expressing dnStat5 from apoptosis, suggesting that during cytokine-dependent differentiation the main function of the protein is to ensure cell survival. Our findings with dnStat5-expressing chicken myeloblasts were confirmed with primary hematopoietic cells from Stat5a/Stat5b-deficient mice. Bone marrow cells from these animals displayed a strong increase in apoptotic cell death during GM-CSF-dependent functional maturation in vitro. The antiapoptotic protein Bcl-x was induced by GM-CSF and IL-3 in a Stat5-dependent fashion. Ectopic expression of Bcl-x rescued Stat5-deficient bone marrow cells from apoptosis, indicating that Stat5 promotes the survival of myeloid progenitor cells through its ability to induce transcription of the bcl-x gene. Finally, the recruitment of myeloid cells to inflammatory sites was found strongly impeded in Stat5-deficient mice. Taken together, our findings suggest that Stat5 may promote cytokine-dependent survival and proliferation of differentiating myeloid progenitor cells in stress or pathological situations, such as inflammation.
Keywords: Stat5, hematopoiesis, apoptosis, myeloid differentiation
During hematopoiesis, multiple types of mature blood cells are generated from a pluripotent hematopoietic stem cell (for review, see Morrison et al. 1997). Stem cells undergo a series of developmental decisions during which their progeny commits towards particular hematopoietic lineages. At the same time, rapid proliferation occurs to expand pools of progenitor cells during their progression to terminal differentiation. Survival, proliferation and differentiation of hematopoietic progenitor cells require the regulation of gene expression by signals originating from cell surface receptors for hematopoietic growth factors (Dexter and Spooncer 1987; Bedi and Sharkis 1995). Most of these receptors belong to the cytokine receptor family lacking intrinsic enzymatic activity that transduces signals using permanently associated Janus nonreceptor tyrosine kinases (Jaks) (Ihle 1995). Immediate targets of receptor-activated Jaks are the signal transducers and activators of transcription (Stats) (Darnell et al. 1994; Leonard and O'Shea 1998). Tyrosine-phosphorylated Stats dimerize via SH2 domains, translocate to the nucleus, and bind to a class of cytokine response elements designated γ interferon-activation site (GAS), after the prototype sequence found in the promoters of IFN-γ-responsive genes (Decker et al. 1997). Jak–Stat signal transduction thus mediates rapid regulation of genes in response to hematopoietins and many other cytokines.
Various cytokines that orchestrate growth, survival, and differentiation of hematopoietic cells from different lineages cause the activation of a particular Stat family member, Stat5. In mammals, two isoforms of this protein, Stat5a and Stat5b, are encoded by closely related, but distinct, genes (Wakao et al. 1994; Azam et al. 1995; Mui et al. 1995). The use of Stat5 in the signal transduction paths originating from multiple hematopoietin receptors suggested that this particular Stat family member might have an essential function during hematopoietic development of multiple cell lineages. This assumption was tested by targeted disruption of both Stat5 genes. In mice Stat5a/b deficiency caused a defect in the proliferation of activated T cells but no overt abnormalities in peripheral erythropoiesis or myelopoiesis, that is, the generation of mature erythrocytes, granulocytes, platelets, and monocytes (Teglund et al. 1998; Moriggl et al. 1999). In vitro, a reduced ability of Stat5a/b-deficient hematopoietic cells to form myeloid colonies in response to cytokines acting via Stat5, such as interleukin-3 (IL-3) and granulocyte–macrophage colony-stimulating factor (GM-CSF), was observed (Teglund et al. 1998).
Studies in hematopoietic cell lines or in T cells from Stat5 knockout mice identified several direct or indirect Stat5 target genes that regulate cell growth or differentiation. Among these bona fide Stat5 target genes are cyclin-dependent kinases (CDKs), inhibitors of CDKs, and G1 cyclins (Matsumura et al. 1997; Moriggl et al. 1999). Similar to Stat3, a family member suggested to play a role in the antiapoptotic activity of IL-6 (Fukada et al. 1996, 1998; Takeda et al. 1998; Catlett-Falcone et al. 1999), Stat5 may regulate cell proliferation also through an inhibitory effect on apoptosis (Zamorano et al. 1998; Nosaka et al. 1999), but this has not been tested in primary hematopoietic cells, and the possible implications for the physiology of humans or animals are not known.
The Bcl-2 family of proteins includes the Bcl-2 and Bcl-xL intracellular inhibitors of apoptosis (Reed 1997). Expression of Bcl-xL, but not of Bcl-2, was found to be regulated by hematopoietic growth factors (Packham et al. 1998). Previous studies investigating the signals needed for Bcl-x expression agreed about an involvement of Janus kinases but differed in finding a requirement for Stats (Packham et al. 1998; Catlett-Falcone et al. 1999; Dumon et al. 1999; Silva et al. 1999).
In the course of our own studies we identified Stat5 as a transcription factor whose activation correlated with macrophage differentiation of human leukemic cell lines in response to various stimuli (Eilers et al. 1994; Woldman et al. 1997). A similar correlation was found between Stat5 activation and myelomonocytic differentiation of primary chicken myeloblasts (Woldman et al. 1997). Infection of chicken bone marrow cells with the ts21E26 retrovirus encoding a temperature-sensitive gag–myb–ets oncogene yields homogenous, differentiation-arrested populations of proliferating myeloblasts at the permissive temperature (37°C). These cells require chicken myelomonocytic growth factor (cMGF) for proliferation. At the nonpermissive temperature (42°C) the temperature-sensitive gag–myb–ets oncogene is inactivated, and terminal differentiation into macrophages occurs within 3–4 days in a cMGF-dependent fashion (Beug et al. 1984, 1987). The outstanding feature of this primary hematopoietic model system is that both progenitor renewal (proliferation without differentiation) and terminal differentiation (again including cell proliferation) can be independently subjected to experimental manipulation. In normal avian and murine progenitors, these two responses to cytokines are intimately linked and inseparable.
In this study we used chicken myeloblasts expressing dominant-negative Stat5 alleles, as well as hematopoietic cells from Stat5-deficient mice, to analyze the role of Stat5 in survival, proliferation, and differentiation of primary myelomonocytic cells. We show that the absence of functional Stat5 results in decreased doubling times and lack of cytokine-dependent protection from apoptotic death during cell differentiation. We propose a mechanism for protection from apoptosis by Stat5, demonstrating a role for bcl-x as an antiapoptotic Stat5 target gene.
Results
Expression of dominant-negative Stat5 alleles in chicken myeloblasts
To functionally inactivate Stat5 in chicken cells, we attempted to express dominant-negative (dn) alleles of the protein. Recent studies from our laboratory demonstrated the activation of a Stat5 chicken homolog (cStat5) in the cMGF-induced signaling pathway of chicken myeloblasts and suggested a considerable extent of similarity between chicken and mammalian Stat5 (Woldman et al. 1997). To ascertain that the known, mammalian dominant-negative alleles of Stat5 would actually function in chicken cells, we cloned chicken Stat5. cStat5 cDNAs were obtained from libraries derived from chicken macrophage- or T-cell lines. Several, independently isolated cDNAs predicted a protein, cStat5b, with 90.4% identity to human Stat5b and 91.4% identity to murine STAT5b (GenBank accession no. AF074248). cStat5 displayed both hallmarks of mammalian Stat5b: (1) the conserved carboxyl terminus of Stat5b, which is 12 amino acids shorter than that of Stat5a and divergent in sequence for another 9 amino acids; and (2) a small stretch of amino acids around position 690 that is present in Stat5b, but absent in Stat5a. A second cStat5 cDNA predicted a protein resembling mammalian Stat5b at the carboxyl terminus but lacking the characteristic amino acid stretch at position 690 (data not shown). Therefore, this second cStat5 isoform represents a hybrid Stat5a–Stat5b protein with regard to mammalian Stat5, suggesting that a Stat5 gene with the Stat5b carboxyl terminus existed prior to the duplication of Stat5 genes during evolution.
These results made it very likely that mammalian dominant-negative Stat5 alleles would function in chicken cells. This was confirmed using clones of ts21E26 myeloblasts infected with retroviral vectors containing either no Stat5 or either one of two dominant-negative alleles of mouse Stat5b, Stat5dC, and Stat5Y699F. The dominant-negative (dn) Stats were tagged with amino-terminal myc epitopes and linked to the green fluorescent protein (GFP) via an internal ribosomal entry site. GFP-positive myeloblast clones expressed mostly high, but sometimes only moderate, levels of the retrovirally transduced Stat5 proteins (data not shown). As expected, activation and DNA binding of endogenous Stat5, as determined by EMSA (Mellitzer et al. 1996), was completely inhibited in clones expressing high levels of Stat5Y699F, whereas those expressing similarly high levels of Stat5dC produced an additional, strongly shifted band (data not shown). This transcriptionally inactive allele acts by displacing endogenous Stat5 from cognate promoter binding sites (Moriggl et al. 1996). As expected, both dnStat5 alleles inhibited cMGF inducibility of a transiently transfected reporter gene regulated by IFP53 GAS sequences, Stat5dC being more effective (Fig. 1A, left panel). Additionally, cMGF-mediated induction of cis, a known Stat5 target gene in mammals (for review, see Yoshimura 1998), was inhibited by Stat5dC (Fig. 1A, right panel). These experiments confirm that mouse Stat5b can be activated in chicken cells and that its dominant-negative alleles similarly interfere with the function of chicken Stat5 as with mammalian Stat5.
Figure 1.
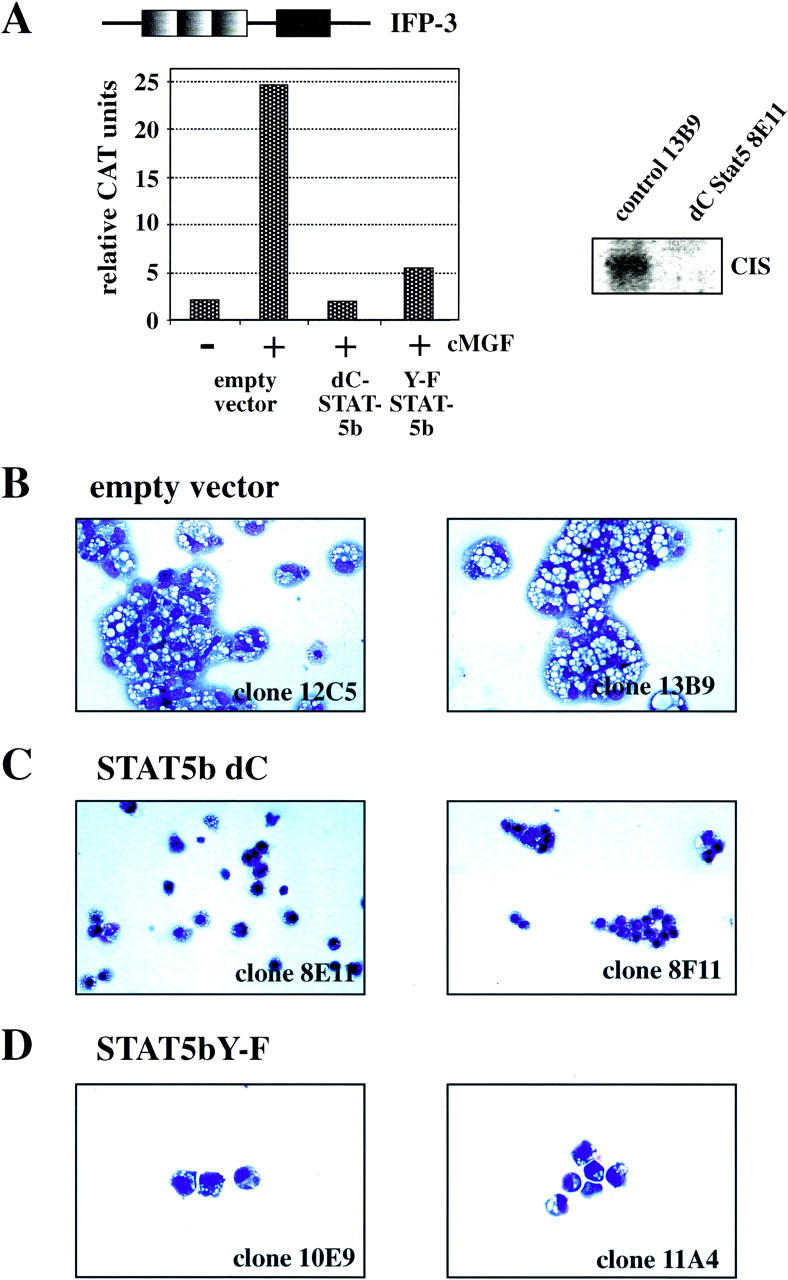
Impaired differentiation and increased cell death in dnStat5-expressing tsE26 myeloblasts after shift to 42°C. (A, left) Stat5dC and Stat5Y-F function as dominant-negative Stat5 versions. Clones verified for high dnStat5 expression by Western blot were transfected with a reporter construct containing three IFP-53 GAS sites (top) as described earlier (Woldman et al. 1997). (Right) Northern blot analysis for CIS mRNA expression, using a control clone and a Stat5dC-expressing clone after stimulation with cMGF. (B–D) Cells from two representative clones expressing empty vector (B), Stat5dC (C), or Stat5Y-F (D) were induced to differentiate at 42°C in the presence of cMGF and IGF-1 for 4 days, cytocentrifuged onto slides, stained with May-Gruenwald-Giemsa solution, and photographed. Note terminally differentiated macrophages with a highly excentric nucleus in B but dead cells (C) or small, partially differentiated cells (D) in the cultures expressing dominant-negative versions of Stat5.
dnStat5 alleles interfere with cMGF-dependent differentiation of chicken myeloblasts by causing apoptosis
To test biological functions of Stat5 in primary cells, five ts21E26 myeloblast clones lacking dnStat5b alleles and more than six clones expressing high levels of Stat5dC and Stat5Y699F were subjected to temperature-induced, cMGF-dependent differentiation into macrophage-like cells (Beug et al. 1984). As described earlier, myeloblasts infected with the ts21E26 retrovirus alone, when shifted from 37°C to 42°C in the presence of cMGF and IGF-1, differentiate into macrophages within 3–4 days (Woldman et al. 1997). ts21E26 clones containing the empty GFP-retrovirus vector behaved identically (shown for two typical clones in Fig. 1B). In contrast, differentiation induction of myeloblasts expressing Stat5dC resulted in a high proportion (>80%) of dead cells after 4 days, showing morphological features of apoptotic cells, like condensed or fragmented nuclei and an “empty” cytoplasm. In addition, a minor proportion of viable, but incompletely differentiated, cells was observed (Fig. 1C). Again, the Stat5dC allele produced a stronger phenotype as compared with Stat5bY699F. In two clones containing the latter Stat5 allele, fewer cells died, and a proportion of incompletely differentiated cells was obtained (Fig. 1D).
To determine whether cell death during cMGF-dependent myeloblast maturation was due to apoptosis, we performed daily TUNEL staining of myeloblast cultures differentiating at 42°C for up to 3 days and analyzed the cells by flow cytometry. FACS diagrams of apoptotic Stat5dC-expressing cells and viable control cells expressing empty vector are shown in Figure 2A. Quantitation of TUNEL-positive cells obtained with five clones derived of tsE26 myeloblasts infected with either empty vector, Stat5dC, or Stat5bY699F cells (Fig. 2B) demonstrated that empty vector cultures were virtually devoid of apoptotic cells (a possible consequence of the addition of IGF-1 to all cultures) (Frampton et al. 1996). In contrast, a large proportion of the cells expressing either one of the two dnStat5 alleles underwent apoptosis within 3 days. The frequency of apoptotic cells increased as differentiation proceeded, and, again, the Stat5dC variant was superior to Stat5bY699F in causing apoptotic cell death (Fig. 2B).
Figure 2.

dnStat5-expressing tsE26 myeloblasts undergo apoptosis during differentiation at 42°C. (A) Combined TUNEL/propidium iodide staining and FACS analysis of representative tsE26 myeloblast clones expressing either empty vector (left) or Stat5dC (right) 3 days after differentiation induction at 42°C. The gate setting for the TUNEL-positive cell population is indicated. (B) Five tsE26 clones each, expressing empty vector (black bars), Stat5Y-F (dotted bars), or Stat5dC (hatched bars) were induced to differentiate at 42°C. TUNEL-positive, apoptotic cells accumulating during differentiation were quantitated daily by TUNEL assay as shown in A. s.d.s are indicated by error bars.
Bcl-2 efficiently reverses the proapoptotic effect of dnStat5
The occurrence of apoptosis during temperature-induced differentiation of dnStat5b-expressing primary myeloblasts precluded a more detailed analysis whether or not dnStat5b would also retard or inhibit their maturation into functional macrophages. In a variety of situations, ectopic expression of the antiapoptotic Bcl-2 gene was shown to substitute for the life-sustaining activity of cytokines (Lagasse and Weissman 1997). We therefore tested, whether or not Bcl-2 expression could overcome the proapoptotic activity of dnStat5 during cMGF-dependent myeloblast differentiation and, if so, whether the cells protected from apoptosis would now undergo normal macrophage differentiation. To this end, chicken bone marrow cells were triple-infected with the transforming retrovirus ts21E26, a dnStat5–GFP retrovirus containing a hygromycin resistance gene and a Bcl-2-encoding virus containing a NeoR cassette. Green colonies, selected in the presence of cMGF, hygromycin, and G418, were expanded and checked for the expression of dnStat5 and Bcl-2. Several clones were obtained that expressed both transgenes. The same infection yielded control clones expressing only Stat5dC. (Fig. 3A).
Figure 3.
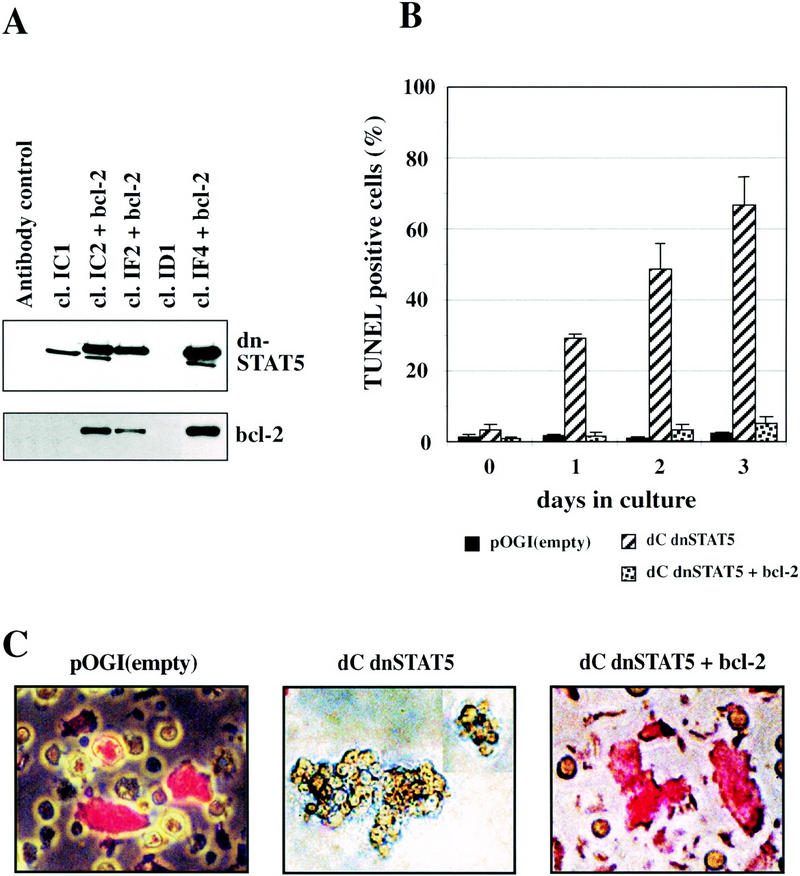
Bcl-2 expression rescues Stat5dC-expressing tsE26 myeloblasts clones from apoptosis during differentiation induction at 42°C. (A) Lysates of several tsE26 clones coinfected with dnStat5dC- and Bcl-2 expressing retroviruses (see Materials and Methods) as well as several control clones were analyzed for myc-tagged Stat5dC and Bcl-2 protein expression by Western blot. The lower molecular weight band detected by the myc antibody in some of the clones is a degradation product. (B) Several clones expressing either empty vector (black bars), dnStat5dC (hatched bars), or dnStat5C plus Bcl-2 (speckled bars) were induced to differentiate at 42°C and analyzed daily for apoptotic cells by TUNEL assay as described in the legend to Fig. 2. (C) Representative clones expressing empty vector (left), dnStat5dC (middle), or dnStat5dC + Bcl2 (right) were induced to differentiate for 3 days at 42°C and allowed to ingest Texas Red-labeled bacteria for 1 hr. Note large cells having phagocytosed bacteria (red fluorescence) in the control and dnStat5dC + Bcl2 expressing cells, whereas cells expressing dnStat5dC alone were nonphagocytic and resembled immature myeloblasts with an apoptotic morphology.
Expression of Bcl-2 in the Stat5dC clones completely rescued them from apoptosis; apoptosis after 3 and 4 days was almost as low as in tsE26 myeloblasts containing only the empty vector. In contrast, the clones expressing Stat5dC alone showed the expected high rate of apoptosis, strongly increasing with time at 42°C (Fig. 3B). After 3 days of differentiation, the cultures expressing both Bcl-2 and dnStat5 contained numerous large, irregularly shaped cells that were highly phagocytic (visualized by ingested Texas Red-labeled bacteria; Fig. 3C), whereas control cells expressing Stat5dC alone formed the expected clumps of small, partially apoptotic myeloblasts. This indicates that myelomonocytic progenitors do not require Stat5 for maturation into phagocytes upon expression of an antiapoptotic protein.
Primary myeloid cells from mice lacking Stat5a/b show a proliferation/differentiation defect accompanied by increased apoptosis
Our studies in primary chicken cells indicated that myeloid progenitors with an impaired function of Stat5 show increased apoptosis during terminal differentiation. Because mice deficient for Stat5a/b (Stat5a/b−/−) have a normal composition of the blood with respect to erythrocytes, platelets, granulocytes, and monocytes (Teglund et al. 1998), we sought to determine whether the lack of Stat5 would also cause apoptosis in primary, differentiating myeloid cells of mouse origin.
Fetal liver or bone marrow cells from Stat5-deficient and wild-type mice were first expanded in a serum-free medium devoid of polypeptide growth factors and hormones (StemPro34), supplemented with SCF, flk2/flt 3 ligand, IL-3, IL-6, GM-CSF, and Dexamethasone for 2 days (expansion factor mix; see Materials and Methods). During this time, aliquots of both cell preparations were infected with a retroviral vector coexpressing GFP and wild-type Stat5a, to directly show that possible alterations in the Stat5a/b−/− cells could be corrected by exogenous Stat5 expression. Thereafter, the cells were seeded in media containing either no growth factors, GM-CSF, M-CSF, or a cocktail of SCF, IL-3, and IL-6. Whereas GM-CSF and IL-3 are very potent stimulators of Stat5 activation, M-CSF activates Stat5 in cell lines overexpressing M-CSF receptor (c-Fms) (Barahmand-pour et al. 1995) but only very weakly in primary macrophages (M. Kieslinger et al., unpubl.).
In the presence of GM-CSF, the Stat5a/b−/− cells multiplied consistently slower, with an almost twofold reduction in cell numbers compared with wild-type cells (Fig. 4A, left). Expression of exogenous Stat5 in 60%–70% of the Stat5a/b−/− cells (GFP expression determined by FACS) restored proliferation to almost wild type (Fig. 4A, left). When the cells were incubated with a mix of myeloid growth factors (SCF, IL-3, IL-6), the difference between wild type and Stat5a/b−/− was further enhanced, amounting to a >2.5-fold reduction in cell numbers obtained from the Stat5a/b−/− cells. Again, the underlying defect was almost completely abolished by retroviral expression of wild-type Stat5.
Figure 4.

Cytokine-dependent proliferation/differentiation of primary myeloid fetal liver cells is impaired in Stat5a/b−/− mice. (A) Fetal liver cells (E12.5) from wild-type (Stat5a/b+/+) and Stat5a/b−/− mice were precultured in StemPro medium containing expansion factor mix; aliquots were infected with a Stat5a-expressing retrovirus (exo Stat5; see Materials and Methods). The cells were then switched to medium containing either GM-CSF (left) or IL3, IL-6, and SCF (right) and subjected to daily counting. Cumulative cell numbers are shown (mean values plus s.d. from three independent determinations). (B) Fetal liver cells from wild-type and Stat5a/b−/− mice were precultured and virus-infected as in A but switched to M-CSF instead of GM-CSF. Cell numbers were normalized to those obtained 3 days after switching to GM-CSF, to correct for the adaptation required from Stat5-dependent growth in expansion factor mix to Stat5-independent proliferation in M-CSF. (□) Stat5a/b−/−; (█) Stat5a/b−/−, exo Stat5; (○) Stat5a/b+/+; (●) Stat5a/b+/+, exo Stat5.
The cells exposed to M-CSF showed a more complex behavior. During the first 2 days, the Stat5a/b−/− cells multiplied more slowly than the Stat5 wild-type cells but then approached wild-type- proliferation rates. Re-expression of Stat5 only abolished this early lag phase in growth rate but did not affect proliferation after day 3 (not shown). We therefore concluded that preincubation of the cells during the infection period with growth factors acting via Stat5 (e.g., IL-3, GM-CSF) caused an initial difference that rapidly levelled off after the cells had adapted to M-CSF. As shown in Figure 4B, no significant differences in proliferation between Stat5 wild-type and Stat5a/b−/− cells were detectable after day 3. As expected, cells from both types of animals were strictly factor dependent, as shown by rapid loss of the viable cell population after cytokine withdrawal (data not shown).
To test whether the absence of Stat5 affected the differentiation phenotype of bone marrow cells after combined proliferation/differentiation induction in GM-CSF, analysis of cell surface markers (flow cytometry) was performed. In each case, ∼90% of the final cell population was positive for the myeloid markers GR-1 and MAC-1/CD11b but negative for erythroid, lymphoid, and multipotent cell markers such as Ter119, B220, CD3, CD34, and CD117/c-KIT/SCF-receptor, respectively (data not shown). Only minor alterations in the myeloid cell types were detected, such as a slightly larger proportion of Gr-1+/Mac-1low cells in the Stat5-deficient mice (data not shown), indicating that Stat5 mainly influenced the quantitative, not the qualitative, result of myelopoiesis.
Prompted by our results in differentiating chicken myeloblasts, we then compared apoptosis occurring in cultures from Stat5 wild-type and Stat5a/b−/− cells. Cells were cultured in GM-CSF, M-CSF, and no factor as described above. After 4 days, aliquots of the culture were subjected to AnnexinV staining, to distinguish early as well as late stages of apoptosis (Zhang et al. 1997; see Fig. 5A). The Stat5 wild-type cells grown in GM-CSF exhibited a relatively high frequency of apoptotic cells (13% total, 5% in late apoptotic stages; Fig. 5A,B). This is explained by the fact that the cultures initially contained cells from diverse lineages, some of which (i.e., erythroid cells) were nonresponsive to GM-CSF and therefore entered apoptosis. Interestingly, and in complete accord with our chicken cell data (see Fig. 2), a significant increase of apoptotic cells was observed in the Stat5a/b−/− cells. These contained >50% apoptotic cells at the time of analysis (Fig. 5A,B). In line with our proliferation data, the cells grown in M-CSF showed very little difference in apoptosis rates between Stat5 wild-type and Stat5a/b−/− cells (Fig. 5B). In summary, these data suggest that Stat5 protects myeloid progenitors from apoptosis during murine and avian hematopoiesis.
Figure 5.
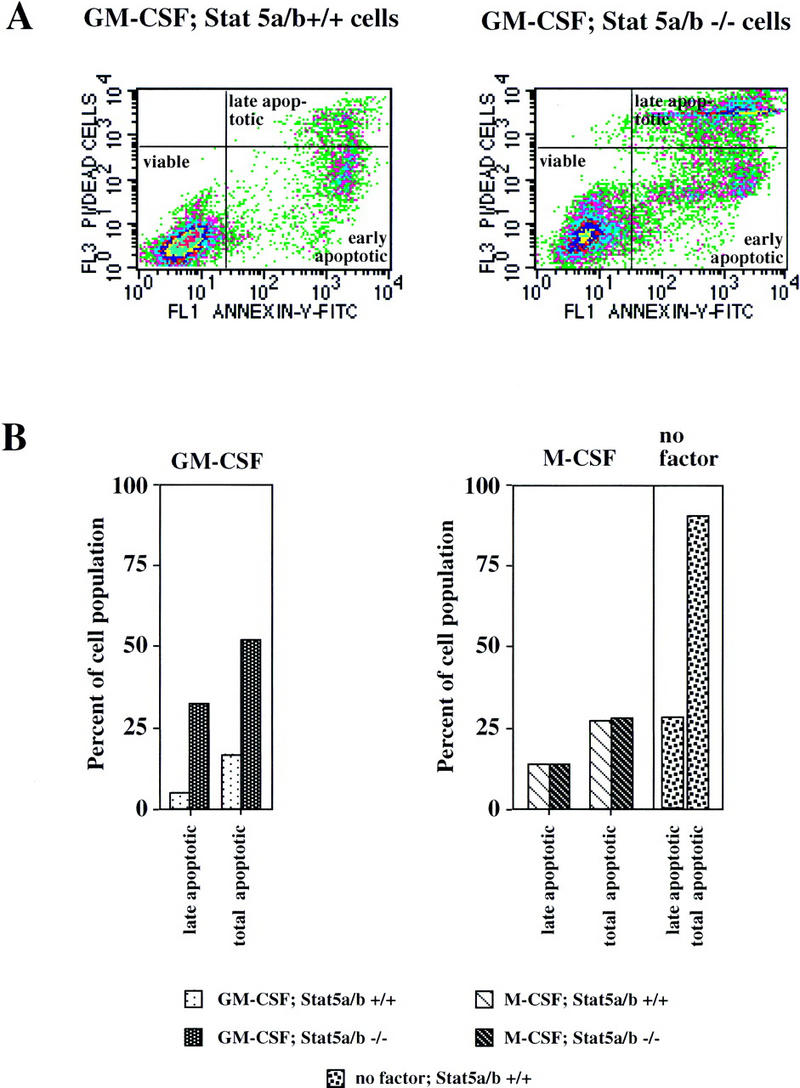
Impairment of cytokine-dependent myeloid cell proliferation/differentiation in Stat5a/b−/− mice is due to increased apoptosis. Cells from wild-type (Stat5a/b+/+) and Stat5a/b−/− mice were cultivated as described in the legend of Fig. 4 and analyzed for apoptotic cells by Annexin V staining and FACS analysis 4 days after switch to GM-CSF, M-CSF, or no factor. (A) Annexin V staining of Stat5a/b+/+ and Stat5a/b−/− cells treated with GM-CSF. This method distinguishes between early (bottom right gate) and late stages of apoptosis (top right gate). (B) Quantitative determination of late apoptotic (bottom right gate) and total apoptotic (bottom plus top right gate) in Stat5a/b+/+ and Stat5a/b−/− cells treated as in A. Note the strong up-regulation of late and total apoptotic cells from Stat5a/b−/− when treated with GM-CSF, no difference between Stat5a/b−/− and wild-type cells in response to M-CSF, and massive apoptosis of wild-type cells after factor withdrawal.
Bcl-x is a Stat5 target gene and promotes the survival of myeloid progenitor cells
In avian myeloblasts, the antiapoptotic protein Bcl-2 was able to prevent cells expressing dnStat5 from apoptosis during differentiation. However, Bcl-2 was not induced by cMGF, indicating that it is not a Stat5 target gene. The related Bcl-xL protein can also protect cells from apoptosis during cytokine responses. Moreover, the bcl-xL gene contains a doublet of functional Stat binding sites in its promoter (Grillot et al. 1997; Silva et al. 1999), and these sequences are strongly conserved between the murine and human genes (Fig. 6A). We therefore assayed murine myelomonocytic progenitor cells after starvation and subsequent stimulation with GM-CSF or IL-3 for cytokine-dependent Bcl-x expression. Both cytokines induced bcl-x mRNA in bone marrow cells from wild-type mice (Fig. 6B). Moreover, the bcl-x gene was directly targeted by IL-3- and GM-CSF-activated transcription factors because mRNA induction occurred in the presence of cycloheximide. Consistent with the mRNA induction, GM-CSF induced the synthesis of Bcl-x protein (Fig. 6B). In Stat5-deficient bone marrow cells, no induction of bcl-x mRNA and protein by IL-3 and GM-CSF was observed (Fig. 6B). Together with the cycloheximide result, this establishes Bcl-x as a Stat5 target gene in GM-CSF and IL-3 responses.
Figure 6.
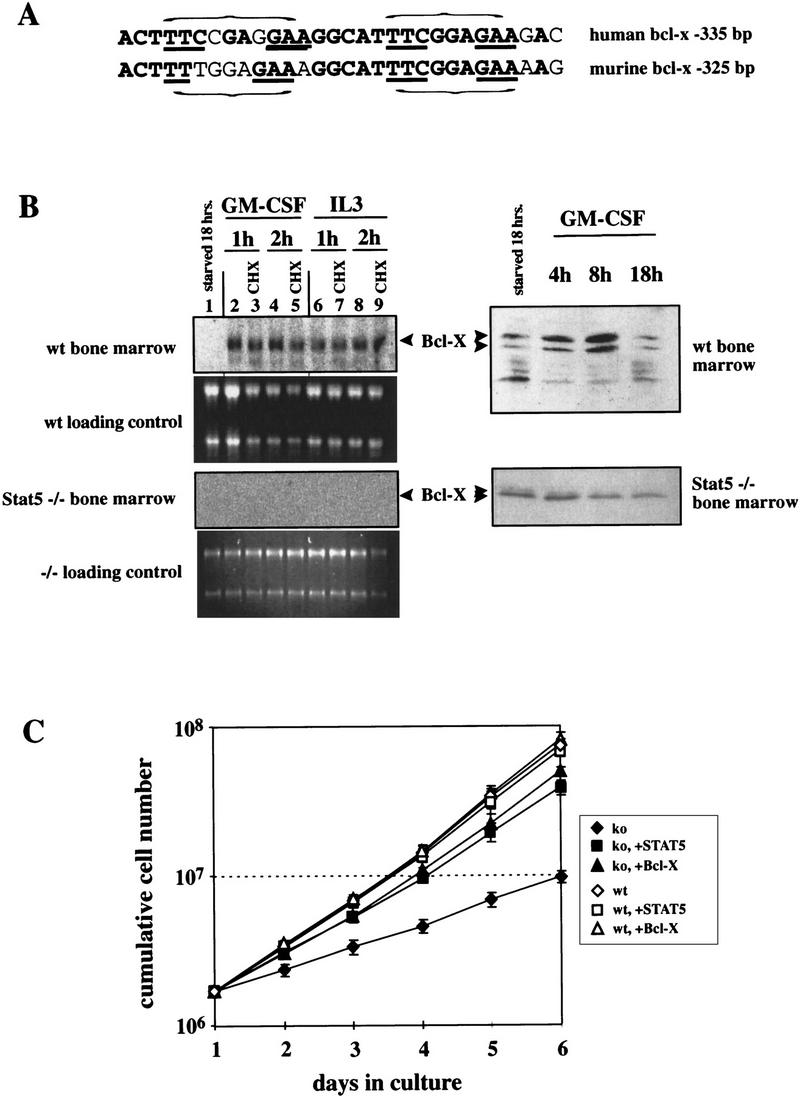
Bcl-x is a Stat5 target gene and mediates survival of differentiating myeloid progenitor cells. (A) Sequence comparison of putative, highly conserved Stat binding regions in the promoters of the human and murine bcl-x genes. (Bold) Homologous regions; (underlined) essential sequence elements of the Stat binding sites (indicated by horizontal brackets). (B) Bone marrow cells from wild-type or Stat5a/b-deficient mice were precultured as described for Fig. 4, withdrawn from factors for 18 hr and restimulated with GM-CSF or IL-3 [minus or plus cycloheximide (CHX) as indicated]. Total RNA was subsequently analyzed by Northern blotting (left) using a Bcl-x probe, and protein extracts were subjected to Western blotting with Bcl-x antibodies. (C) Bone marrow cells from wild-type or Stat5a/b-deficient mice were infected with retrovirus expressing Stat5 or Bcl-x while proliferating in expansion factor mix (see Materials and Methods). The infected cells were transferred to GM-CSF-containing medium and cumulative cell numbers were determined at the times indicated.
To further investigate a causal relationship between the absence of Bcl-x induction in Stat5-deficient hematopoietic progenitor cells and the induction of apoptosis during differentiation, a genetic rescue of precultured bone marrow cells was performed. Cells from Stat5-deficient mice produced only ∼15% of the progeny observed with cells from wild-type mice after 6 days of culture in GM-CSF. After infection with recombinant retrovirus, this defect was corrected equally well by either constitutive Stat5 or Bcl-x expression (Fig. 6B), whereas empty vector was without any effect (data not shown). In accordance with our data obtained with hematopoietic progenitor cells from chicken, the results of Figure 6 indicate that Bcl-x rescues differentiating cultures of Stat5a/b−/− mouse bone marrow cells by preventing their apoptosis.
Effect of Stat5 deficiency on the proliferation of immature hematopoietic progenitors
So far, our data did not address the question of whether the reduced proliferation rate of Stat 5−/− cells differentiating in GM-CSF or Il-3/Il-6/SCF is entirely due to increased apoptosis or whether the proliferation rate of viable cells is also affected. In the differentiation-arrested, immature avian myeloblasts transformed by temperature-sensitive gag–myb–ets at 37°C (Beug et al. 1984, 1987), dnStat5 (Stat5dC) slowed down proliferation (doubling time increased from 19 to 32 hr) but completely failed to induce significant apoptosis. The same was true in chicken myeloblasts coexpressing exogenous Stat5dC and Bcl-2 (data not shown). To investigate whether this was an effect due to oncogenic transformation or also occurred in nontransformed, immature progenitors, fetal liver cells from Stat5a/b−/− or wild-type mice were cultivated in expansion factor mix for 6 days (SCF, flk2/flt 3 ligand, IL-3, IL-6, GM-CSF, and dexamethasone). As will be published elsewhere, this factor combination caused sustained proliferation of immature cells (c-Kit/CD34/Sca1 positive), able to differentiate into multiple lineages after factor withdrawal. Such cultures from Stat5−/− mice exhibited an approximately twofold lower proliferation rate than wild-type cultures (doubling time increased from 21 to 39 hr) but showed only mildly enhanced apoptosis (wild-type, 9/15% late/total apoptosis; −/−: 15/21% late/total apoptosis; see legend to Fig. 4). Apoptosis occurred almost exclusively in differentiating, lineage marker positive cells, because immature cells, gated for CD117/c-Kithigh; Ter119 and Mac-1low cells, showed only 2% and 3% total apoptotic cells in wild-type and Stat5−/− populations, respectively. Our data suggest that Stat5 also plays a role in proliferation induction of immature hematopoietic cells prior to lineage commitment or terminal differentiation. Final proof of this idea will have to await the analysis of pure, clonal populations of such immature progenitors (as available in the chicken by gag–myb–ets oncogene transformation).
Effect of Stat5 deficiency on bone marrow mobilization during inflammation
Based on the clear effects of Stat5 deficiency on the generation of mature myeloid cells in vitro, one might expect a limited ability of Stat5-deficient mice to produce mature macrophages and granulocytes in situations of severe challenge, for example, during acute inflammation. We tested this assumption by intraperitoneal injection of thioglycollate. This treatment causes acute peritonitis and formation of an exudate that contains inflammatory cells recruited from the bone marrow. Mobilization of progenitor cells is very rapid; 3–4 days after thioglycollate injection, the exudate contains predominantly mature macrophages, granulocytes, and some lymphocytes (Daems and Koerten 1978). Peritoneal exudates from Stat5a/b-deficient mice contained almost three times less cells than exudates of wild-type mice (Fig. 7A). When exudate cells from wild-type mice were analyzed by flow cytometry for phagocytosis and cell surface marker expression, the gate characteristic of myeloid cells contained mostly phagocytes expressing high levels of the Mac-1/CD11b surface antigen, an attribute of mature macrophages (Fig. 7B). The myeloid gate in exudates from Stat5a/b-deficient mice also contained predominantly phagocytic cells, but with a higher proportion of cells expressing lower levels of Mac-1/CD11b, most likely granulocytes (Fig. 7B). Quantitative analysis of the myeloid gated cells showed that exudates of Stat5-deficient mice contained 10 times less mature, phagocytic myeloid cells than exudates of wild-type mice. In contrast, the number of lymphoid cells was reduced less than twofold in the Stat5a/b−/− exudates (Fig. 7C). These data show a decreased ability of Stat5-deficient mice to rapidly deliver inflammatory cells to a peripheral site of inflammation.
Figure 7.
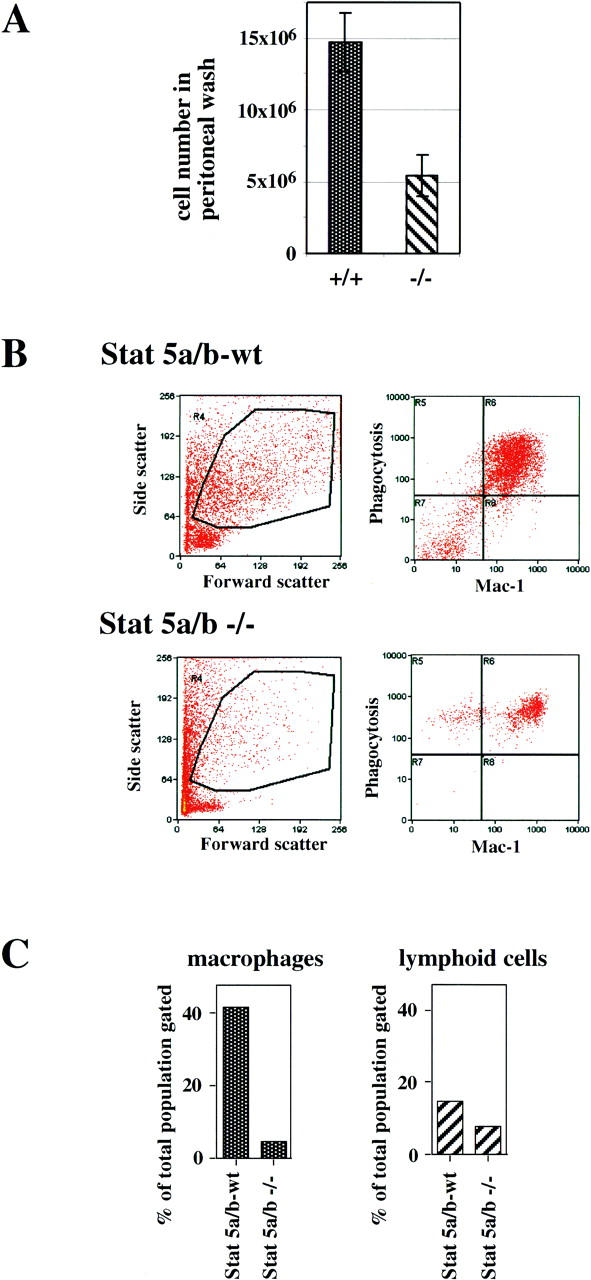
Recruitment of inflammatory myeloid cells to the peritoneal cavity of wild-type and Stat5a/b-deficient mice. Wild-type and Stat5a/b-deficient mice (three each) were injected intraperitoneally with thioglycollate (4% wt/vol). Three days later, inflammatory cells were collected by peritoneal lavage and analyzed. (A) Total cell numbers isolated from the peritoneal cavity (see Materials and Methods). (B) Analysis of peritoneal cells by flow cytometry: Cells were gated for the myeloid cell population (left) and analyzed for phagocytosis of Texas Red-labeled bacteria plus expression of the Mac-1 surface marker (right). (C, left) Total numbers of Mac-1/phagocytosis-positive myeloid cells (macrophages, calculated from top right quadrants in B). (Right) Total numbers of lymphoid-gated cells (low forward and side scatter, not shown).
Discussion
Experiments in established cell lines suggested a potentially important role for Stat5 in hematopoiesis driven by soluble hematopoietins (for review, see Mui 1999). However, the data raised in Stat5a/b−/− mice indicated that Stat5 plays no major role during steady-state hematopoiesis (Teglund et al. 1998). Therefore, it remained an open question whether and how Stat5 affects proliferation, differentiation, and/or apoptosis of primary hematopoietic cells. In an attempt to study the function of Stat5 in primary bone marrow, we investigated avian myelomonocytic progenitors expressing dominant-negative Stat5 alleles as well as primary progenitors from mice in which the Stat5a/b genes had been ablated by targeted gene disruption. In avian myeloblasts transformed by a temperature-sensitive oncoprotein (temperature-sensitive p135gag–myb–ets) progenitor renewal (proliferation without differentiation; at 37°C) or terminal differentiation (shift to 42°C) can be induced at will. Thus, homogenous populations of cells synchronously undergoing terminal differentiation can be obtained, to separate effects of Stat5 on differentiation from those on proliferation. Because primary mouse myeloid progenitors proliferate and differentiate at the same (Dexter and Spooncer 1987), the effects observed in the wild-type and Stat5−/− murine cells are best compared with those seen in the tsE26 cells differentiating at 42°C in the presence of cMGF. We show here that Stat5 loss of function abolishes protection from apoptosis during cytokine-dependent terminal differentiation in both chicken myeloblasts and mouse bone marrow cells.
Differentiation of ts21E26 myeloblasts in the presence of cMGF alone was reported to be accompanied by apoptotic death of a major fraction of the cells (Frampton et al. 1996). In our experiments, the addition of IGF-1 and cMGF during differentiation caused the vast majority of cells to survive, and only the lack of a functional Stat5 protein caused significant apoptosis instead of terminal differentiation. Of note, IGF-1 was reported to enhance Epo-mediated Stat5 tyrosine phosphorylation (Okajima et al. 1998). A similar IGF-1-induced enhancement of Stat5 activation by cMGF may explain why ts21E26 myeloblast differentiation occurs without any signs of apoptosis in the presence of both cytokines and also the failure of IGF-1 to protect the cells expressing dnStat5 from apoptosis during differentiation.
Bcl-2 was able to rescue differentiating cells expressing dnStat5, allowing them to mature into large, highly phagocytic cells. This indicates that Stat5 is primarily involved in keeping cells alive during differentiation. Unlike lineage-determining transcription factors, for example, from the GATA or Ets families (Bedi and Sharkis 1995), Stat5 is probably not required for lineage decisions or functional maturation during commitment and differentiation. Rather, it may control the number of myeloid progenitors and, possibly, those from other lineages by supporting rapid growth and survival during differentiation. Bcl-2, although able to revert the apoptotic effect of dnStat5, is not induced by cMGF and therefore not a Stat5 target gene (data not shown). In contrast, the Bcl-xL protein was shown to be a Stat5 target gene in mouse hematopoietic cells treated with IL-3 or GM-CSF. Moreover, Bcl-x was able to rescue Stat5-deficient cells from apoptosis in a similar fashion as the reintroduction of Stat5. Based on these results, we propose that lack of Bcl-xL induction by IL-3 or GM-CSF is mainly responsible for the phenotype resulting from Stat5 deficiency in myeloid progenitor cells.
Earlier findings by others concerning a potential role of Stat5 in cell proliferation and differentiation are contradictory. In Stat5-deficient mice, no gross growth defects or lack in the production of differentiated blood cells was observed. The minor growth retardation observed in cultured mature macrophages from Stat5a−/− mice (Feldman et al. 1997) is inconclusive, because the assay used did not discriminate between a true increase in cell numbers versus a size increase of the essentially nonproliferating macrophages during terminal differentiation. Studies using transfected mutants of various cytokine receptors lacking the docking sites required for activation of Stat5 reported little effect on the proliferation of some very immature hematopoietic cell lines if the cells were stimulated with saturating concentrations of cytokines (Damen et al. 1995; Fujii et al. 1995; Quelle et al. 1996). In contrast, a reduction of growth rate occurred in some of these mutant receptor cells when exposed to suboptimal cytokine levels (Damen et al. 1995; Chretien et al. 1996). Moreover, dnStat5 was able to reduce, but not suppress, factor-dependent growth of hematopoietic cell lines (Mui et al. 1996). Our data suggest that in myeloid progenitor cells activation of Stat5 is not a necessary requirement for cell proliferation and that apoptosis during differentiation was clearly the major reason for the reduced increase in cell numbers of Stat5-deficient progenitors exposed to myeloid growth/differentiation factors. However, Stat5 may serve as an enhancer of proliferative signals, enhancing the ability of cells to grow in nonsaturating doses of cytokines and other regulators. This is suggested by dnStat5 reducing the proliferation rate of gag–myb–ets transformed avian myeloblasts and by the impaired proliferation of very immature progenitors from Stat5−/− mice. In both cases, increased apoptosis was not involved, and changes in cyclin D2 or CDK6 mRNA levels on IL-3 or GM-CSF treatment were not observed (M. Kieslinger et al., unpubl.). In this respect, primary myeloid cells may differ from primary T cells that display a more absolute requirement for Stat5 in IL-2-mediated proliferation (Moriggl et al. 1999).
The role of Stat5 as an accelerator rather than an essential prerequisite of myelopoiesis may explain the apparent contradiction between in vitro cultures and in vivo homeostasis of hematopoietic cells with regard to the effect of Stat5 deficiency. Alternatively (and additionally), in vitro and in vivo data are reconciled by the concept that secreted cytokines play a minor, if any, role during steady-state hematopoiesis in the healthy organism. Cytokines may exert major, pleiotropic effects on the generation of myeloid cells in vitro but are nevertheless dispensable for steady state hematopoiesis in vivo (Ihle 1992; Dranoff et al. 1994; Stanley et al. 1994). Furthermore, mature macrophages could be generated in vivo in mice lacking both GM-CSF and CSF-1 (Lieschke et al. 1994). Therefore, Stat5 may be a transcription factor dispensable for steady state hematopoiesis but important for stress/pathological situations in which rapid mobilization of progenitor cells from hematopoietic organs involves secreted cytokines like GM-CSF. In line with this assumption, Stat5a/b-deficient mice exhibited a severely impaired influx of progenitor cells from bone marrow into the peritoneum under conditions of inflammation (Fig. 9). Several findings argue against the assumption that this impaired inflammatory cell recruitment is secondary to the T-cell defect seen in older Stat5a/b-deficient mice. First, experiments were solely performed with young mice not yet displaying overt T-cell abnormalities. Moreover, earlier findings demonstrated that the influx of monocytes and macrophages to the peritoneum in response to sterile inflammatory agents was normal in the absence of functional T cells (Stinnett and Majeski 1980). However, the possibility that abnormal, Stat5-deficient T cells create a suppressive environment with regard to bone marrow mobilization elsewhere in the body needs to be further investigated.
In conclusion, we suggest that the frequent use of Stat5 by receptors of hematopoietic growth factors does not indicate a general requirement for the protein during hematopoiesis. Rather, Stat5 may be instrumental in multiple hematopoietic lineages by promoting cell survival, and, possibly, as an accelerator of progenitor cell proliferation during commitment and differentiation in abnormal/pathological situations, such as stress, inflammation, etc., where a massive recruitment of progenitor cells to particular sites of the body occurs.
Materials and methods
Cells and cell culture
Cell culture of E26-virus-transformed primary avian myeloblasts was performed as described previously (Beug et al. 1984; Woldman et al. 1997). Bone marrow cells from C57BL/6 and STAT5a/b−/− mice (Teglund et al. 1998) were prepared from femurs of 6- to 8-week old mice, lacking apparent signs of T cell related abnormalities like splenomegaly. Fetal liver cells were prepared from day 12 to day 14 embryos. Briefly, fetal livers were removed from the embryos, suspended in StemPro Medium, passed several times through a micropipette, and filtered through a cell strainer (70 μm, Falcon). All mouse cells were cultivated in StemPro-34 SFM (serum-free medium) (Life Technologies). Recombinant murine GM-CSF, SCF, IL-3, flt 3 ligand, and human IL-6 were obtained from R&D Systems, recombinant human M-CSF from Genzyme, and Dexamethasone (Dex) from Sigma. Concentrations of cytokines used were as follows: 10 ng/ml IL-3; 100 ng/ml SCF; 5 ng/ml IL-6; 20 ng/ml flt 3 ligand; 15 ng/ml M-CSF; 1 ng/ml GM-CSF; and 40 ng/ml IGF-1. For initial expansion (and for long-term propagation of immature progenitors), cells were maintained in SCF, flt 3 ligand, GM-CSF, IL-3, IL-6, dexamethasome, and IGF-1. Combined myeloid cell proliferation and differentiation was induced either in GM-CSF, M-CSF, or GM-CSF plus IL-3 and SCF.
To obtain peritoneal cells, 8-week-old C57BL/6 and STAT5a/b−/− mice were injected intraperitoneally with 1 ml of sterile, 4% thioglycollate. After 3 days, mice were sacrificed; the peritoneum was flushed with 8 ml of PBS. Five milliliters of the resulting cell suspensions was used for further analysis. Total number of cells were counted using a CASY I cell analyser (Schaerfe Systems, Inc., Germany).
Phagocytosis assay
Cells (1 × 106) were incubated with sonicated, Texas Red-conjugated Escherichia coli (Molecular Probes) for 1 hr, stained with FITC-conjugated Mac-1 antibody and analyzed as described above. Avian macrophages were photographed after phagocytosis as above in either white light (to reveal histological details) or monochromatic green light suitable to excite Texas Red fluorescence (546 nm). These photographs were then overlaid by use of Adobe Photoshop software.
Analysis of cell proliferation
Murine bone marrow or fetal liver cells were maintained in the factor combinations indicated, counted daily, and appropriately diluted to keep cell numbers between 1.5 × 106 and 4 × 106 cells/ml. Cumulative cell numbers were calculated using the dilution factors of individual samples.
Antibodies
Previously described antibodies against human STAT5a and STAT5b were used at a final concentrations of 1/5000 for Western Blot analysis. Antibodies against Bcl-x (B22620) were purchased from Transduction Laboratories and used at dilutions of 1/500. Supernatants of 9E10 hybridoma cells were used to obtain anti-myc-tag antibodies. For FACS analysis, the following antibodies were purchased from Pharmingen: phycoerythrin (PE)-conjugated-anti-B220 (RA3-6B2), PE-conjugated-anti-TER119, PE-conjugated-anti-CD3 (145-2C11), fluorescein isothiocyanate (FITC)-conjugated-anti-CD117 (2B8), FITC-conjugated-anti-GR-1 (RB6-8C5), FITC-conjugated-anti-Mac-1 (M1/70), and FITC-conjugated-anti-CD34 (RAM34).
Cloning of cStat5 cDNAs
The cDNA containing the 5′-incomplete coding sequence for the homolog cStat5 was cloned from 1 of 10 pools of a chicken macrophage library (HD11 cells). The complete coding sequence of cStat5b was isolated from one of six pools of a T-cell library (Weining et al. 1996). In both cases, PCR screening of the pools prior to analytical gel electrophoresis and Southern blotting was performed to identify a pool presumably containing a 5′-complete coding sequence. For this purpose the 5′-end of the murine Stat5b ORF was labeled with [32P]dCTP by PCR, and the radioactive probe was purified using Sephadex G50 beads. The same protocol to generate the probe was used for the subsequent colony hybridization screen of the respective transformed cDNA pool according to standard protocols. After four steps of colony hybridization, the cDNA inserts in independent DNA preparations from three colonies scoring positively in the screen for the cStat5 cDNA were sequenced completely. The sequences were identical and encoded the amino-terminally incomplete Stat5 protein shown in the multiple sequence alignment. The cStat5b cDNA was obtained after five steps of colony hybridization. Again, the inserts in independent DNA preparations from three colonies were sequenced. One cDNA contained the full-length ORF for cStat5b. The other two clones yielded the same 5′-incomplete cDNAs starting at position 43 of the coding sequence but were otherwise identical as compared with the full-length clone.
Construction of plasmids, viral producer cells, and infection
For infection of avian cells, murine dominant-negative STAT5 (dC and Y699F) was amino-terminally fused to 9× myc tag and cloned into an avian retroviral vector containing EGFP-IRES (Clontech) (Oft et al. 1998) as follows: pOli (Wessely et al. 1999) was digested with HindIII and XbaI, and both ends were filled in with Klenow polymerase. The digested vector was then ligated to the blunted AseI EcoRI fragment of pEGFP-C1 (Clontech) to create a retroviral vector that drives EGFP from an internal CMV promoter. Subsequently, the vector was redigested with BsrgI, blunted, and ligated to the blunted 594-bp EcoR1 fragment of EMCV IRES.
In this vector, expression of inserted genes is driven from the CMV promoter. Because this vector contains no other viral sequences besides the LTR and part of gag for packaging, helper virus DNA (RCAS) (Tran Quang et al. 1997) was used for cotransfection into chicken embryonic fibroblasts. The chicken bcl-2 gene was cloned into RCAS; (J. Ghysdael, unpubl.). Bone marrow cells were doubly infected with these retroviruses and E26 by cocultivation with mitomycin C-arrested fibroblasts and E26-transformed producer myeloblasts for 72 hr and seeded in semisolid methylcellulose medium (Methocel). Successfully infected single colonies could be isolated from medium by virtue of their GFP expression (green fluorescence).
For infection of murine cells, a bicistronic retroviral vector was used. Expression of Stat5a or Bcl-x was coupled to GFP via an IRES sequence and was driven by the murine stem cell virus (MCSV) long terminal repeats. The methods used for preparation of the constructs and for obtaining high titer viral producer cell lines have been described in detail elsewhere (Persons et al. 1997, 1998). Bone marrow or fetal liver cells were isolated as described and infected during initial expansion by coculture on irradiated (1500 rads) ectopic producer cell lines for 48 hr. Infection efficiency was determined by FACS analysis.
Western blot analysis
Cell extracts were prepared and subjected to Western blot analysis with the antibodies indicated as described earlier (Woldman et al. 1997).
RNA isolation and Northern blot
Cells were factor-withdrawn and restimulated with GM-CSF or IL-3 as indicated. Cycloheximide (Sigma) was added shortly before addition of cytokines at a final concentration of 20 μg/ml. Total RNA was isolated using Trizol reagent (GIBCO BRL) according to the manufacturer's instructions. Northern blot analysis was performed according to standard procedures (using 15 μg of total RNA). Probes used were 32P-labeled murine full-length CIS cDNA (a gift from A. Yoshimura, Institute of Life Science, Kurume University, Kurume 839-0861, Japan) and 32P-labeled full-length murine Bcl-x cDNA (Packham et al. 1998).
Apoptosis assays
Apoptosis was measured by staining with Annexin V FITC (Clontech) followed by flow cytometry analysis according to manufacturers instructions. Alternatively, the TUNEL assay was performed: Paraformaldehyde fixed cells were labeled with Cy5-dCTP (Amersham) and terminal transferase (Boehringer Mannheim), again followed by flow cytometry.
FACS analysis
Expression of cell surface markers, sometimes combined with Annexin V staining, was analyzed on a FACSCalibur or FACSVantage and data processed using CellQuest software (Becton Dickinson).
Transient transfections and CAT assay
tsE26 transformed myeloblast clones containing or lacking dnStat5 were transfected with Lipofectamine (GIBCO BRL) as described (Mellitzer et al. 1996), followed by cultivation in the presence or absence of cMGF. Cytoplasmic extracts were prepared, and CAT amounts were determined using a CAT ELISA kit (Boehringer Mannheim). Transfection efficiency was assessed by transfection of a β-galactosidase gene, driven by the RSV promoter and determination of β-galactosidase activity with a chemiluminescent substrate (Tropix). Results were normalized to transfection efficiency and cell numbers.
Acknowledgments
We thank Peter Steinlein for performing flow cytometry and Bill Walker, Christen Rothammer, and Veronika Sexl for help with performing the mouse experiments. We also thank Peter Staeheli for providing chicken cDNA libraries and Manuela Baccarini for critical reading of the manuscript. This work was funded by the Austrian Research Foundation (FWF; grant P12946-Gen to T.D.), the FWF Austria (SFB-F006 to H.B.), and the Forschungsförderungsfonds der gewerblichen Wirtschaft (H.B.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL beug@nt.imp.univie.ac.at; FAX 43-1-7989390.
References
- Azam M, Erdjument-Bromage H, Kreider BL, Xia M, Quelle F, Basu R, Saris C, Tempst P, Ihle JN, Schindler C. Interleukin-3 signals through multiple isoforms of Stat5. EMBO J. 1995;14:1402–1411. doi: 10.1002/j.1460-2075.1995.tb07126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barahmand-pour F, Meinke A, Eilers A, Gouilleux F, Groner B, Decker T. Colony-stimulating factors and interferon-gamma activate a protein related to MGF-Stat 5 to cause formation of the differentiation-induced factors in myeloid cells. FEBS Letters. 1995;360:29–33. doi: 10.1016/0014-5793(95)00072-h. [DOI] [PubMed] [Google Scholar]
- Bedi A, Sharkis SJ. Mechanisms of cell commitment in myeloid cell differentiation. Curr Opin Hematol. 1995;2:12–21. doi: 10.1097/00062752-199502010-00003. [DOI] [PubMed] [Google Scholar]
- Beug H, Leutz A, Kahn P, Graf T. Ts mutants of E26 leukemia virus allow transformed myeloblasts, but not erythroblasts or fibroblasts, to differentiate at the nonpermissive temperature. Cell. 1984;39:579–588. doi: 10.1016/0092-8674(84)90465-3. [DOI] [PubMed] [Google Scholar]
- Beug H, Blundell PA, Graf T. Reversibility of differentiation and proliferative capacity in avian myelomonocytic cells transformed by tsE26 leukemia virus. Genes & Dev. 1987;1:277–286. doi: 10.1101/gad.1.3.277. [DOI] [PubMed] [Google Scholar]
- Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunẽz G, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- Chretien S, Varlet P, Verdier F, Gobert S, Cartron JP, Gisselbrecht S, Mayeux P, Lacombe C. Erythropoietin-induced erythroid differentiation of the human erythroleukemia cell line TF-1 correlates with impaired STAT5 activation. EMBO J. 1996;15:4174–4181. [PMC free article] [PubMed] [Google Scholar]
- Daems WT, Koerten HK. The effects of various stimuli on the cellular composition of peritoneal exudates in the mouse. Cell Tissue Res. 1978;190:47–60. doi: 10.1007/BF00210035. [DOI] [PubMed] [Google Scholar]
- Damen JE, Wakao H, Miyajima A, Krosl J, Humphries RK, Cutler RL, Krystal G. Tyrosine 343 in the erythropoietin receptor positively regulates erythropoietin-induced cell proliferation and Stat5 activation. EMBO J. 1995;14:5557–5568. doi: 10.1002/j.1460-2075.1995.tb00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Decker T, Kovarik P, Meinke A. Gas elements: A few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res. 1997;17:121–134. doi: 10.1089/jir.1997.17.121. [DOI] [PubMed] [Google Scholar]
- Dexter TM, Spooncer E. Growth and differentiation in the hemopoietic system. Annu Rev Cell Biol. 1987;3:423–441. doi: 10.1146/annurev.cb.03.110187.002231. [DOI] [PubMed] [Google Scholar]
- Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. 1994;264:713–716. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- Dumon S, Santos SC, Debierre-Grockiego F, Gouilleux-Gruart V, Cocault L, Boucheron C, Mollat P, Gisselbrecht S, Gouilleux F. IL-3 dependent regulation of Bcl-xL gene expression by STAT5 in a bone marrow derived cell line. Oncogene. 1999;18:4191–4199. doi: 10.1038/sj.onc.1202796. [DOI] [PubMed] [Google Scholar]
- Eilers A, Baccarini M, Horn F, Hipskind RA, Schindler C, Decker T. A factor induced by differentiation signals in cells of macrophage lineage binds to the gamma interferon activation site. Mol Cell Biol. 1994;14:1364–1373. doi: 10.1128/mcb.14.2.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman GM, Rosenthal LA, Liu X, Hayes MP, Wynshaw-Boris A, Leonard WJ, Hennighausen L, Finbloom DS. Stat5a-deficient mice demonstrate a defect in granulocyte-macrophage colony-stimulating factor-induced proliferation and gene expression. Blood. 1997;90:1768–1776. [PubMed] [Google Scholar]
- Frampton J, Ramqvist T, Graf T. v-Myb of E26 leukemia virus up-regulates bcl-2 and suppresses apoptosis in myeloid cells. Genes & Dev. 1996;10:2720–2731. doi: 10.1101/gad.10.21.2720. [DOI] [PubMed] [Google Scholar]
- Fujii H, Nakagawa Y, Schindler U, Kawahara A, Mori H, Gouilleux F, Groner B, Ihle JN, Minami Y, Miyazaki T, et al. Activation of Stat5 by interleukin 2 requires a carboxyl-terminal region of the interleukin 2 receptor beta chain but is not essential for the proliferative signal transmission. Proc Natl Acad Sci. 1995;92:5482–5486. doi: 10.1073/pnas.92.12.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fujitani Y, Yamaguchi T, Nakajima K, Hirano T. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: Involvement of STAT3 in anti-apoptosis. Immunity. 1996;5:449–460. doi: 10.1016/s1074-7613(00)80501-4. [DOI] [PubMed] [Google Scholar]
- Fukada T, Ohtani T, Yoshida Y, Shirogane T, Nishida K, Nakajima K, Hibi M, Hirano T. STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. EMBO J. 1998;17:6670–6677. doi: 10.1093/emboj/17.22.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillot DA, Gonzalez-Garcia M, Ekhterae D, Duan L, Inohara N, Ohta S, Seldin MF, Nuñez G. Genomic organization, promoter region analysis, and chromosome localization of the mouse bcl-x gene. J Immunol. 1997;158:4750–4757. [PubMed] [Google Scholar]
- Ihle JN. Interleukin-3 and hematopoiesis. Chem Immunol. 1992;51:65–106. doi: 10.1159/000420755. [DOI] [PubMed] [Google Scholar]
- ————— Cytokine receptor signalling. Nature. 1995;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- Lagasse E, Weissman IL. Enforced expression of Bcl-2 in monocytes rescues macrophages and partially reverses osteopetrosis in op/op mice. Cell. 1997;89:1021–1031. doi: 10.1016/s0092-8674(00)80290-1. [DOI] [PubMed] [Google Scholar]
- Leonard WJ, O'Shea JJ. Jaks and STATs: Biological implications. Annu Rev Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- Lieschke GJ, Stanley E, Grail D, Hodgson G, Sinickas V, Gall JA, Sinclair RA, Dunn AR. Mice lacking both macrophage- and granulocyte-macrophage colony-stimulating factor have macrophages and coexistent osteopetrosis and severe lung disease. Blood. 1994;84:27–35. [PubMed] [Google Scholar]
- Matsumura I, Ishikawa J, Nakajima K, Oritani K, Tomiyama Y, Miyagawa J, Kato T, Miyazaki H, Matsuzawa Y, Kanakura Y. Thrombopoietin-induced differentiation of a human megakaryoblastic leukemia cell line, CMK, involves transcriptional activation of p21(WAF1/Cip1) by STAT5. Mol Cell Biol. 1997;17:2933–2943. doi: 10.1128/mcb.17.5.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellitzer G, Wessely O, Decker T, Eilers A, Hayman MJ, Beug H. Activation of Stat 5b in erythroid progenitors correlates with the ability of ErbB to induce sustained cell proliferation. Proc Natl Acad Sci. 1996;93:9600–9605. doi: 10.1073/pnas.93.18.9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriggl R, Gouilleux Gruart V, Jahne R, Berchtold S, Gartmann C, Liu X, Hennighausen L, Sotiropoulos A, Groner B, Gouilleux F. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol Cell Biol. 1996;16:5691–5700. doi: 10.1128/mcb.16.10.5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriggl R, Topham DJ, Teglund S, Sexl V, McKay C, Wang D, Hoffmeyer A, van Deursen J, Sangster MY, Bunting KD, et al. Stat5 is required for IL-2 induced cell cycle progression of peripheral T cells. Immunity. 1999;10:249–259. doi: 10.1016/s1074-7613(00)80025-4. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Shah NM, Anderson DJ. Regulatory mechanisms in stem cell biology. Cell. 1997;88:287–298. doi: 10.1016/s0092-8674(00)81867-x. [DOI] [PubMed] [Google Scholar]
- Mui A. The role of Stats in proliferation, differentiation and apoptosis. Cell Mol Life Sci. 1999;55:1547–1558. doi: 10.1007/s000180050394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mui AL, Wakao H, O'Farrell AM, Harada N, Miyajima A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J. 1995;14:1166–1175. doi: 10.1002/j.1460-2075.1995.tb07100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mui AL-F, Wakao H, Kinoshita T, Kitamura T, Miyajima A. Suppression of interleukin-3-induced gene expression by a C-terminal truncated Stat: Role of Stat5 in proliferation. EMBO J. 1996;15:2425–2431. [PMC free article] [PubMed] [Google Scholar]
- Nosaka T, Kawashima T, Misawa K, Ikuta K, Mui AL, Kitamura T. STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. 1999;18:4754–4765. doi: 10.1093/emboj/18.17.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oft M, Heider KH, Beug H. TGFβ signalling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- Okajima Y, Matsumura I, Nishiura T, Hashimoto K, Yoshida H, Ishikawa J, Wakao H, Yoshimura A, Kanakura Y, Tomiyama Y, Matsuzawa Y. Insulin-like growth factor-I augments erythropoietin-induced proliferation through enhanced tyrosine phosphorylation of STAT5. J Biol Chem. 1998;273:22877–22883. doi: 10.1074/jbc.273.36.22877. [DOI] [PubMed] [Google Scholar]
- Packham G, White EL, Eischen CM, Yang H, Parganas E, Ihle JN, Grillot DA, Zambetti GP, Nuñez G, Cleveland JL. Selective regulation of Bcl-XL by a Jak kinase-dependent pathway is bypassed in murine hematopoietic malignancies. Genes & Dev. 1998;12:2475–2487. doi: 10.1101/gad.12.16.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persons DA, Allay JA, Allay ER, Smeyne RJ, Ashmun RA, Sorrentino BP, Nienhuis AW. Retroviral-mediated transfer of the green fluorescent protein gene into murine hematopoietic cells facilitates scoring and selection of transduced progenitors in vitro and identification of genetically modified cells in vivo. Blood. 1997;90:1777–1786. [PubMed] [Google Scholar]
- Persons DA, Mehaffey MG, Kaleko M, Nienhuis AW, Vanin EF. An improved method for generating retroviral producer clones for vectors lacking a selectable marker gene. Blood Cells Mol Dis. 1998;24:167–182. doi: 10.1006/bcmd.1998.0184. [DOI] [PubMed] [Google Scholar]
- Quelle FW, Wang D, Nosaka T, Thierfelder WE, Stravopodis D, Weinstein Y, Ihle JN. Erythropoietin induces activation of Stat5 through association with specific tyrosines on the receptor that are not required for a mitogenic response. Mol Cell Biol. 1996;16:1622–1631. doi: 10.1128/mcb.16.4.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- Silva M, Benito A, Sanz C, Prosper F, Ekhterae D, Nuñez G, Fernandez-Luna JL. Erythropoietin can induce the expression of Bcl-xL through Stat5 in erythropoietin-dependent progenitor cell lines. J Biol Chem. 1999;274:22165–22169. doi: 10.1074/jbc.274.32.22165. [DOI] [PubMed] [Google Scholar]
- Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, Maher DW, Cebon J, Sinickas V, Dunn AR. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci. 1994;91:5592–5596. doi: 10.1073/pnas.91.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinnett JD, Majeski JA. Macrophage activation and mobilization in nude mice by Corynebacterium parvum and pyran: A functional and histologic study. J Surg Oncol. 1980;14:327–332. doi: 10.1002/jso.2930140406. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: Generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–4660. [PubMed] [Google Scholar]
- Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- Tran Quang C, Wessely O, Pironin M, Beug H, Ghysdael J. Cooperation of SPI-1/PU.1 with an activated erythropoietin receptor inhibits apoptosis and EPO-dependent differentiation in primary erythroblasts and induces their KIT-ligand dependent proliferation. EMBO J. 1997;16:5639–5653. doi: 10.1093/emboj/16.18.5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakao H, Gouilleux F, Groner B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J1994, May. 1994;13(9):2182–2191. doi: 10.1002/j.1460-2075.1994.tb06495.x. . [Published erratum appeared in EMBO J.1995 Feb. 15 14(4): 854-855.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weining KC, Schultz U, Munster U, Kaspers B, Staeheli P. Biological properties of recombinant chicken interferon-gamma. Eur J Immunol. 1996;26:2440–2447. doi: 10.1002/eji.1830261026. [DOI] [PubMed] [Google Scholar]
- Wessely O, Bauer A, Quang CT, Deiner EM, von Lindern M, Mellitzer G, Steinlein P, Ghysdael J, Beug H. A novel way to induce erythroid progenitor self renewal: Cooperation of c-Kit with the erythropoietin receptor. Biol Chem. 1999;380:187–202. doi: 10.1515/BC.1999.027. [DOI] [PubMed] [Google Scholar]
- Woldman I, Mellitzer G, Kieslinger M, Buchhart D, Meinke A, Beug H, Decker T. Stat5 involvement in the differentiation response of primary chicken myeloid progenitor cells to chicken myelomonocytic growth factor. J Immunol. 1997;159:877–886. [PubMed] [Google Scholar]
- Yoshimura A. The CIS family: Negative regulators of JAK-STAT signaling. Cytokine Growth Factor Rev. 1998;9:197–204. doi: 10.1016/s1359-6101(98)00019-7. [DOI] [PubMed] [Google Scholar]
- Zamorano J, Wang HY, Wang R, Shi Y, Longmore GD, Keegan AD. Regulation of cell growth by IL-2: Role of STAT5 in protection from apoptosis but not in cell cycle progression. J Immunol. 1998;160:3502–3512. [PubMed] [Google Scholar]
- Zhang G, Gurtu V, Kain SR, Yan G. Early detection of apoptosis using a fluorescent conjugate of Annexin V. BioTechniques. 1997;23:525–531. doi: 10.2144/97233pf01. [DOI] [PubMed] [Google Scholar]