

Abstract
Microphthalmia (Mi) is a bHLHZip transcription factor that is essential for melanocyte development and postnatal function. It is thought to regulate both differentiated features of melanocytes such as pigmentation as well as proliferation/survival, based on phenotypes of mutant mouse alleles. Mi activity is controlled by at least two signaling pathways. Melanocyte-stimulating hormone (MSH) promotes transcription of the Mi gene through cAMP elevation, resulting in sustained Mi up-regulation over many hours. c-Kit signaling up-regulates Mi function through MAP kinase phosphorylation of Mi, thereby recruiting the p300 transcriptional coactivator. The current study reveals that c-Kit signaling triggers two phosphorylation events on Mi, which up-regulate transactivation potential yet simultaneously target Mi for ubiquitin-dependent proteolysis. The specific activation/degradation signals derive from MAPK/ERK targeting of serine 73, whereas serine 409 serves as a substrate for p90 Rsk-1. An unphosphorylatable double mutant at these two residues is at once profoundly stable and transcriptionally inert. These c-Kit-induced phosphorylations couple transactivation to proteasome-mediated degradation. c-Kit signaling thus triggers short-lived Mi activation and net Mi degradation, in contrast to the profoundly increased Mi expression after MSH signaling, potentially explaining the functional diversity of this transcription factor in regulating proliferation, survival, and differentiation in melanocytes.
Keywords: Microphthalmia, c-Kit, steel factor, MAPK, p90 Rsk, ubiquitin
Recruitment of RNA polymerase II to eukaryotic promoters requires a complex integration of molecular events including chromatin perturbation and the combinatorial and potentially cooperative assembly of sequence-specific DNA-binding factors (Roeder 1998; Zaman et al. 1998; Berk 1999). The post-translational modification of many of the proteins participating in this process is recognized to be of critical importance in gene expression (for review, see Armstrong and Emerson 1998; Kornberg and Lorch 1999). Microphthalmia (Mi) is an essential basic helix–loop–helix–leucine-zipper (bHLHZip) transcription factor for the melanocyte lineage. Its absence results in complete lack of neural crest-derived pigment cells (Silvers 1979). In humans, heterozygous mutations in Mi produce Waardenburg syndrome IIA (Hughes et al. 1994; Tassabehji et al. 1994; Tachibana 1997), manifesting pigment cell abnormalities and deafness due to defective inner ear melanocytes. In addition, in contrast to many melanocytic markers, which are lost during transformation to malignant melanoma, Mi expression appears to be very highly conserved in human melanoma (King et al. 1999).
The transcriptional activity of Mi has been found to be modified by mitogen-activated protein kinase (MAPK) phosphorylation (Hemesath et al. 1998). Steel factor (Sl) stimulation of the receptor tyrosine kinase c-Kit initiates a signaling cascade that culminates in MAP kinase-mediated phosphorylation of Mi and an increase in its transactivation potential. Signal-dependent transcriptional up-regulation occurs through selective recruitment of the transcriptional coactivator p300 by MAPK-phosphorylated Mi (Price et al. 1998a). This signaling pathway and post-translational modification may contribute to the phenotypic overlap seen among mice carrying germ-line mutations in the cytokine Sl, its receptor c-Kit, and the transcription factor Mi (Russell 1979; Witte 1990; Dubreuil et al. 1991; Moore 1995; Hemesath et al. 1998).
In melanocytes, Mi is thought to be required for both differentiation/pigmentation as well as proliferation/survival. The role of Mi in pigmentation has emerged from evidence that its expression is potently induced by the pigmenting factor melanocyte-stimulating hormone (MSH) (Bertolotto et al. 1998a; Price et al. 1998b) as well as recognition that Mi can up-regulate transcription through a conserved consensus element in the promoters of the major pigment enzyme genes (Bentley et al. 1994; Hemesath et al. 1994; Yasumoto et al. 1994). Evidence of a major role for Mi in survival/proliferation of melanocytes comes from the mivit mouse mutation that displays nearly normal neonatal melanocyte numbers followed by precocious melanocyte loss over several months of age (premature gray/white) (Lerner et al. 1986). This phenotype is consistent with an essential role for Mi in post-developmental melanocyte proliferation or survival. In addition, the position of Mi downstream of Steel/c-Kit signaling is consistent with mitogenic or survival signals known to be stimulated by Steel/c-Kit in a variety of contexts (Andrews et al. 1994; Hassan and Zander 1996; Sykora et al. 1997). Mi has also been shown to regulate c-Kit expression transcriptionally in mast cells (Tsujimura et al. 1996), suggesting the possibility of homeostatic regulation among these factors.
As a target of at least two signaling pathways, MSH and c-Kit, Mi may reside at a pivotal position for its ability to trigger alternative transcriptional programs. Although much remains to be learned about the spectrum of genes activated by Mi in melanocytes, it is plausible that different genes are targeted in distinct contexts and that the transcriptional activity of Mi may, therefore, be tightly regulated in a signal-dependent fashion.
Both the MSH and c-Kit signaling pathways up-regulate the transcriptional activity of Mi, but they do so in very different ways. MSH stimulation up-regulates cAMP and stimulates new transcription of Mi through a cAMP response element (CRE) in the Mi promoter in melanocytes. Thus, MSH stimulation profoundly increases Mi protein expression over the course of hours (Bertolotto et al. 1998a; Price et al. 1998b). In contrast, c-Kit stimulation produces very rapid MAPK-mediated phosphorylation of Mi, producing enhanced recruitment of p300/CBP (CREB-binding protein) (Price et al. 1998a), the coactivator family that interacts with and modulates the transcriptional activity of Mi (Sato et al. 1997)—all occurring over the course of minutes. The kinetic differences between these alternative means of up-regulating Mi are significant and could contribute to the different biological consequences of stimulating these signaling pathways. In other settings, transcriptional activity has been suggested to rely on proteolytic degradation of nuclear receptors (Nawaz et al. 1999), suggesting such phenomena could be of widespread importance.
In the current study we investigated the consequences of c-Kit signaling on Mi stability and function. We show that Mi is targeted for rapid ubiquitin-dependent proteolysis with Steel factor stimulation. The specific signals were found to be phosphorylation by either MAPK at serine 73 or Rsk-1 at serine 409. Double serine-to-alanine mutations at these two residues result in a protein that is both profoundly stable and transcriptionally inactive. These c-Kit-induced phosphorylations thus produce coupled, short-lived activation–destruction signals on the nuclear target Mi.
Results
Mi is degraded after c-Kit stimulation
The observation that Mi protein is degraded after c-Kit signaling was first made in the study of Sl stimulation of human melanoma cells. Western blots using a Mi-specific monoclonal antibody showed that Sl stimulation produced an initial mobility shift of Mi due to MAPK/ERK phosphorylation on serine 73 as previously described (Hemesath et al. 1998). Subsequent to this shift, Mi protein levels appeared to diminish over time (Fig. 1A, left). Previous studies demonstrated that Steel/c-Kit signals are transmitted to Mi through MAPK/ERK (Hemesath et al. 1998). To test whether this same pathway was triggering Mi degradation, the MAPK/ERK pathway inhibitor PD98059 was used (Fig. 1) and was found to prevent both the c-Kit-dependent mobility shift and c-Kit-induced Mi degradation. This observation suggested that phosphorylation of ser-73 or another MAPK-dependent signal was responsible for Mi degradation. To clarify the fate of Mi protein after Sl signaling, de novo protein synthesis was inhibited by incubation with cycloheximide and the kinetics of Mi degradation were analyzed. With cycloheximide, Sl produced the same MAPK-dependent mobility shift in Mi, followed by more pronounced degradation of Mi (Fig. 1B, left). There was no reappearance of the lower band with arresting protein synthesis in these cells (Fig. 1B, left). The drug PD98059 again stabilized Mi protein levels over time (Fig. 1B, right). Pulse/chase analysis showed the same changes in Mi stability as cycloheximide decay kinetics (data not shown).
Figure 1.

c-Kit activation produces MAPK-dependent Mi degradation. (A) Human melanoma cells (501 Mel) were stimulated at the c-Kit receptor with recombinant Steel factor (Sl) in the presence or absence of the MEK inhibitor drug PD98059 and whole cell extracts probed with monoclonal antibody against Mi (doublet) or α-tubulin-loading control at the time points indicated. (B) c-Kit stimulation was also carried out in the presence of cycloheximide (CHX) to reveal Mi degradation kinetics.
Mi degradation is proteasome dependent
Mi from melanoma cells (501 Mel or MeWo) and melanocytes was examined to determine whether Mi instability involved proteasome-mediated degradation. Proteasome inhibition using MG132 or lactacystin (Fig. 2A–E) prevented Sl/c-Kit-induced Mi degradation in melanoma cells as well as primary human melanocytes (Fig 2A–C). The proteasome inhibitor MG115 showed the same effect (data not shown). Inhibition of Mi degradation traps Mi in the upper phosphorylated form. Over time there is appearance of the lower band suggesting a dephosphorylation event. Treatment of cells with okadaic acid, a phosphatase 1 and 2A inhibitor, prevented the appearance of the lower migrating Mi isoform after 2 hr of Sl stimulation (Fig. 2C, right), suggesting that phospho-Mi may be subject to both degradation and dephosphorylation events in these cells. The same results were found with either murine melanoma cells (Fig. 2D) or murine melanocytes (Fig. 2E). For murine cells, MAPK stimulation was triggered with phorbol ester (TPA) in lieu of Sl because these cells lack functional c-Kit receptor. The results from multiple cell systems suggest that Sl/c-Kit- and MAPK-dependent destabilization of Mi involves targeting of Mi to the proteasome degradation pathway.
Figure 2.

c-Kit-induced Mi degradation is blocked by proteasome inhibition. Presence of a Mi dephosphorylating activity. Mi degradation kinetics were followed using cycloheximide (CHX). (A) Human melanoma cells (501 Mel) were stimulated by recombinant Steel factor (SI) in the presence of cycloheximide (Sl + CHX) and with and without the proteasome inhibitor MG132. α-Tubulin was probed as internal loading control. (B) Lactacystin was used as proteasome inhibitor on SI-stimulated human melanoma cells. (C) Primary human melanocytes were stimulated with recombinant SI in the presence of cycloheximide (Sl + CHX) in the absence or presence of Mg132. Reappearance of the lower migrating Mi isoform at later time points was reversed by treatment of cells with the phosphatase inhibitor okadaic acid in the human melanoma MeWo. (D) Mouse melanoma cells (B16) were treated with TPA to activate MAPK as these cells lack functional c-Kit receptor. Mi levels were compared in the absence or presence of Mg132 at the time points indicated. (E) Mouse melanocytes were treated with TPA as above and Mi levels assessed by Western blotting.
c-Kit stimulation triggers ubiquitination of Mi
Proteins that are degraded by the proteasome machinery in cells are first marked for this fate by the covalent addition of polyubiquitin chains to specific lysine residues (Chau et al. 1989; Goldberg and Rock 1992; Hershko and Ciechanover 1992; Mahaffey et al. 1993; Palombella et al. 1994; Goldberg 1995). To assess whether Mi undergoes covalent ubiquitination, two types of experiments were done. In the first, the BHK kidney cell line was transfected with Mi in the presence or absence of a 6× histidine–ubiquitin-encoding plasmid. Cellular lysates were passed over a nickel resin to bind His-tagged ubiquitin, and bound proteins were analyzed by Western blot using anti-Mi antibody. As shown in Figure 3A, Mi was identified in pull-downs only from lysates in which tagged ubiquitin and Mi had been expressed together. A limitation of this experiment was that misexpression of Mi in kidney cells may not reflect events in pigment cells.
Figure 3.
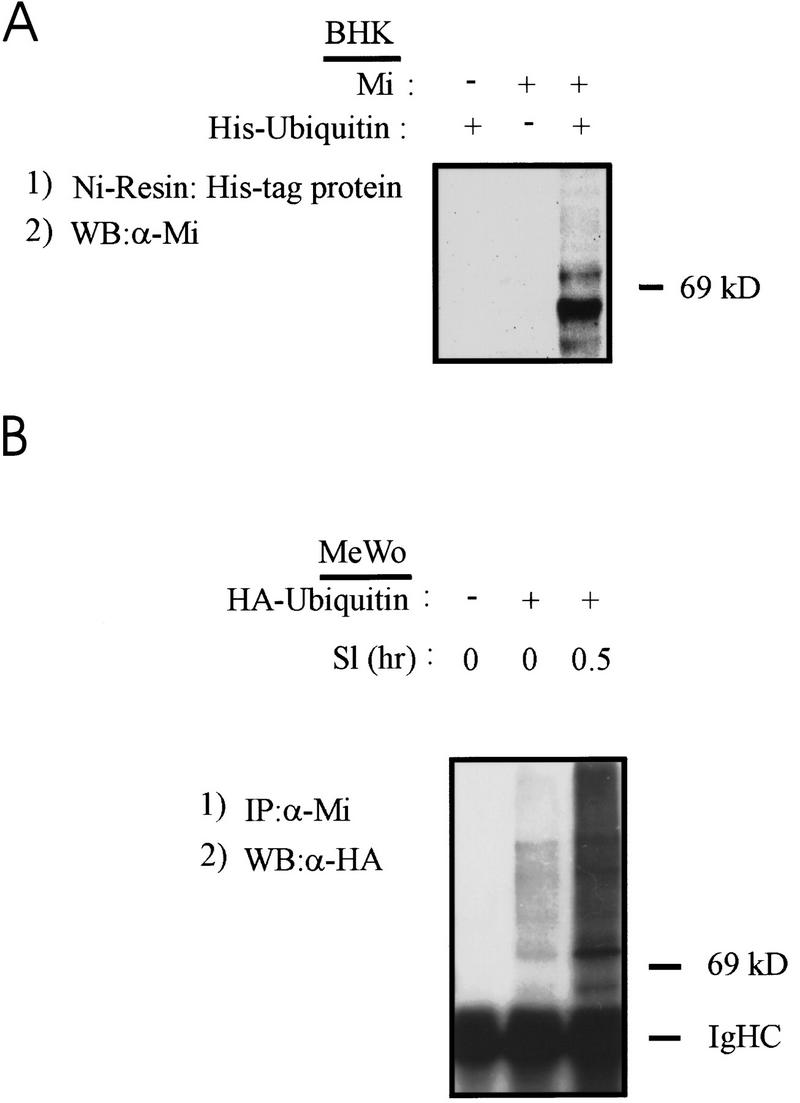
Mi is covalently ubiquitinated in a c-Kit responsive fashion. (A) Plasmids encoding Mi or His-tagged ubiquitin were transfected transiently into baby hamster kidney cells (BHK) and cell lysates were made 24 hr later. His-tagged protein (and associated protein) was affinity purified on nickel resin, proteins eluted and resolved by SDS-PAGE, and Western blotted for Mi. (B) HA-tagged ubiquitin plasmid was transfected into human melanoma cells (as indicated). Twenty-four hours later cells were stimulated with recombinant Steel factor (Sl, as indicated) and extracted 0.5 hr later. Lysates were immunoprecipitated with anti-Mi antibody followed by Western blotting for HA (ubiquitin). Endogenous Mi covalently associates with ubiquitin in a SI-inducible fashion.
Therefore, ubiquitination of endogenous Mi was tested with or without Sl stimulation of human melanoma cells transfected with HA-tagged ubiquitin. Cellular lysates were immunoprecipitated with Mi antibody, resolved by SDS-PAGE, transferred to nitrocellulose, and probed for ubiquitin with anti-HA antibody. A significant increase in Mi/ubiquitin conjugates was detected after stimulation by Sl (Fig. 3B). In both of these experiments it is notable that some endogenous MAPK activity exists even in the absence of exogenous Steel factor addition, likely explaining the presence of modest but detectable ubiquitination of Mi in unstimulated MeWo (Fig. 3B). These studies show that Mi (either endogenous or exogenous) can be modified covalently by ubiquitin in response to Sl/c-Kit signaling.
Ser-73 is dispensable for Mi degradation
The presence of functional Mi was next measured by assessing DNA-binding activity in nuclear extracts of melanoma cells after stimulation by Sl, with or without proteasome inhibition. To assess simultaneously the role of specific phosphorylation and degradation of Mi, wild-type and specific Mi mutants were engineered into adenoviral expression vectors and used to infect (>90%) melanoma cells efficiently before examination of DNA-binding activity in nuclear extracts. Virally expressed, epitope-tagged Mi-specific DNA binding was identified by monoclonal antibody supershift in an electrophoretic mobility shift assay (EMSA) using radiolabelled DNA probe. Mi DNA-binding activity declined within 2 hr in cells exposed to Sl, whereas no loss of DNA binding was seen in cells treated with Sl plus MG132 (Fig. 4, left). Because c-Kit signaling results in Mi phosphorylation at ser-73, the role of this serine residue in destabilization was examined. As previously shown (Price et al. 1998a), DNA-binding activity is retained with mutation of ser-73 to alanine. However, the S73A and wild-type proteins were found to be equally labile after Sl stimulation (Fig. 4, right). Furthermore, DNA-binding activity of the S73A mutant was also stabilized by proteasome inhibition. These results indicate that although MAPK phosphorylation of ser-73 occurs after Sl stimulation, it is not absolutely required for Sl/c-Kit-induced destabilization of Mi. Therefore, another consequence of c-Kit signaling must contribute to the observed Mi destabilization.
Figure 4.

Ser-73 is not essential to c-Kit-induced Mi degradation. Adenoviruses encoding wild-type Mi or ser-73 mutated to alanine (S73A) were used to infect human melanoma cells (501 Mel), which were subsequently stimulated with Steel factor (SI) in the presence or absence of cycloheximide and Mg132 proteasome inhibitor. Nuclear extracts from these cells were used in gel shift assays and the Mi-specific DNA-binding activity was identified by supershifting with anti-HA antibody addition (as indicated). Both wild-type and S73A mutant Mi DNA-binding activities diminish after SI stimulation.
Because Sl/kit-induced degradation of Mi is blocked by MAPK inhibition (Fig. 1), the mediators of this signal must reside at the level of MAPK or downstream. One possible explanation for the unperturbed degradation of the S73A mutant was that a second phosphorylation could contribute to ubiquitin-dependent proteolysis. Mi contains a number of potential phosphorylation targets but no additional MAPK consensus. One of these, S409, resides within a consensus sequence for the Rsk family of serine/threonine kinases (RRXS/T) (Leighton et al. 1995). Rsk also represents a known downstream target of MAPK activation (Tsai et al. 1993; Dalby et al. 1998; Gavin and Nebreda 1999), potentially explaining why MEK inhibition could stabilize Mi in the context of Sl treatment.
Phosphorylation of Mi by Rsk-1 in response to c-Kit stimulation
To test whether Rsk may phosphorylate Mi directly at ser-409, initial studies examined in vitro kinase (IVK) activity. As shown in Figure 5A, immunoprecipitates of Rsk-1 from Sl-stimulated melanoma cells could phosphorylate recombinant Gst–Mi fusion protein, but not a Mi fusion carrying the S409A mutation. This activity was strongly induced by Sl stimulation of the melanoma cells. Evidence of autophosphorylation by Rsk in response to c-Kit stimulation was also seen, as indicated (Fig. 5A). An unidentified substrate protein that coprecipitates with Rsk is indicated by an asterisk. The kinetics of Rsk and MAPK/ERK activation after Sl stimulation were also examined in side-by-side IVK reactions followed by PhosphorImager quantitation. Peak Rsk activation paralleled but slightly lagged the induction of MAPK/ERK activity in melanoma cells after Sl stimulation (Fig. 5B), as anticipated for a kinase that is, itself, a substrate for (and activated by) MAPK (Blenis 1993). Therefore, Rsk-1 is capable of directly phosphorylating recombinant Mi on ser-409 in vitro in a manner regulated by Sl stimulation with kinetics that recall those of the ser-73 Mi kinase ERK.
Figure 5.
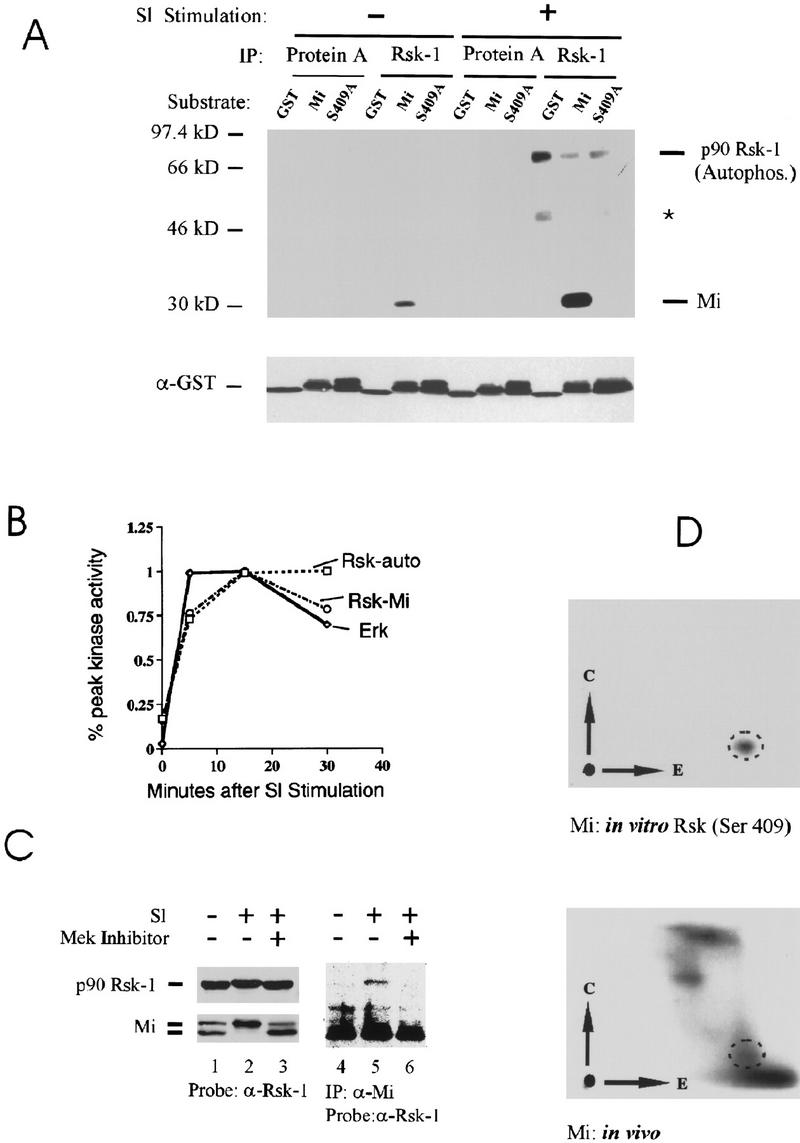
Involvement of Rsk-1 in phosphorylation of ser-409. (A) In vitro kinase assays were carried out using immunoprecipitated Rsk-1 (or protein A control) from unstimulated or Steel factor-stimulated (SI) melanoma cells. Kinase substrates consisted of GST (control), GST fusion to carboxy-terminal fragment of wild-type Mi (“Mi”), or GST fusion to carboxy-terminal fragment of Mi mutated at ser-409 to alanine (“S409A”). GST blot is shown at the bottom. Rsk-1 autophosphorylation is indicated and a weakly coprecipitating second Rsk substrate is indicated by asterisk. (B) Kinetics of Rsk and MAPK/ERK activation after SI stimulation. Kinase assays as described in A were quantitated by PhosphorImager and plotted as percent of maximal activity, indicating that Rsk activation lags ERK activation slightly. (C) Rsk-1 associates with Mi upon SI stimulation. Human melanoma cells were stimulated with SI (or unstimulated) in the presence or absence of MEK inhibitor drug (PD98059). Cell lysates were either probed directly for p90–Rsk-1 or Mi (lanes 1–3). MEK inhibition prevents a Steel-induced mobility shift in both Mi and a subtle mobility shift in Rsk-1. Mi was also immunoprecipitated from cell lysates followed by Western blotting for Rsk-1. Rsk-1 associated with Mi after Steel stimulation only if MAP kinase was not inhibited. (D) Two-dimensional phosphotryptic mapping indicates presence of phosphoserine 409 within endogenous cellular Mi. The two-dimensional mobility of phosphoserine 409 was determined by analyzing in vitro phosphorylated recombinant Mi (as in Fig. 5A). 32P-labeled melanoma cells were stimulated by SI and extracted; endogenous Mi was immunoprecipitated and subjected to tryptic digestion and two-dimensional analysis (simultaneously run with in vitro phosphorylated recombinant Mi). A spot was observed (dotted circles), which displays superimposable migration relative to the phosphoserine 409 spot generated by Rsk-1 phosphorylation of ser-409 in vitro.
To determine whether Rsk-1 and Mi interact in vivo, we asked whether the endogenous forms of the two proteins are associated with one another in melanoma cells. Lysates of Sl-stimulated (presence or absence of MEK inhibitor) or unstimulated human melanoma cells were immunoprecipitated with Mi antibody and examined by Western blot for coprecipitation of Rsk. As shown in Figure 5C, the mobility shift of Mi is prevented by MAPK inhibition (lanes 1–3). Probing of the same blot for Rsk-1 reveals equal abundance of the protein, although with some evidence for a slight mobility shift with Sl stimulation (Fig. 5C, lanes 1–3). From these same lysates, endogenous Rsk-1 specifically coimmunoprecipitates with endogenous Mi only after Sl stimulation (lanes 4,5). Moreover, MAPK inhibition prevents this Rsk-1:Mi association (lane 6). Thus, endogenous Mi and Rsk proteins appear to physically associate with one another after Sl stimulation in a manner that is dependent on MAPK activation.
We then examined whether endogenous Mi was phosphorylated at ser-409 in vivo using two-dimensional phospho-tryptic mapping. To identify the ser-409 phosphopeptide, recombinant Mi was phosphorylated in vitro by Rsk-1 as above, and a tryptic digest was resolved by electrophoresis and thin layer chromatography using an apparatus producing inverted replicas (Fig. 5D). For comparison, endogenous cellular 32P-labeled Mi protein was immunoprecipitated from melanoma cells, digested, and resolved in the same way. Several 32P-labeled spots were apparent, one of which perfectly comigrates with the ser-409 Rsk-phosphorylated recombinant species based on superimposing the images (encircled in Fig. 5D). Although these experiments cannot directly prove that Rsk phosphorylates Mi in vivo, several observations support this possibility: (1) the presence of a Rsk target consensus site at ser-409; (2) the stimulation of endogenous Rsk activity in response to Sl with the same kinetics as those of ERK; (3) the physical association of Rsk with endogenous Mi in response to Sl stimulation; (4) the profound stabilization of Mi in response to MAPK inhibition; and (5) the presence of a phosphopeptide from endogenous Mi that is indistinguishable from a Rsk-phosphorylated Mi peptide. These observations are consistent with the possibility that Rsk phosphorylates Mi in cells at ser-409 in response to Sl signals.
Double mutations of MAP kinase and Rsk target serines profoundly stabilize Mi
Mutation of S409 to alanine was examined for its impact on the signal-dependent proteolysis described above. NIH 3T3 cells were transfected with wild-type Mi, single serine-to-alanine mutants at positions 73 or 409 (S73A and S409A), or a double mutant at both serines 73 and 409 (S73/409A), and stabilities were tested with stimulation with TPA. As shown in Figure 6A, wild-type protein and the two single point mutants were all degraded rapidly. In contrast the double mutant at both S73 and S409 was significantly stabilized. Similarly, transfection into human melanoma cells followed by c-Kit stimulation produced short-lived wild-type and single mutant proteins (S73A or S409A), whereas the S73/409A double mutant was profoundly stabilized (Fig. 6B).
Figure 6.


Simultaneous mutation of ser-73 and ser-409 protects Mi from c-Kit-induced proteolysis. (A) Plasmids encoding wild-type Mi (“Mi”) or individual mutations at ser-73 or ser-409 or the double mutant at both sites (S73/409A) were transfected into NIH 3T3 cells. Twenty-four hours later MAPK was activated by TPA in the presence of cycloheximide (CHX), and Mi degradation was followed using Western blot analysis. Tubulin control is indicated. (B) The same constructs as in A were introduced into 501 Mel human melanoma cells, followed by assessment of Mi degradation after Steel stimulation (SI).
Coupled activation/degradation: S73/409A mutant Mi is transcriptionally incompetent although intact for DNA binding
Transactivation potentials of the mutant Mi proteins were assessed by transfection/reporter assay. This is of particular importance because Sl stimulation has been shown previously to superactivate Mi function, and mutation of ser-73 partially abrogates transactivation (Hemesath et al. 1998). The current observations suggest that the identical modification that activates Mi also targets the protein for degradation. If so, the stabilized double mutant S73/409A might be a poor transcriptional activator despite its greater stability. To test this, Mi expression plasmids were cotransfected into NIH 3T3 as well as human melanoma cells with a luciferase reporter gene driven by the tyrosinase promoter, a Mi-responsive target (Bentley et al. 1994; Hemesath et al. 1994; Yasumoto et al. 1994; Bertolotto et al. 1996). As shown in Figure 7A, wild-type Mi stimulated the tyrosinase promoter, in agreement with prior results (Hemesath et al. 1998). The S73A and S409A single mutants were less efficient transactivators than wild-type Mi. However, the double mutant was devoid of transactivation potential despite its greater stability. The lack of activity for the double mutant is specific for this reporter, as all data are normalized to a cotransfected constitutive reporter. Of note, the double mutant reproducibly diminishes promoter activity below the basal (vector control) levels, consistent with the possibility that it acts dominant negatively with respect to other E box factors in these cells. If the double mutant exhibits dominant negative function, it would be expected to retain DNA-binding activity, as predicted from the location of the two mutations (which are remote from the bHLHZip motif).
Figure 7.

Double mutant Mi, resistant to MAPK and Rsk, is transcriptionally inactive despite enhanced stability and intact DNA binding. (A) Transactivation assays were performed using wild-type Mi and single or double mutants at ser-73 and ser-409. NIH 3T3 or 501 Mel melanoma cells were cotransfected with pEBB (empty vector) or indicated Mi constructs and luciferase reporter under control of the tyrosinase promoter. Data are normalized to cotransfected sea pansy luciferase plasmid. (B) Gel shifts of BHK cells transfected with the same Mi constructs as in A after MAPK activation with TPA and cycloheximide (CHX). Mi-specific DNA-binding activity is identified using supershifting antibody.
DNA-binding activity from nuclear extracts of cells transfected transiently with either wild-type Mi or the S73/409A double mutant were examined by EMSA after MAPK stimulation. The Mi/DNA complex was again identified using supershifting monoclonal antibody. Whereas wild-type Mi displayed a significant loss of DNA-binding activity after MAPK stimulation, the S73/409A double mutant was resistant to this reduction in DNA binding (Fig. 7B). DNA-binding patterns closely recapitulated the direct protein levels seen by Western blot after transfection of the Mi constructs. These data indicate that mutations of serines 73 and 409 do not perturb DNA binding of the Mi protein, and that the stability seen with the double mutant in protein blots is reflected in functional protein isolated from the nuclei of cells. Because simultaneous mutations of both serines 73 and 409 were necessary to produce Mi protein resistant to this degradation pathway, phosphorylation at either ser-73 or ser-409 in response to c-Kit stimulation appears to be sufficient to signal ubiquitin-dependent proteolysis of Mi. These experiments demonstrate that simultaneous mutation of serines 73 and 409 prevents signal-induced Mi degradation and produces a stable protein, but at the same time cripples the transactivation potential of Mi. Thus, the signals that produce transcriptional activation and protein degradation of Mi are functionally coupled in melanocytes.
Discussion
The data presented here suggest that the Sl/c-Kit signaling pathway in melanocytes targets the transcription factor Mi simultaneously for activation and proteolytic degradation. This conclusion is based on evidence that the half-life of Mi is shortened by c-Kit-triggered MAPK activity. Because MAPK inhibition profoundly stabilizes Mi protein, the necessary events that signal Mi degradation appear to reside at or downstream of MAPK. On the basis of mutational and functional studies these include MAPK itself and likely Rsk-1 (although other kinases might potentially substitute for Rsk-1; see below). These kinases appear to phosphorylate Mi at serines 73 and 409, respectively, either of which is sufficient to target Mi for degradation. Sl stimulation induces the association of Rsk-1 with Mi, and Rsk-1 activity is measurably up-regulated in these cells with a kinetic profile that closely follows that of MAPK. Covalent modification by ubiquitin also suggests that the degradation pathway involves ubiquitin-dependent proteolysis, likely mediated by proteasome activity that was repressible by several proteasome inhibitors. Because simultaneous alanine substitutions at both serines 73 and 409 produce highly stabilized, but transcriptionally inactive Mi, the stimulation and degradation signals appear to be biochemically coupled.
The existence of such tightly coupled activation/degradation signals likely creates a circumstance in which up-regulatory stimuli are received at target gene promoters, while simultaneously limiting their duration. This potential scenario is of interest, in part, because of the multiple activities with which Mi is associated. For example, in contrast to the net degradation of Mi after Sl/kit stimulation, the MSH signaling pathway profoundly up-regulates Mi gene expression, producing significantly elevated Mi protein levels over many hours (Bertolotto et al. 1998a; Price et al. 1998b). Whereas Mi protein may be depleted with Sl/c-Kit stimulation, prolonged transcription of the Mi gene after MSH results in sustained levels, despite the presence of constitutive basal MAPK activity (Price et al. 1998b). Thus, the short-lived effects of Sl/kit signaling on Mi may provide a mechanistic basis for analyzing the diverse roles of Mi within melanocytes.
One implication of Mi degradation after Sl/kit stimulation is that in tumor cells (melanomas) pathologic c-Kit activation could produce net diminished Mi levels and function. This prediction is of interest because of numerous reports that c-Kit expression is frequently absent in melanomas, and that Steel factor stimulation of melanomas, which retain c-Kit, may result in paradoxically antiproliferative effects (Zakut et al. 1993). However, intermittent subcutaneous Steel factor injection into humans clearly induces melanocytic proliferation (Costa et al. 1996), as predicted from the known function of c-Kit as a mitogenic receptor tyrosine kinase and proto-oncogene in other contexts (Williams et al. 1990). The net inhibition of melanoma growth upon sustained c-Kit stimulation is a behavior that may correlate with Mi degradation. A recent clinical series (King et al. 1999) demonstrated nuclear Mi expression in 100% of 76 consecutively accessioned melanoma specimens, despite concomitant loss of other melanocytic markers such as pigment, c-Kit, pmel/17 (HMB 45), and other melanocytic markers (Carrel and Rimoldi 1993; Zakut et al. 1993; Halaban et al. 1997; Kaufmann et al. 1998). Because Mi plays an essential post-developmental role in melanocyte proliferation or survival (based on the Mivit mutant), it is possible that degradation of Mi with persistent c-Kit stimulation might be inhibitory to melanoma growth.
The observation that activation and degradation appear to be coupled to the identical phosphorylation events on Mi raises the question of how phospho-Mi is recognized for degradation. The above data demonstrate a role for ubiquitin-mediated proteolysis and proteasomal activity. It is interesting to speculate that p300/CBP, which is selectively recruited to Mi by MAPK-mediated phosphorylation (Price et al. 1998a), might in some manner directly facilitate Mi degradation as well. Such a role for p300/CBP has been suggested in the regulation of p53 degradation (Grossman et al. 1998). This could involve p300/CBP-dependent recruitment of a separate ubiquitin ligase or might reflect an intrinsic ubiquitin ligase activity of p300/CBP itself. It has been suggested that signal mediated degradation of a transcription factor bound to a promoter is required for dissociation of the pro-initiation complex and transcript elongation (Nawaz et al. 1999). Mechanistically, it is possible that promoter-bound, phosphorylated Mi may eventually impede transcriptional initiation or even elongation, and for this reason it could be advantageous to degrade (or dephosphorylate) the activated protein. Such a model could operate for many signal-responsive transcriptional pathways.
Another paradoxical observation, which might be explained by the coupled activation/degradation signals described here, involves regulation of pigmentation. Mi recognizes and, in experimental systems, has been shown to transactivate the tyrosinase, TRP-1, and TRP-2 pigment enzyme gene promoters (Bentley et al. 1994; Hemesath et al. 1994; Yasumoto et al. 1994; Bertolotto et al. 1996, 1998b). This transactivation activity is measurably enhanced by MAPK-mediated activation of Mi (Hemesath et al. 1998). However, dominant active Ras has been found to diminish pigmentation and dominant negative Ras to increase it (Englaro et al. 1998) despite evidence that c-Kit up-regulates Mi through MAPK (Hemesath et al. 1998) and can enhance pigmentation in vivo (Costa et al. 1996). One explanation for these findings is that the same signaling pathway may target Mi for activation as well as proteolytic degradation. Homeostatic down-regulation is a hallmark of receptor tyrosine kinase signaling and occurs at multiple levels including cytokine/receptor internalization, dephosphorylation, and proteolysis. The current study extends such homeostatic down-regulation to a nuclear target of c-Kit.
Mi phosphorylation at ser-73 is strongly established as a target of MAPK/ERK on the basis of in vitro and in vivo biochemical and inhibitor data (Hemesath et al. 1998). Significant correlative data suggest that Rsk-1 may target ser-409, although lack of Rsk inhibitor reagents renders this point incompletely proven in vivo. Moreover, ser-409 might serve as a target substrate for other kinases, such as protein kinase A (PKA), whose consensus sequence overlaps. Indeed, in vitro kinase assays demonstrate the ability of PKA to phosphorylate ser-409 (data not shown). Although phosphorylations of serines 73 and 409 may be coupled in the c-Kit signaling pathway, it is possible that phosphorylation at ser-409 may occur independently of MAPK activation in other signaling contexts, and such events should be sufficient to destabilize the Mi protein.
The kinases ERK-2 and p90 Rsk are physically associated with one another in the cytoplasm of many cell types (Scimeca et al. 1992) including 501Mel cells (data not shown) and translocate to the nucleus together upon MEK-mediated activation of ERK. ERK has been shown to phosphorylate Rsk and stimulate the activity of its amino-terminal kinase domain (Sturgill et al. 1988; Blenis 1993; Grove et al. 1993). We found that the kinetics of ERK and Rsk activation in response to c-Kit signaling closely paralleled one another in melanoma cells. This is the first example of these two regulated kinases acting in concert to modulate the stability and the cofactor coupling of a sequence-specific transcription factor. The only other common phosphorylation target of the kinases ERK and Rsk is c-Fos, which together with c-Jun forms the transcription factor complex AP-1. Phosphorylations at the extreme carboxyl terminus of c-Fos by ERK and Rsk may result in an enhancement of the growth-promoting properties of c-Fos in fibroblast cells (Chen et al. 1993, 1996), although the exact molecular mechanisms are unclear.
IκB, a cytosolic inhibitor of the transcription factor NFκB, undergoes phosphorylation-dependent ubiquitin-mediated degradation in response to a number of extracellular stimuli. Rsk is one of several kinases that may be involved in triggering NFκB nuclear translocation by the phosphorylation of ser-32 in IκB, targeting the latter for degradation (Ghoda et al. 1997; Schouten et al. 1997). There are other kinases triggered by distinct receptor systems that serve the same function in NFκB induction. Ubiquitin-mediated degradation of Stat proteins also produce signal-responsive regulation for this transcription factor family (Kim and Maniatis 1996).
The transcriptional effects observed with phosphorylation of Mi in this system appear to involve p300/CBP coactivator recruitment (Sato et al. 1997; Price et al. 1998a). It is important to consider, however, that the involvement of these coactivators may vary at different transcriptional targets. Thus, some promoters may use Mi for p300-dependent transcription, whereas others might not require p300 for Mi-driven transcription. The discovery of transcriptional targets genes of Mi, particularly in the contexts of these distinct signaling pathways, may shed important light on both the biological output of the signals and the mechanisms whereby Mi as a common transcriptional intermediate may modulate diverse functional pathways in human disease.
Materials and methods
Cell culture and treatments
Human melanoma cell lines 501 MEL and MeWo (gifts of Dr. Ruth Halaban, Yale Medical School, New Haven, CT; Zakut et al. 1993) were grown in F10 media (GIBCO-BRL) with 10% fetal bovine serum plus penicillin/streptomycin/glutamine (GIBCO-BRL). Human primary melanocytes purchased from Clonetics (CA) were cultured in MBM2 medium according to the manufacturer's protocol. Murine melanocytes (gift of Dr. Ruth Halaban) and melanoma line B16, as well as NIH 3T3 (mouse fibroblasts), BHK (hamster kidney cell line), and COS cells were grown in DMEM plus 10% fetal bovine serum. As indicated, 501 MEL cells were treated with 20 ng/ml recombinant Steel factor (stem cell factor, R & D Systems) with or without 15 μg/ml cyclohexamide (Sigma), whereas B16 and other cell lines that either lost or do not express c-Kit receptor were treated with 100 nm TPA (Sigma) plus or minus cyclohexamide. Various drugs, as described in the text, were used at the indicated concentrations: 20 μm MEK inhibitor PD 98059 (New England BioLab), 25 μm proteasome inhibitor MG132 (CalBiochem), 25 μm lactacystin, and 20 nm okadaic acid (GIBCO-BRL).
Transfection, infection, and Mi constructs
501 MEL, BHK, COS, and 3T3 cells were transfected using FuGENE 6 (Boehringer Mannheim) following the manufacturer's protocol with wild-type human Mi (MITF) vector (Hemesath et al. 1998) and its derivative MITF mutants. A utation from serine to alanine at position 73 generated S73A (Hemesath et al. 1998), whereas one mutated at ser-409 termed S409A, and a dual mutation at both sites produced S73/409A. HA-tagged (MT 123) or His-tagged (MT 107) ubiquitin expression vectors (gift from Dr. Dirk Bohmann, European Molecular Biology Laboratory, Heidelberg, Germany) were also used in transfecting MeWo and BHK cells.
501 MEL cells were also infected with E1A- and E1B-deleted adenovirus type 5, which contained human wild-type MITF and ser-73 to alanine-mutated MITF, respectively, both of which were HA tagged. The MITF insert was cloned into a unique XbaI site located 452 bp upstream from the 5′ end of the vector backbone. The insert was driven by an elongation factor α promotor (Mizushima and Nagata 1990). Subconfluent 501 MEL cells were incubated with the adenovirus constructs in serum-free F10 supplemented with 10 mm MgCl2 for 30 min at a multiplicity of infection of 100. After the infection the medium was replaced by fresh F10 medium plus 10% fetal bovine serum, and cells were cultured for another 62 hr followed by human Steel factor stimulation in the presence or absence of cyclohexamide and the proteasome inhibitor MG132.
Immunoblotting and immunoprecipitation
The monoclonal antibody D5 was raised against a histidine fusion protein expressed from the amino-terminal Taq–Sac fragment of human Mi cDNA (Tachibana et al. 1994) and produces a specific supershift of a Mi/DNA complex, but does not react with any closely related family members (not shown). An alternative Mi monoclonal antibody C5 (Weilbaecher et al. 1998) was used to detect murine MITF. High-affinity rat HA antibody (Boehringer Mannheim) was used to detect HA-tagged Mi protein, and antibody against α-tubulin (Sigma) for protein-loading control. For immunoblot analysis, cells were lysed in lysis buffer [50 mm Tris (pH 7.6), 150 mm NaCl, 1% Triton-X 100] plus protease inhibitors (Boehringer Mannheim) and phosphatase inhibitors (20 mm NaPP, 10 mm NaF, and 1 mm Na3VO4). Protein concentrations were determined by using the Dc protein assay kit (Bio-Rad). Samples were solubilized in SDS sample buffer plus 50 mm DTT and boiled for 5 min. After SDS-PAGE and transfer to nitrocellulose, blots were blocked in 5% milk plus 0.05% Tween-20 in Tris-buffered saline before antibody incubation. Antibody complexes were detected with peroxidase-conjugated secondary antibody (Cappel) and chemiluminescence reagents (Amersham).
For immunoprecipitation, cells were lysed with lysis buffer as above or with RIPA buffer [10 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% Nonidet P-40, 0.1% SDS, 1% sodium sarcosyl, 1 mm DTT] plus protease inhibitors. The soluble fraction was incubated overnight at 4°C with primary antibodies and subsequently protein G agarose beads (GIBCO-BRL) were added to the solution and incubated for an additional 1 hr at 4°C. Beads were washed three times with cold PBS or RIPA buffer, resuspended in SDS sample buffer, and boiled for 5 min. The eluted proteins were resolved on SDS-PAGE and immunoblot analysis performed as described above. For His-tagged protein analysis, washed Ni–NTA agarose beads (Qiagen) were added to the cell lysates and incubated at 4°C for 4 hr. Eluted proteins were analyzed as above.
Supershift EMSA
Cells were washed twice in cold PBS and resuspended in buffer A [10 mm HEPES (pH 7.9), 1.5 mm MgCl2, 10 mm KCl, and 0.5 mm DTT plus protease inhibitors] and incubated on ice for 10 min. The incubations were agitated for 10 sec and centrifuged (14,000 rpm for 10 sec using a microcentrifuge), and the resulting pellet was resuspended in two packed-nuclei volumes of buffer C [20 mm HEPES (pH 7.9), 420 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.5 mm DTT, 25% glycerol] plus protease inhibitors and further incubated on ice for 30 min. The suspension was centrifuged and the supernatant was quantitated, and equal amount of protein used to perform gel mobility assay. Probe labeling and DNA-binding reactions were performed as described (Hemesath et al. 1998). For supershift EMSA, 1 μl of high affinity HA antibody (Boehringer Mannheim) was added concurrently with other components of the binding reaction.
Transactivation assay
Transfections of 501 MEL or 3T3 cells were carried out in 24-well plates using FuGENE 6 as above according to manufacturer's recommendations. Each experiment was performed in triplicate, and transfection efficiency was normalized to cotransfected sea pansy luciferase plasmid (Promega). Cells in each well were transfected with 0.1 μg of tyrosinase reporter (Hemesath et al. 1998), 0.5 μg of MITF expression plasmid or its derivative mutant vectors, and 0.1 μg of sea pansy luciferase plasmid. After 48 hr of transfection, cells were lysed in 150 μl of passive lysis buffer as per manufacturer's recommendations (Promega) and incubated for 30 min at room temperature. An aliquot of the lysates was used to perform luciferase assays using dual luciferase reagents (Promega). An equal amount of lysate was subjected to immunoblotting analysis for expression of the MITF (wild type or its derivatives).
Phosphotryptic mapping
For in vivo labeling of MITF, 501 MEL cells were washed in phosphate-free, serum-free DMEM (GIBCO) and then labeled in vivo with 1 mCi/ml of inorganic 32P in a 10-cm dish for 8 hr. Cells were washed with ice-cold PBS, then lysed in l% Triton, 170 mm NaCl, 20 mm Tris (pH 7.4) plus protease and phosphatase inhibitors. Lysates were immunoprecipitated with anti-MITF monoclonal antibody (C5) (Weilbaecher et al. 1998) and protein G agarose beads (GIBCO) at 4°C overnight. Beads were washed and boiled in loading buffer (3% SDS, 10% glycerol, 120 mm Tris, and 0.1 m DTT). Proteins were separated on SDS-PAGE and transferred to nitrocellulose membranes. Bands were visualized by autoradiography, excised, and digested for 20 hr at 37°C with 25 μg of TPCK-treated trypsin (Sigma). Phosphopeptide mapping was carried out as described (Hemesath et al. 1998) with the Pharmacia LKB Multiphor II apparatus, which generates inverted replica blots, and using ammonium carbonate (pH 8.9).
For two-dimensional phosphotryptic analysis of in vitro Rsk-1 labeled MITF, a recombinant fragment of GST–MITF fusion protein spanning amino acid residues 370–419 was used. 501 MEL cells were starved in serum-free medium for 2 hr and then stimulated with 20 ng/ml of recombinant human Steel factor (R & D Systems). Cells were washed with ice-cold PBS, then lysed in 1% Triton, 170 mm NaCl, 20 mm Tris (pH 7.4) plus protease and phosphatase inhibitors. Lysates were immunoprecipitated with polyclonal antibody to Rsk-1 (Santa Cruz) and protein G agarose beads (GIBCO) at 4°C overnight. Beads were washed twice in PBS and 10 μCi of [32P]ATP was added to 100 ng of recombinant GST–MITF in 20 μl of IVK buffer [50 mm HEPES (pH 7.6), 10 mm MgCl2, 2 mm NaVO4, 0.5 mg/ml Pefabloc, 2 mm DTT, and 50 μm ATP] and incubated at 32°C for 30 min. Loading buffer was added to the sample, boiled, then resolved on SDS-PAGE, transferred to nitrocellulose, and subjected to tryptic digestion and two-dimensional analysis as described above.
In vitro kinase assay
Cells were Steel-stimulated and lysed as described above and 4 μl of anti-Erk-2 or anti-Rsk-1 antisera (Santa Cruz) plus protein G agarose beads were added to the lysates and mixed overnight at 4°C. Beads were washed three times with lysis buffer and once with IVK buffer [50 mm HEPES (pH 7.6), 2 mm NaVO4, 10 mm MgCl2, 1 mm PMSF, 2 mm DTT, and 50 μm ATP]. For each reaction, 440 μl of IVK buffer, 10 μCi [32P]ATP, and recombinant phosphoacceptor proteins were added. Mi histidine fusion proteins spanning residues 16–185 containing ser-73 (Tachibana et al. 1994) or GST fusion protein spanning residues 389–419 containing ser-409 were added as substrates and incubated at 30°C for 30 min. Reactions were stopped by addition of loading buffer and analyzed by immunoblotting and autoradiography.
Acknowledgments
We gratefully acknowledge Dr. Ruth Halaban for the gifts of human and mouse melanocytes as well as human melanoma cell lines and useful discussions, Dr. Dirk Bohmann for ubiquitin expression plasmids, and Dr. John Blenis for useful discussions early in the project. This work was supported by National Institutes of Health grants HL03113 (DZF) and AR43369 (DEF).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL david_fisher@dfci.harvard.edu; FAX (617) 632-2085.
References
- Andrews RG, Briddell RA, Appelbaum FR, McNiece IK. Stimulation of hematopoiesis in vivo by stem cell factor. Curr Opin Hematol. 1994;1:187–196. [PubMed] [Google Scholar]
- Armstrong JA, Emerson BM. Transcription of chromatin: These are complex times. Curr Opin Genet Dev. 1998;8:165–172. doi: 10.1016/s0959-437x(98)80137-8. [DOI] [PubMed] [Google Scholar]
- Bentley NJ, Eisen T, Goding CR. Melanocyte-specific expression of the human tyrosinase promoter: Activation by the microphthalmia gene product and role of the initiator. Mol Cell Biol. 1994;14:7996–8006. doi: 10.1128/mcb.14.12.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk AJ. Activation of RNA polymerase II transcription. Curr Opin Cell Biol. 1999;11:330–335. doi: 10.1016/S0955-0674(99)80045-3. [DOI] [PubMed] [Google Scholar]
- Bertolotto C, Bille K, Ortonne J, Ballotti R. Regulation of tyrosinase gene expression by cAMP in B16 melanoma cells involves two CATGTG motifs surrounding the TATA box: Implication of the microphthalmia gene product. J Cell Biol. 1996;134:747–755. doi: 10.1083/jcb.134.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotto C, Abbe P, Hemesath TJ, Bille K, Fisher DE, Ortonne JP, Ballotti R. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J Cell Biol. 1998a;142:827–835. doi: 10.1083/jcb.142.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotto C, Busca R, Abbe P, Bille K, Aberdam E, Ortonne JP, Ballotti R. Different cis-acting elements are involved in the regulation of TRP1 and TRP2 promoter activities by cyclic AMP: Pivotal role of M boxes (GTCATGTGCT) and of microphthalmia. Mol Cell Biol. 1998b;18:694–702. doi: 10.1128/mcb.18.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blenis J. Signal transduction via the MAP kinases: Proceed at your own RSK. Proc Natl Acad Sci. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel S, Rimoldi D. Melanoma-associated antigens. Eur J Cancer. 1993;13:1903–1907. doi: 10.1016/0959-8049(93)90548-t. [DOI] [PubMed] [Google Scholar]
- Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- Chen RH, Tung R, Abate C, Blenis J. Cytoplasmic to nuclear signal transduction by mitogen-activated protein kinase and 90 kDa ribosomal S6 kinase. Biochem Soc Trans. 1993;21:895–900. doi: 10.1042/bst0210895. [DOI] [PubMed] [Google Scholar]
- Chen RH, Juo PC, Curran T, Blenis J. Phosphorylation of c-Fos at the C-terminus enhances its transforming activity. Oncogene. 1996;12:1493–1502. [PubMed] [Google Scholar]
- Costa J, Demetri G, Harrist T, Dvorak A, Hayes D, Merica E, Menchaca D, Gringeri A, Schwartz L, Galli S. Recombinant human stem cell factor (kit ligand) promotes human mast cell and melanocyte hyperplasia and functional activation in vivo. J Exp Med. 1996;183:2681–2686. doi: 10.1084/jem.183.6.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby KN, Morrice N, Caudwell FB, Avruch J, Cohen P. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J Biol Chem. 1998;273:1496–1505. doi: 10.1074/jbc.273.3.1496. [DOI] [PubMed] [Google Scholar]
- Dubreuil P, Forrester L, Rottapel R, Reedijk M, Fujita J, Bernstein A. The c-fms gene complements the mitogenic defect in mast cells derived from mutant W mice but not mi (microphthalmia) mice. Proc Natl Acad Sci. 1991;88:2341–2345. doi: 10.1073/pnas.88.6.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englaro W, Bertolotto C, Busca R, Brunet A, Pages G, Ortonne JP, Ballotti R. Inhibition of the mitogen-activated protein kinase pathway triggers B16 melanoma cell differentiation. J Biol Chem. 1998;273:9966–9970. doi: 10.1074/jbc.273.16.9966. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Nebreda AR. A MAP kinase docking site is required for phosphorylation and activation of p90(rsk)/MAPKAP kinase-1. Curr Biol. 1999;9:281–284. doi: 10.1016/s0960-9822(99)80120-1. [DOI] [PubMed] [Google Scholar]
- Ghoda L, Lin X, Greene WC. The 90-kDa ribosomal S6 kinase (pp90rsk) phosphorylates the N-terminal regulatory domain of IkappaBalpha and stimulates its degradation in vitro. J Biol Chem. 1997;272:21281–21288. doi: 10.1074/jbc.272.34.21281. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Functions of the proteasome: The lysis at the end of the tunnel [Comment] Science. 1995;268:522–523. doi: 10.1126/science.7725095. [DOI] [PubMed] [Google Scholar]
- Goldberg AL, Rock KL. Proteolysis, proteasomes and antigen presentation. Nature. 1992;357:375–379. doi: 10.1038/357375a0. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Perez M, Kung AL, Joseph M, Mansur C, Xiao ZX, Kumar S, Howley PM, Livingston DM. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell. 1998;2:405–415. doi: 10.1016/s1097-2765(00)80140-9. [DOI] [PubMed] [Google Scholar]
- Grove JR, Price DJ, Banerjee P, Balasubramanyam A, Ahmad MF, Avruch J. Regulation of an epitope-tagged recombinant Rsk-1 S6 kinase by phorbol ester and erk/MAP kinase. Biochemistry. 1993;32:7727–7738. doi: 10.1021/bi00081a018. [DOI] [PubMed] [Google Scholar]
- Halaban R, Cheng E, Zhang Y, Moellmann G, Hanlon D, Michalak M, Setaluri V, Hebert DN. Aberrant retention of tyrosinase in the endoplasmic reticulum mediates accelerated degradation of the enzyme and contributes to the dedifferentiated phenotype of amelanotic melanoma cells. Proc Natl Acad Sci. 1997;94:6210–6215. doi: 10.1073/pnas.94.12.6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan HT, Zander A. Stem cell factor as a survival and growth factor in human normal and malignant hematopoiesis. Acta Haematol. 1996;95:257–262. doi: 10.1159/000203893. [DOI] [PubMed] [Google Scholar]
- Hemesath TJ, Steingrimsson E, McGill G, Hansen MJ, Vaught J, Hodgkinson CA, Arnheiter H, Copeland NG, Jenkins NA, Fisher DE. Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes & Dev. 1994;8:2770–2780. doi: 10.1101/gad.8.22.2770. [DOI] [PubMed] [Google Scholar]
- Hemesath TJ, Price ER, Takemoto C, Badalian T, Fisher DE. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature. 1998;391:298–301. doi: 10.1038/34681. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system for protein degradation. Annu Rev Biochem. 1992;61:761–807. doi: 10.1146/annurev.bi.61.070192.003553. [DOI] [PubMed] [Google Scholar]
- Hughes AE, Newton VE, Liu XZ, Read AP. A gene for Waardenburg syndrome type 2 maps close to the human homologue of the microphthalmia gene at chromosome 3p12-p14.1. Nat Genet. 1994;7:509–512. doi: 10.1038/ng0894-509. [DOI] [PubMed] [Google Scholar]
- Kaufmann O, Koch S, Burghardt J, Audring H, Dietel M. Tyrosinase, melan-A, and KBA62 as markers for the immunohistochemical identification of metastatic amelanotic melanomas on paraffin sections. Mod Pathol. 1998;11:740–746. [PubMed] [Google Scholar]
- Kim TK, Maniatis T. Regulation of interferon-gamma-activated STAT1 by the ubiquitin–proteasome pathway. Science. 1996;273:1717–1719. doi: 10.1126/science.273.5282.1717. [DOI] [PubMed] [Google Scholar]
- King R, Weilbaecher KN, McGill G, Cooley E, Mihm M, Fisher DE. Microphthalmia transcription factor. A sensitive and specific melanocyte marker for Melanoma diagnosis. Am J Pathol. 1999;155:731–738. doi: 10.1016/S0002-9440(10)65172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. Chromatin-modifying and -remodeling complexes. Curr Opin Genet Dev. 1999;9:148–151. doi: 10.1016/S0959-437X(99)80022-7. [DOI] [PubMed] [Google Scholar]
- Leighton IA, Dalby KN, Caudwell FB, Cohen PT, Cohen P. Comparison of the specificities of p70 S6 kinase and MAPKAP kinase-1 identifies a relatively specific substrate for p70 S6 kinase: The N-terminal kinase domain of MAPKAP kinase-1 is essential for peptide phosphorylation. FEBS Lett. 1995;375:289–293. doi: 10.1016/0014-5793(95)01170-j. [DOI] [PubMed] [Google Scholar]
- Lerner AB, Shiohara T, Boissy RE, Jacobson KA, Lamoreux ML, Moellmann GE. A mouse model for vitiligo. J Invest Dermatol. 1986;87:299–304. doi: 10.1111/1523-1747.ep12524353. [DOI] [PubMed] [Google Scholar]
- Mahaffey D, Yoo Y, Rechsteiner M. Ubiquitin metabolism in cycling Xenopus egg extracts. J Biol Chem. 1993;268:21205–21211. [PubMed] [Google Scholar]
- Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ. Insight into the microphthalmia gene. Trends Genet. 1995;11:442–448. doi: 10.1016/s0168-9525(00)89143-x. [DOI] [PubMed] [Google Scholar]
- Nawaz Z, Lonard DM, Dennis AP, Smith CL, O'Malley BW. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci. 1999;96:1858–1862. doi: 10.1073/pnas.96.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- Price ER, Ding HF, Badalian T, Bhattacharya S, Takemoto C, Yao TP, Hemesath TJ, Fisher DE. Lineage-specific signaling in melanocytes. C-kit stimulation recruits p300/CBP to microphthalmia. J Biol Chem. 1998a;273:17983–17986. doi: 10.1074/jbc.273.29.17983. [DOI] [PubMed] [Google Scholar]
- Price ER, Horstmann MA, Wells AG, Weilbaecher KN, Takemoto CM, Landis MW, Fisher DE. alpha-Melanocyte-stimulating hormone signaling regulates expression of microphthalmia, a gene deficient in Waardenburg syndrome. J Biol Chem. 1998b;273:33042–33047. doi: 10.1074/jbc.273.49.33042. [DOI] [PubMed] [Google Scholar]
- Roeder RG. Role of general and gene-specific cofactors in the regulation of eukaryotic transcription. Cold Spring Harbor Symp Quant Biol. 1998;63:201–218. doi: 10.1101/sqb.1998.63.201. [DOI] [PubMed] [Google Scholar]
- Russell E. Hereditary anemias of the mouse: A review for geneticists. Adv Genet. 1979;20:357–459. [PubMed] [Google Scholar]
- Sato S, Roberts K, Gambino G, Cook A, Kouzarides T, Goding CR. CBP/p300 as a co-factor for the Microphthalmia transcription factor. Oncogene. 1997;14:3083–3092. doi: 10.1038/sj.onc.1201298. [DOI] [PubMed] [Google Scholar]
- Schouten GJ, Vertegaal AC, Whiteside ST, Israel A, Toebes M, Dorsman JC, van der Eb AJ, Zantema A. IkappaB alpha is a target for the mitogen-activated 90 kDa ribosomal S6 kinase. EMBO J. 1997;16:3133–3144. doi: 10.1093/emboj/16.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimeca JC, Nguyen TT, Filloux C, Van Obberghen E. Nerve growth factor-induced phosphorylation cascade in PC12 pheochromocytoma cells. Association of S6 kinase II with the microtubule-associated protein kinase, ERK1. J Biol Chem. 1992;267:17369–17374. [PubMed] [Google Scholar]
- Silvers WK. The coat colors of mice: A model for mammalian gene action and interaction. New York, NY: Spinger-Verlag; 1979. [Google Scholar]
- Sturgill TW, Ray LB, Erikson E, Maller JL. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature. 1988;334:715–718. doi: 10.1038/334715a0. [DOI] [PubMed] [Google Scholar]
- Sykora KW, Tomeczkowski J, Reiter A. C-kit receptors in childhood malignant lymphoblastic cells. Leuk Lymphoma. 1997;25:201–216. doi: 10.3109/10428199709114160. [DOI] [PubMed] [Google Scholar]
- Tachibana M. Evidence to suggest that expression of MITF induces melanocyte differentiation and haploinsufficiency of MITF causes Waardenburg syndrome type 2A. Pigment Cell Res. 1997;10:25–33. doi: 10.1111/j.1600-0749.1997.tb00462.x. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Perez-Jurado LA, Nakayama A, Hodgkinson CA, Li X, Schneider M, Miki T, Fex J, Francke U, Arnheiter H. Cloning of MITF, the human homolog of the mouse microphthalmia gene and assignment to chromosome 3p14.1-p12.3. Hum Mol Genet. 1994;3:553–557. doi: 10.1093/hmg/3.4.553. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8:251–255. doi: 10.1038/ng1194-251. [DOI] [PubMed] [Google Scholar]
- Tsai M, Chen RH, Tam SY, Blenis J, Galli SJ. Activation of MAP kinases, pp90rsk and pp70-S6 kinases in mouse mast cells by signaling through the c-kit receptor tyrosine kinase or Fc epsilon RI: Rapamycin inhibits activation of pp70-S6 kinase and proliferation in mouse mast cells. Eur J Immunol. 1993;23:3286–3291. doi: 10.1002/eji.1830231234. [DOI] [PubMed] [Google Scholar]
- Tsujimura T, Morii E, Nozaki M, Hashimoto K, Moriyama Y, Takebayashi K, Kondo T, Kanakura Y, Kitamura Y. Involvement of transcription factor encoded by the mi locus in the expression of c-kit receptor tyrosine kinase in cultured mast cells of mice. Blood. 1996;88:1225–1233. [PubMed] [Google Scholar]
- Weilbaecher KN, Hershey CL, Takemoto CM, Horstmann MA, Hemesath TJ, Tashjian AH, Fisher DE. Age-resolving osteopetrosis: A rat model implicating microphthalmia and the related transcription factor TFE3. J Exp Med. 1998;187:775–785. doi: 10.1084/jem.187.5.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DE, Eisenman J, Baird A, Rauch C, Van Ness K, March CJ, Park LS, Martin U, Mochizuki DY, Boswell HS, Burgess GS, Cosman D, Lyman SD. Identification of a ligand for the c-kit proto-oncogene. Cell. 1990;63:167–174. doi: 10.1016/0092-8674(90)90297-r. [DOI] [PubMed] [Google Scholar]
- Witte ON. Steel locus defines new multipotent growth factor. Cell. 1990;63:5–6. doi: 10.1016/0092-8674(90)90280-r. [DOI] [PubMed] [Google Scholar]
- Yasumoto K, Yokoyama K, Shibata K, Tomita Y, Shibahara S. Microphthalmia-associated transcription factor as a regulator for melanocyte-specific transcription of the human tyrosinase gene. Mol Cell Biol. 1994;14:8058–8070. doi: 10.1128/mcb.14.12.8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakut R, Perlis R, Eliyahu S, Yarden Y, Givol D, Lyman SD, Halaban R. KIT ligand (mast cell growth factor) inhibits the growth of KIT-expressing melanoma cells. Oncogene. 1993;8:2221–2229. [PubMed] [Google Scholar]
- Zaman Z, Ansari AZ, Gaudreau L, Nevado J, Ptashne M. Gene transcription by recruitment. Cold Spring Harbor Symp Quant Biol. 1998;63:167–171. doi: 10.1101/sqb.1998.63.167. [DOI] [PubMed] [Google Scholar]