

Abstract
During human nucleotide excision repair, damage is recognized, two incisions are made flanking a DNA lesion, and residues are replaced by repair synthesis. A set of proteins required for repair of most lesions is RPA, XPA, TFIIH, XPC–hHR23B, XPG, and ERCC1–XPF, but additional components have not been excluded. The most complex and difficult to analyze factor is TFIIH, which has a 6-subunit core (XPB, XPD, p44, p34, p52, p62) and a 3-subunit kinase (CAK). TFIIH has roles both in basal transcription initiation and in DNA repair, and several inherited human disorders are associated with mutations in TFIIH subunits. To identify the forms of TFIIH that can function in repair, recombinant XPA, RPA, XPC–hHR23B, XPG, and ERCC1–XPF were combined with TFIIH fractions purified from HeLa cells. Repair activity coeluted with the peak of TFIIH and with transcription activity. TFIIH from cells with XPB or XPD mutations was defective in supporting repair, whereas TFIIH from spinal muscular atrophy cells with a deletion of one p44 gene was active. Recombinant TFIIH also functioned in repair, both a 6- and a 9-subunit form containing CAK. The CAK kinase inhibitor H-8 improved repair efficiency, indicating that CAK can negatively regulate NER by phosphorylation. The 15 recombinant polypeptides define the minimal set of proteins required for dual incision of DNA containing a cisplatin adduct. Complete repair was achieved by including highly purified human DNA polymerase δ or ε, PCNA, RFC, and DNA ligase I in reaction mixtures, reconstituting adduct repair for the first time with recombinant incision factors and human replication proteins.
Keywords: Transcription, DNA repair, cisplatin, kinase, DNA polymerase, xeroderma pigmentosum
The process of nucleotide excision repair (NER) removes lesions in DNA by locating damage, introducing incisions on the damaged strand on each side of a lesion, excising an oligonucleotide of 24–32 residues, and filling in the gap by repair synthesis and ligation (de Laat et al. 1999). Considerable progress has been made in recent years toward determining the core components that carry out nucleotide excision repair in eukaryotes. In human cells, the proteins necessary for the incision reaction include XPA, XPC–hHR23B complex, RPA, TFIIH, and the nucleases XPG and ERCC1–XPF (Aboussekhra et al. 1995; Mu et al. 1995, 1996). In the yeast Saccharomyces cerevisiae, incision of UV lesions can be accomplished by the homologous set of proteins Rad14, Rad4–Rad23, RPA, and TFIIH and the nucleases Rad2 and Rad1–Rad10 (Guzder et al. 1995; He et al. 1996). Uncertainties remain as to whether this list of proteins represents truly the minimal set of polypeptides involved in incision. An additional factor in yeast is composed of the Rad7 and Rad16 polypeptides and was designated nucleotide excision repair factor 4 (Guzder et al. 1997). It significantly increases incision efficiency in the yeast system and was suggested to be either an ATP-dependent damage recognition component (Guzder et al. 1998) or necessary for postincision events (Reed et al. 1998). Yeast cells with Rad7 or Rad16 mutations show little repair of pyrimidine dimers in nontranscribed DNA strands (Verhage et al. 1994), and extracts have much diminished repair in vitro (Wang et al. 1997). During reconstitution of NER with UV-irradiated DNA, using Escherichia coli DNA polymerase I to perform damage dependent DNA synthesis, we found an additional factor designated IF7 that significantly stimulated repair (Aboussekhra et al. 1995). Furthermore, the UV–DDB protein complex, defective in xeroderma pigmentosum group E, is necessary in cells for efficient repair of cyclobutane pyrimidine dimers in nontranscribed DNA (Hwang et al. 1999). Another factor with a marked effect on yeast NER in vivo is the MMS19 gene product, which appears to activate TFIIH for repair (Lauder et al. 1996; Lombaerts et al. 1997). So far, experiments to reconstitute the NER incision reaction have used a mixture of recombinant components and proteins purified from human cell lysates. This has left open the possibility that unidentified factors copurified with some of the components required for the core repair reaction. To reach a more definitive conclusion, we set out here to assemble the dual incision reaction exclusively with recombinant, highly purified components including TFIIH.
TFIIH is remarkable in that it functions both in basal transcription initiation of mRNA as well as in nucleotide excision repair (Svejstrup et al. 1996; de Laat et al. 1999). Mammalian TFIIH includes a 6-subunit core (XPB, XPD, p62, p52, p44, and p34) and three additional components (Cdk7, cyclin H, and MAT1). The latter subunits comprise the CDK-activating kinase (CAK) complex, which phosphorylates the carboxy-terminal domain of RNA polymerase II during transcription initiation (Svejstrup et al. 1996). The TFIIH helicase subunits XPB and XPD are responsible for catalyzing the open complex formation that takes place around a promoter during transcription initiation and around a lesion during NER (Evans et al. 1997b; Mu et al. 1997; Tirode et al. 1999; Winkler et al. 2000). Particular mutations in XPB and XPD lead to different human syndromes with a surprising range of photosensitivities, cancer susceptibilities, and developmental abnormalities (Taylor et al. 1997; Botta et al. 1998; de Laat et al. 1999). These include xeroderma pigmentosum (XP), Cockayne syndrome (CS), and trichothiodystrophy (TTD).
An outstanding question is which forms of the transcription-repair complex TFIIH are active in repair. Evidence exists that in yeast there are interchangeable TFIIH forms with different principal tasks, one form containing CAK that works mainly in transcription and another without CAK components for repair (Svejstrup et al. 1995). Depending on the method used to isolate the TFIIH complex from human cells, differences in the subunit composition can be observed, and some free CAK kinase complex may exist (Feaver et al. 1994; Adamczewski et al. 1996; Rossignol et al. 1997). By taking advantage of the recent preparation of recombinant TFIIH (Tirode et al. 1999) it is possible to clearly determine whether the 6- and 9-subunit forms of TFIIH are both functional in repair. Moreover, the activity of TFIIH purified from cells containing mutations in different subunits can be assessed in a defined system. Finally, we have asked whether all of the recombinant damage recognition and incision components can be combined with highly purified human DNA synthesis proteins to achieve complete repair.
Results
Dual incision with recombinant factors and TFIIH fractions from HeLa cells
To monitor the dual incision reaction of NER, we used a closed-circular plasmid containing a single 1,3 intrastrand d(GpTpG) cisplatin–DNA cross-link. This DNA was combined with the purified recombinant proteins shown in Figure 1A. XPA, heterotrimeric RPA, and ERCC1–XPF complex were produced in E. coli, whereas XPG and XPC–hHR23B complex were produced in insect cells.
Figure 1.


Purified human proteins for NER. (A) Incision factors (top line) and repair synthesis factors (bottom line). PCNA, Lig1, and all incision factors are recombinant polypeptides except for TFIIH Hap fr. VI. The XPA, RPA, XPG, PCNA, and Lig1 gels were stained with Coomassie blue and the others with silver. Asterisks represent identified subunits of human RFC, Pol δ, and Pol ε. (B) Recombinant TFIIH. Silver-stained gels (left) and immunoblots (right); equivalent amounts of rIIH6 or rIIH9 were loaded. The six core subunits are present in similar amounts in the two preparations, and the CAK subunits in rIIH9 appear to be roughly stoichiometric with the core subunits.
To define the form of TFIIH active in repair and to assess the need for any further incision proteins, these five recombinant factors were incubated with DNA containing the cisplatin lesion and TFIIH purified from HeLa cells through various steps as outlined in Figure 2A. NER activity was followed by direct end-labeling of the oligonucleotides produced by dual incision. Throughout the purification procedure, TFIIH activity was followed by assaying for transcription initiation activity, helicase and ATPase activities, and immunoblotting for known TFIIH components (Gerard et al. 1991; Marinoni et al. 1997).
Figure 2.

Transcription and NER activities of TFIIH copurify. (A) Scheme for TFIIH purification from HeLa cells; TFIIH Hep is the result of step IV, heparin–HPLC and TFIIH Hap is the result of step VI, hydroxyapatite–HPLC. (B) (Top panel) In vitro runoff transcription assays (Tirode et al. 1999), using 1 μl of each fraction; (middle panel) 5 μl of each step V fraction was separated by 10% SDS-PAGE and immunoblotted against the three indicated subunits of TFIIH; (bottom panel) 3 μl of each step V fraction was used in a nucleotide excision repair assay containing all the recombinant proteins needed for dual incision formation (except TFIIH); 1.5 μl of TFIIH Hep fr. IV was used where indicated. Only the area of the gel containing NER excision products is shown. (C) As in B, for step VI TFIIH fractions. (M) 35- and 27-nucleotide molecular weight markers from a labeled MspI digest of pBR322.
TFIIH purified from HeLa cells over four chromatographic steps, TFIIH Hep fr. IV, is active in NER in combination with the other purified components (Fig. 2B, first two lanes). Fraction IV was subjected to two further HPLC purification steps, a phenyl-5PW and a hydroxyapatite column (Fig. 2A). TFIIH purified over five chromatographic steps, TFIIH Phe fr. V, was also active in transcription and in dual incision formation. The peak of protein, detected by immunoblotting, coincides with both activities (Fig. 2B). For NER assays, we found it particularly important to use TFIIH fractions that were as concentrated as possible.
Fraction V was purified further through a hydroxyapatite column, yielding a highly purified TFIIH Hap fr. VI (Fig. 1A). NER activity of fraction VI also peaked in the same fraction as transcription activity (Fig. 2C). Individual omission of any of the other factors (Fig. 3) abolished repair, showing that the reaction is totally dependent on the addition of RPA, XPA, XPC–hHR23B, XPG, and ERCC1–XPF and that these repair factors are not present in the TFIIH Hap fr. VI.
Figure 3.

Dual incision with highly purified TFIIH from human cells. NER was performed in the presence of recombinant RPA, XPA, XPC–hHR23B, XPG, ERCC1–XPF, and TFIIH fr. IV or fr. VI as indicated. Repair factors were individually omitted as shown. Amounts of TFIIH used were 2 μl of Hap fr. VI (lanes 2–7), 1.5 μl of Hep fr. IV (lane 1), and 2 μl of Hap fr. VI from a different purification (lane 8); TFIIH Hap fr. VI is fraction 6 from Fig. 2C.
Dual incision with recombinant TFIIH
The above results with TFIIH purified from HeLa cells did not rule out the possibility that another polypeptide is needed for the reaction and copurifies with TFIIH. TFIIH Hap fr. VI, for example, still contains some unknown polypeptides detectable by silver staining (Fig. 1A), and as outlined in the introductory section there are suggestions that additional factors may be involved.
To further define the components involved in the dual incision stage of NER, two different recombinant TFIIH complexes were produced in insect cells. A 9-subunit TFIIH containing XPB, XPD, p62, p52, p44, p34, cdk7, cyclin H, and MAT1 was designated rIIH9, and TFIIH lacking the latter three CAK subunits was designated rIIH6. The p44 and cyclin H subunits were His-tagged (Tirode et al. 1999). Both complexes were purified to homogeneity over a heparin–Sepharose column and a metal chelate affinity column (Fig. 1B) and are active in transcription initiation, rIIH9 having a higher transcription activity than rIIH6 due to the presence of the CAK components (Tirode et al. 1999).
We found that both rIIH9 (Fig. 4, lanes 4–6) and rIIH6 (Fig. 4, lanes 7–9) are active in NER. This shows that although TFIIH containing the CAK subcomplex functions in repair, CAK is not needed for dual incision formation in vitro. The concentration of these two complexes is approximately the same as measured by immunoblotting (Fig. 1B), but the response of the reaction appeared to be nonlinear in terms of the amount of TFIIH added (Fig. 4). For this reason, and because of the different purification schemes and possible post-translational modifications, a direct comparison with the activity of native TFIIH cannot be made confidently. However, the possibly higher activity attained with rIIH6 than with rIIH9 suggested that the presence or activity of the CAK subunits might have an inhibitory effect on the NER reaction.
Figure 4.

Dual incision reconstituted with recombinant proteins: Recombinant TFIIH is active in NER. Different amounts of rIIH6 and rIIH9 were used in a nucleotide excision repair assay containing all the recombinant proteins needed for dual incision formation, as indicated. The TFIIH Hep fr. IV lane used 1.5 μl of fraction, and the HeLa lane used 20 μg of whole cell extract protein.
Stimulation of NER by inhibition of CAK activity
To directly examine the influence of CAK subunits on NER, recombinant CAK complex (rCAK) was produced in insect cells as described (Rossignol et al. 1997). We tested the activity of TFIIH complexes containing 6 subunits (rIIH6) with and without the addition of rCAK. Under the conditions used in this assay, there were no detectable differences in the NER activity of TFIIH upon adding CAK (Fig. 5A, lanes 3–5). We did notice, however, a significant effect of CAK when an ATP-regenerating system was included in reaction mixtures. Such an ATP-regenerating system is not necessary when using purified proteins (Fig. 5A), but we often include it in reaction mixtures because it is needed for optimal activity when using cell extracts and cruder fractions (Wood et al. 1988). With an ATP-regenerating system present, addition of CAK to rIIH6 inhibited the reconstituted NER reaction (Fig. 5A, lanes 6–8). This suggested that the higher steady-state levels of ATP maintained by regeneration might lead to an inhibitory effect of CAK kinase, by phosphorylating a component of the reaction.
Figure 5.

Role of CAK in dual incision formation. (A) rIIH6 (2 μl) was tested in a dual incision assay together with increasing amounts of rCAK in the presence (lanes 6–9) or absence (lanes 1–5) of an ATP-regenerating system. The effect of H-8 in the presence of CAK was measured by adding the inhibitor to a final concentration of 100 μm (lanes 3,7). Data were normalized to the amount of repair obtained in reaction mixtures containing TFIIH Hep fr. IV instead of rIIH6. (B) TFIIH Hep fr. IV (1.5 μl) was tested for dual incision formation in the presence (lanes 6–10) or absence (lanes 1–5) of the same ATP-regenerating system as in A; increasing amounts of H-8 kinase inhibitor were added as indicated. (C) TFIIH purified from HeLa cells without the CAK subunit [TFIIH (−CAK), 2 μl] was tested in a dual incision assay in the presence of ATP-regenerating system. TFIIH (−CAK) was prepared by disrupting TFIIH complex with buffer containing 1.2 m KCl and depleting the CAK by immunopurification with a cdk7 antibody (Rossignol et al. 1997), and so is less concentrated than TFIIH Hep fr. IV. Increasing amounts of H-8 were added as indicated (lanes 4–6); 1 μl of rCAK was added to a reaction mixture in the absence of H-8 (lane 7). (D) TFIIH Hap fr. VI (1.5 μl) was tested in a dual incision assay in the presence of the ATP-regenerating system and increasing amounts of H-8 as indicated (lanes 3–5). In all panels 1.5 μl of Hep fr. IV was used. Quantification was done on a PhosphorImager with ImageQuant. The density of the bands corresponding to the 26- to 30-mer products was quantified and divided by the value corresponding to the reactions performed in the presence of TFIIH Hep fr. IV to give the relative counts plotted in the graph.
Using these conditions we tested a chemical inhibitor of CAK kinase activity, H-8. This isoquinoline sulfonamide derivative inhibits the kinase activity of cdk7 and its phosphorylation of the RNA Pol II carboxy-terminal domain during transcription (Dubois et al. 1994). This inhibitor was used in dual incision assays with various TFIIH fractions, in the presence of an ATP-regenerating system. Addition of H-8 to reactions containing TFIIH Hep fraction IV increased its activity in NER by about threefold (Fig. 5B, lanes 7–10). H-8 did not increase the NER activity when this fraction IV TFIIH was used in reaction mixtures without an ATP-regenerating system (Fig. 5B, lanes 2–5). In reaction mixtures with the purest TFIIH fraction from HeLa cells (TFIIH Hap fr. VI), addition of increasing amounts of H-8 could stimulate repair by up to fivefold (Fig. 5D, lanes 2–5). Addition of 100 μm H-8 to reactions containing rIIH6 and rCAK overcame some of the inhibition caused by addition of the kinase complex (Fig. 5A, lane 9). When TFIIH was used that was depleted of CAK subcomplex (Rossignol et al. 1997), H-8 did not stimulate repair, even with ATP regeneration (Fig. 5C). These results indicate that NER activity can be partially inhibited by CAK if ATP concentration is continually maintained.
NER activity of purified TFIIH containing mutated subunits
In view of the association with human syndromes it was important to take advantage of the reconstituted NER system and examine the activity of TFIIH purified from cell lines carrying alterations in various subunits. In addition to the association of XPB and XPD with the disorders XP, CS, and TTD, the human syndrome spinal muscular atrophy (SMA) is also connected with alterations in a TFIIH subunit gene. The p44 gene is duplicated in the human chromosome 5q13 region. The more telomeric of the genes (p44t) is located in a region associated with SMA, whereas the more centromeric (p44c) is not. The two p44 gene products differ by three amino acids (Burglen et al. 1997; Tirode et al. 1999). The interaction of p44 with XPD was shown to be important for helicase activity (Coin et al. 1998).
To analyze the activity of TFIIH containing known mutations, the complex was immunopurified using an antibody against the p44 subunit (Coin et al. 1999). The activity of TFIIH from the repair-proficient cell lines HeLa and MRC5 [an SV40-transformed human fibroblast, e.g., see Mackenney et al. (1997)] was compared with TFIIH from an XP-B cell line, an XP-D cell line, a TTD group A cell line, and two cell lines derived from patients with SMA.
As expected, TFIIH purified from HeLa or MRC5 cells was active in dual incision (Fig. 6, lanes 3,5). TFIIH from the XP-B cell line GM2252 (patient XP11BE) was completely defective in dual incision (Fig. 6, lane 10), whereas TFIIH from GM1855, a cell line derived from the asymptomatic mother of patient XP11BE (Hwang et al. 1996), was active in NER (Fig. 6, lane 9). TFIIH from HD2 cells with an XPD mutation supported only a very low level of NER (Fig. 6, lane 6).
Figure 6.

NER activity of TFIIH containing mutations in different subunits. TFIIH samples from repair-proficient cell lines (MRC5, HeLa, and GM1855, heterozygous normal parent of XP11BE), XP-B (GM2252, patient XP11BE), HD2 (XP-D mutation R683W), TTD-A (TTD1BR), SMA line RA (TFIIH p44c), and SMA line DJ [TFIIH p44(t+c)] were tested for dual incision activity in combination with the other recombinant core incision components as indicated. The TFIIH concentration of each sample was estimated by immunoblotting (Coin et al. 1999), and equivalent amounts of the different preparations were used in the incision assays.
The activities of TFIIH from RA cells containing only the p44c subunit (TFIIH p44c, Fig. 6, lane 7) and a TFIIH from DJ cells containing both p44t and p44c [TFIIH p44(t+c), Fig. 6, lane 8] were tested. TFIIH activity in NER was not affected by the absence of p44t. Finally, we examined repair in TFIIH from cells of TTD1BR, in the TTD-A group. Reactions containing TFIIH from TTD1BR cells formed low levels of dual incisions (Fig. 6, lane 4), consistent with what was observed previously with whole cell extract from TTD1BR (Evans et al. 1997b). Implications of these results are discussed below.
Complete reconstitution of repair synthesis with purified human factors
Nucleotide excision repair synthesis has been examined with UV-irradiated DNA and a mixture of proteins purified from HeLa cells, bacteria, and insect cells, and DNA polymerase components purified from calf thymus (Aboussekhra et al. 1995). It was found that calf thymus DNA Pol ε could function in repair in conjunction with the recognition–incision proteins. The availability of highly purified recombinant incision components allowed us to determine whether these proteins could be combined with repair synthesis factors to obtain full repair. Instead of using UV-irradiated DNA containing a mixture of lesions, we used DNA containing a single, specifically located cisplatin lesion. For DNA polymerase components, we used only highly purified human enzymes. Finally, we checked systems using both DNA Pol δ and Pol ε. Although model studies have suggested that either polymerase may fill NER-generated incision gaps (Shivji et al. 1995), it was possible that only one of the polymerases would function in a highly defined coupled system.
For this purpose the same DNA molecule containing a single adduct was used as in the incision assays presented above, but reaction mixtures included a 32P-labeled deoxynucleotide so that patches would be radiolabeled during repair synthesis. Cleavage of the closed circular M13 containing the cisplatin adduct with BstNI restriction enzyme generates a 33-nucleotide fragment that includes the repair site and several larger fragments (Moggs et al. 1996). Synthesis arising specifically from filling the 24- to 32-nucleotide gap during NER should be largely confined to the 33-nucleotide fragment (labeled C in Fig. 7), with some specific synthesis in the 68-nucleotide fragment (labeled B in Fig. 7) because some mapped 5′ incision sites fall within this fragment (Fig. 7, top).
Figure 7.
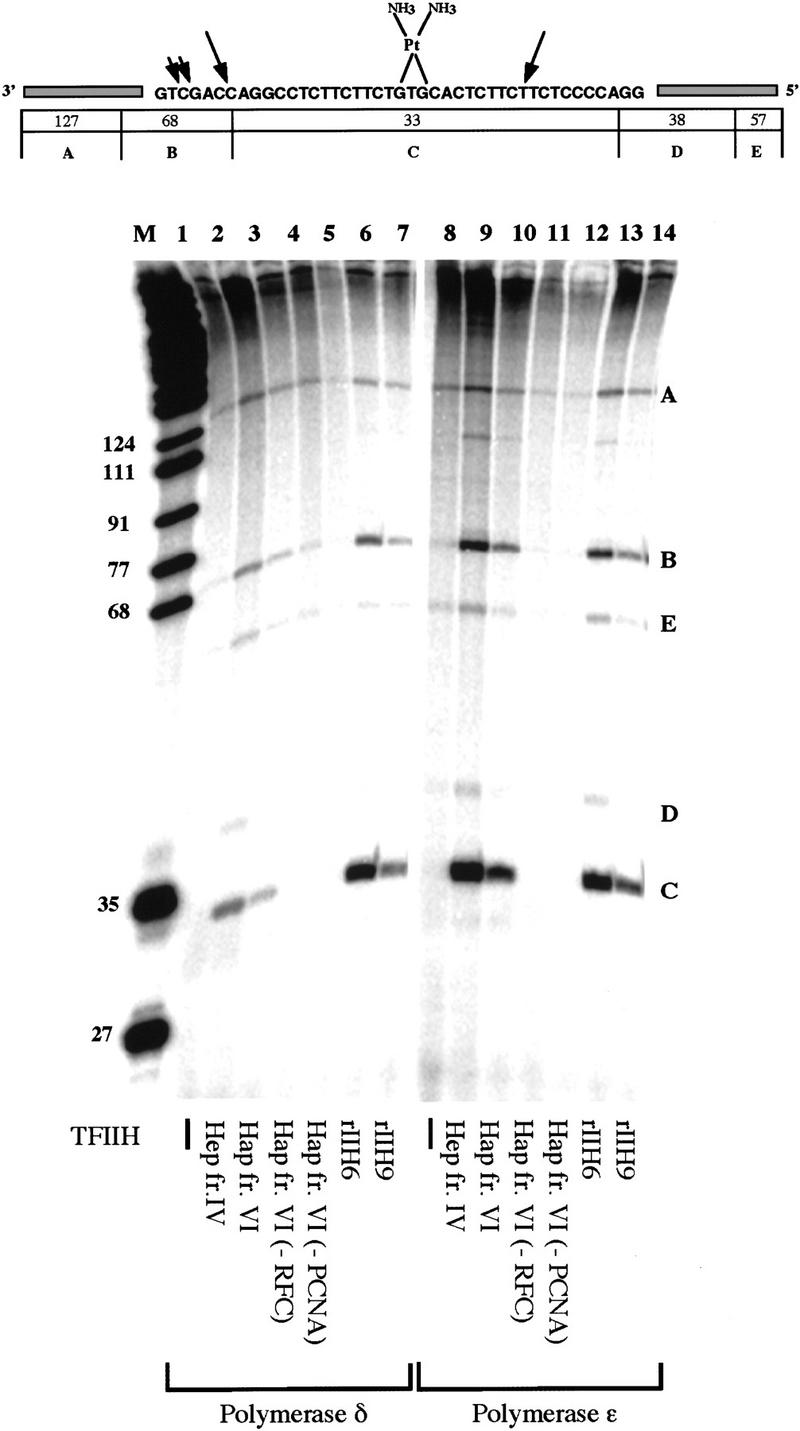
Reconstitution of incision and repair synthesis with recombinant and human purified factors. (Top) Schematic representation of the DNA substrate containing a single 1,3 intrastrand d(GpTpG) cisplatin cross-link. The sizes of the fragments generated by BstNI restriction digestion are shown underneath. Arrows indicate mapped positions of NER incisions. (Bottom) Reconstitution of repair synthesis on a plasmid containing a single cisplatin adduct. Different TFIIH preparations were used in reactions performed in the presence of Pol δ (lanes 1–7) or Pol ε (lanes 8–14); lanes 2 and 9 contain 1.5 μl of TFIIH Hep fr. IV, lanes 3–5 and 10–12 contain 3 μl of TFIIH Hap fr. VI, lanes 6 and 13 contain 3 μl of rIIH6, and lanes 7 and 14 contain 3 μl of rIIH9. Reactions in lanes 4 and 11 were done in the absence of RFC, and reactions in lanes 5 and 12 were done in the absence of PCNA.
To test for repair synthesis activity, all of the proteins anticipated to be needed for NER were combined in the same 12.5-μl volume. The reaction mixtures included recombinant RPA, XPA, XPC–hHR23B, XPG, ERCC1–XPF, TFIIH (with 6 or 9 subunits), PCNA, DNA ligase I, with RFC and human DNA Pol δ or Pol ε purified from human cells (Fig. 7). Both Pol δ and Pol ε could function in repair. Activity was observed with TFIIH purified from HeLa cells (Fig. 7 lanes 2,3,9,10), as well as with 6-subunit recombinant TFIIH (Fig. 7, lanes 6,13) and 9-subunit recombinant TFIIH (Fig. 7, lanes 7,14). No repair was observed when TFIIH was omitted (Fig. 7, lanes 1,8). Reaction mixtures with Pol δ also included FEN1 (DNase IV), as this was found useful in limiting strand-displacement during NER synthesis (Shivji et al. 1995). Under these conditions (∼70 mm salt), both the Pol δ and Pol ε reactions required both PCNA (Fig. 7, lanes 5,12) and the ATP-dependent PCNA loading factor RFC (Fig. 7, lanes 4,11).
Discussion
Minimal sets of human proteins for dual incision and repair synthesis
To unequivocally identify the minimum number of factors involved in the dual incision reaction of NER, we used exclusively recombinant proteins in conjunction with a specific and sensitive method that detects dual incision in a plasmid containing a single cisplatin lesion. As recombinant RPA, XPA, XPC–hHR23B, XPG, and TFIIH complex were sufficient for the reconstitution of the first stage of NER, the conclusion can be reached that only these six factors are needed. These comprise 15 or 18 polypeptides, depending on whether TFIIH with CAK subunits is used. To date, this is the most definitive way to rule out the participation of additional essential factors in the NER reaction on naked DNA in vitro. This does not exclude roles for other stimulatory proteins that are likely to be needed in cells, such as MMS19, UV–DDB, homologs of Rad7 and Rad16, or enzymes yet to be identified. Some factors may, for example, assist with damage recognition in chromatin.
In addition, we reconstituted the complete NER repair synthesis reaction by combining these recombinant incision factors for the first time with recombinant and highly purified human DNA polymerase components. Overall the full repair reaction (dual incision plus repair synthesis) can be achieved in vitro by 10 components, many with multiple subunits: RPA (involved in both steps), XPA, XPC–hHR23B, XPG, ERCC1–XPF, TFIIH complex (with 6 or 9 subunits), DNA Pol δ or Pol ε, RFC, PCNA, and DNA ligase I (and FEN1 for reaction mixtures including DNA Pol δ). Repair synthesis occurred on a singly adducted DNA substrate with all proteins in the same reaction mixture, indicating that no additional factor is required for coupling repair synthesis with dual incision. Repair synthesis under these reaction conditions depends on PCNA for both DNA Pol δ and Pol ε, consistent with previous studies using model DNA substrates containing short gaps (Podust and Hübscher 1993; Shivji et al. 1995, 1998). The human DNA Pol ε preparation used here contained a full-length 261-kD catalytic subunit and a 59-kD small subunit (Fig. 1A). This is in contrast to most previous work with the enzyme, which has been done with preparations containing a stable 140-kD fragment of the catalytic subunit of the enzyme (Aboussekhra et al. 1995; Chui and Linn 1995). With both polymerases, the RFC complex that loads PCNA onto DNA in an ATP-dependent reaction was also required for repair synthesis on this circular template.
Active 6- and 9-subunit forms of TFIIH and modulation of repair activity by CAK
In a complementation assay for TFIIH repair activity using five recombinant incision factors, we found that repair activity eluted in a peak coinciding with TFIIH protein and transcription activities. This suggested that the same form of TFIIH can function in both transcription and in NER. In support of this, we found that a 9-subunit form of recombinant TFIIH containing the CAK subunits is active in dual incision. However, CAK is dispensable for NER in vitro, as shown here with the 6-subunit form of recombinant TFIIH and as shown by complementation of S. cerevisiae cell lysates (Svejstrup et al. 1995) or of partially purified human proteins (Mu et al. 1996). In yeast, evidence has been presented that TFIIH without the corresponding CAK subunits may be specialized for repair, as it is found physically associated with some of the other NER components in a preassembled repairosome (Svejstrup et al. 1995). Definitive evidence for a corresponding high molecular weight functional association in mammalian cells is presently lacking (Araújo and Wood 1999).
TFIIH easily separates into several subcomplexes upon purification, and it is uncertain which forms are the natural ones in cells and which are created during purification. Nine-subunit TFIIH containing CAK can be isolated with gentle methods, but forms without the CAK subunits are also found during purification, particularly a 5-subunit complex without the CAK and XPD subunits (Drapkin et al. 1996; Reardon et al. 1996; Rossignol et al. 1997). XPD helicase activity is necessary for nucleotide excision repair (Winkler et al. 2000) and TFIIH lacking the XPD subunit is inactive (S.J. Araújo, R.D. Wood, and J.M. Egly, unpubl.). It remains to be shown whether the 6- or 9-subunit form, or both, actually participate in NER in vivo. Microinjection of an anti-cdk7 antibody can inhibit NER in cells (Roy et al. 1994), suggesting either that the 9-subunit version normally participates in repair or that the antibody inhibits its conversion into a form active in repair (Svejstrup et al. 1995).
We made the unexpected finding that although the physical presence of the CAK subunits is not necessarily detrimental to NER, inclusion of CAK inhibited repair when reaction mixtures included an ATP regenerating system. Consistent with this effect, inhibition of CAK activity by H-8 kinase inhibitor could stimulate repair reactions and partially reverse the inhibitory effect of CAK. It seems likely that when ATP levels are sufficiently high, the CAK inhibits repair by phosphorylating one or more components of the reaction. It is clear that excessive phosphorylation can inhibit repair, as shown by the fact that inhibitors of protein phosphatases caused dramatic suppression of repair activity in cell extracts (Ariza et al. 1996). The targets of such kinase inhibition are unknown; a potential substrate is the XPG protein, which is readily phosphorylated (S.J. Araújo and R. Ariza, unpubl.). It is noteworthy that the kinase activity of TFIIH decreases after UV irradiation of mammalian cells, suggesting that cells may have a mechanism to suppress CAK activity when DNA repair is urgently required (Adamczewski et al. 1996). The present result emphasizes the potential for regulation of NER by phosphorylation. Because CAK is needed for transcription but not for repair, and can even inhibit repair under some conditions, it is tempting to speculate that a 6-subunit form may be specialized for NER. However, we find that essentially all TFIIH in cell extracts is associated with CAK (S.J. Araújo and R. Wood, unpubl.).
XPB, XPD, and p44 TFIIH mutants in the defined repair system
The defined in vitro system provides an ideal tool to study the influence of different TFIIH mutations on dual incision formation. To initiate such studies, TFIIH was immunopurified from cell extracts containing known naturally occuring mutations in various subunits. The XP individual XP11BE was the first XP-B patient identified and was a severely affected patient with both XP and Cockayne syndrome. The causative mutation in XP11BE alters the carboxy-terminal 42 amino acid residues of XPB, outside the conserved helicase domains (Weeda et al. 1990). The transcription activity of XP11BE TFIIH is partially impaired, showing ∼15% of the activity displayed by normal TFIIH (Coin et al. 1999). NER experiments carried out with cell extracts from XP11BE showed that this TFIIH still functions to form an open bubble intermediate and a normal 3′ incision, but no 5′ incision is carried out (Evans et al. 1997b). We found that XP11BE TFIIH could not support dual incision in the reconstituted system, consistent with its specific defect in 5′ incision activity.
The immortalized cell line HD2 was derived by fusing XP102LO XP-D cells with HeLa cells and selection of a hybrid clone with low repair capacity (Johnson et al. 1985). XP102LO cells carry a R683W mutation in one allele, and both an L461V substitution and a deletion of amino acids 716–730 in another allele (Takayama et al. 1995). The causative mutation in HD2 is assumed to be R683W as this mutation determines the phenotype in patient XP102LO (Taylor et al. 1997). The R683W amino acid substitution is in a region of the XPD gene in which many mutations are present (Taylor et al. 1997). This region of the protein is involved in the interaction between XPD and p44 (Coin et al. 1998), and there is a lower fraction of XPD subunit in HD2 TFIIH, leading to ∼50% of normal transcription assay. We observed a low level of dual incision in reactions with HD2 TFIIH. Repair in HD2 cells was originally measured by survival after UV irradiation and by UV-induced unscheduled DNA synthesis in hybrids with known XP-D cell lines. Both indices were reported to be slightly higher than that of the XP-D parental cell line XP102LO (Johnson et al. 1985). It seems plausible that residual activity might be contributed by a low level of expression of a normal TFIIH allele in the HD2 hybrid.
In the SMA cell lines, the helicase activity of XPD was shown to be normal, and TFIIH from both cell lines had equivalent transcription activity (Coin et al. 1999). Furthermore, recombinant TFIIH containing either p44c or p44t supports transcription (Tirode et al. 1999). The data presented here show that TFIIH containing only p44c is also active in repair, a result consistent with the absence of clinical symptoms related to NER in SMA patients (Burglen et al. 1997).
The defect giving rise to TTD group A is thought to reside in a TFIIH-associated factor, because the TTD1BR repair defect can be complemented by microinjection of TFIIH complex (Stefanini et al. 1993; Vermeulen et al. 1994). However, unlike other TTD cell lines, mutations in a known TFIIH subunit have not been found in TTD1BR. The present results are consistent with this, in that TFIIH from TTD1BR cells is capable of functioning in NER. However, the TFIIH from TTD1BR had a lower activity. The reason for this is still unknown but could be caused, for example, by an altered post-translational modification such as phosphorylation in TTD-A cells.
The ability to specifically measure TFIIH activity in a pure system, as described here, coupled with the means to distinguish defects in open complex formation and 3′ and 5′ incisions (Evans et al. 1997b; Constantinou et al. 1999) opens the way to dissection of the defects in other naturally occurring and directed mutants.
Materials and methods
Proteins
His-tagged recombinant XPA protein was produced in E. coli, purified as described (Jones and Wood 1993), and had a concentration of 450 μg/ml. Recombinant heterotrimeric RPA was produced in E. coli, purified as described (Henricksen et al. 1994), and had a concentration of 500 μg/ml. Recombinant ERCC1–XPF complex was produced in E. coli, purified (Sijbers et al. 1996), and had a concentration of 20 μg/ml. Recombinant XPG protein was produced in insect cells, purified to homogeneity as described (Evans et al. 1997a), and had a concentration of 45 μg/ml. Recombinant XPC–hHR23B complex was expressed and purified from insect cells (Masutani et al. 1997) and the final concentration of the complex was 15 μg/ml. TFIIH was purified from HeLa cells (Marinoni et al. 1997) or in recombinant form from insect cells (Tirode et al. 1999).
DNA Pol δ and RFC were purified from HeLa cells (Weiser et al. 1991; Hübscher et al. 1999). Human Pol ε was purified as outlined (Syväoja and Linn 1989), except that the glycerol gradient centrifugation was performed in the presence of 20 mm potassium phosphate (pH 7.5). Recombinant DNA ligase I (Mackenney et al. 1997) and DNase IV (FEN1) (Robins et al. 1994) were provided by T. Lindahl (ICRF, UK). The recombinant PCNA was purified from E. coli cells (Biggerstaff and Wood 1999) and had a final concentration of 600 μg/ml.
Nucleotide excision repair–dual incision assay
Covalently closed circular DNA containing a single 1,3-intrastrand d(GpTpG) cisplatin–DNA crosslink (Pt–GTG) was prepared as described (Shivji et al. 1999). Repair was carried out in 8.5 μl reaction mixtures for the fully defined reactions. Reactions were carried out in a buffer containing 45 mm HEPES–KOH (pH 7.8), 70 mm KCl, 5 mm MgCl2, 1 mm DTT, 0.3 mm EDTA, 10% glycerol, 2.5 μg of BSA, and 2 mm ATP. Where indicated, reactions were supplemented with an ATP-regenerating system consisting of 40 mm phosphocreatine (di-Tris salt) and 0.5 μg of creatine phosphokinase (type I). Each reaction contained 50 ng of RPA, 22.5 ng of XPA, 10 ng of XPC–hHR23B complex, 50 ng of XPG, 20 ng of ERCC1–XPF complex, and either 1.5 μl of HeLa TFIIH [Hep fr. IV (Marinoni et al. 1997)] or the indicated amounts of other TFIIH fractions. Following preincubation for 10 min at 30°C, 50 ng Pt–GTG was added and reactions were continued for 90 min at 30°C. Rapid freezing stopped the reactions. 6 ng of an oligonucleotide complementary to the excised DNA fragment (and containing four extra G residues at the 5′ end) was annealed to the excision products. Sequenase v. 2.0 polymerase (0.1 unit) and 1 μCi of [α-32P]dCTP were used to add four radiolabeled C residues to each excision product. Products were separated on a denaturing 14% polyacrylamide gel and visualized by autoradiography as described (Shivji et al. 1999).
Nucleotide excision repair–repair synthesis assay
Single lesion (Pt–GTG) plasmid was used as in the previous section for the formation of dual incisions. All reaction mixtures (12.5 μl) were supplemented with 40 mm phosphocreatine (di-Tris salt) and 0.5 μg of creatine phosphokinase (type I), and synthesis was performed in the presence of 2 μCi of [α-32P]dCTP (3000 Ci/mmole), 5 μm dCTP, and of 20 μm of each: dATP, dGTP, and TTP. Repair synthesis was performed simultaneously with the incision step in fully reconstituted reactions. Proteins involved in the dual incision step are in the same amounts as described above and proteins involved in the synthesis step were 60 ng of recombinant PCNA, 50 ng of recombinant DNA ligase I, 0.8 unit of RFC, and 0.9 unit of Pol ε or 0.1 unit of Pol δ. One RFC unit is defined as the incorporation of 10 pmoles of dNMP into acid-insoluble material in 60 min at 37°C in an RFC dependent replication reaction containing primed circular DNA, RP-A, Pol δ, and PCNA (Hübscher et al. 1999); 0.8 unit corresponds to 0.5 μl of the RFC used. Reactions performed with Pol δ also contained 15 ng of recombinant FEN1. Following preincubation for 15 min at 30°C, 50 ng of Pt–GTG was added and reactions were continued for 3 hr at 30°C. DNA was digested in 15-μl reactions with 5 units of BstNI at 60°C for 2 hr prior to electrophoresis in denaturing 14% polyacrylamide gels as described (Shivji et al. 1998).
Acknowledgments
We thank Dawn Batty for providing the XPC–hHR23B protein, Elena Ferrari for expert assistance and advice on the RFC preparation, and G. Daly, T. Lindahl, M. Shivji, and other members of our laboratories for reagents and advice. S.J.A. is recipient of a Portuguese Programa Gulbenkian de Doutoramento em Biologia e Medicina (PGDBM) fellowship. The work of S.J.A. and R.D.W. was supported by the Imperial Cancer Research Fund. M.S. and U.H. were funded by the Swiss Cancer League and the Kanton of Zurich. The work of J.E.S. is supported by a grant from the Research Council for the Environment and Natural Resources, Academy of Finland. J.-M.E., F.T., and F.C. received support from the Institut National de la Santé et de la Recherche Médicale, the CNRS, and the Ministère de la Recherche de l'Enseignement Supérieur.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL wood@icrf.icnet.uk; FAX 44 20 7269 3803.
References
- Aboussekhra A, Biggerstaff M, Shivji MKK, Vilpo JA, Moncollin V, Podust VN, Protic' M, Hübscher U, Egly J-M, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- Adamczewski JP, Rossignol M, Tassan JP, Nigg EA, Moncollin V, Egly JM. MAT1, cdk7 and cyclin H form a kinase complex which is UV light-sensitive upon association with TFIIH. EMBO J. 1996;15:1877–1884. [PMC free article] [PubMed] [Google Scholar]
- Araújo SJ, Wood RD. Protein complexes in nucleotide excision repair. Mutat Res. 1999;435:23–33. doi: 10.1016/s0921-8777(99)00042-7. [DOI] [PubMed] [Google Scholar]
- Ariza RR, Keyse SM, Moggs JG, Wood RD. Reversible protein phosphorylation modulates nucleotide excision repair of damaged DNA by human cell extracts. Nucleic Acids Res. 1996;24:433–440. doi: 10.1093/nar/24.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggerstaff M, Wood RD. Assay for nucleotide excision repair proteins using mammalian cell extracts and UV damaged plasmid DNA. In: Henderson DS, editor. DNA repair protocols: Eukaryotic systems. Totowa, NJ: Humana Press; 1999. pp. 357–372. [Google Scholar]
- Botta E, Nardo T, Broughton BC, Marinoni S, Lehmann AR, Stefanini M. Analysis of mutations in the XPD gene in Italian patients with trichothiodystrophy—Site of mutation correlates with repair deficiency, but gene dosage appears to determine clinical severity. Am J Hum Genet. 1998;63:1036–1048. doi: 10.1086/302063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burglen L, Seroz T, Miniou P, Lefebvre S, Burlet P, Munnich A, Pequignot E, Egly J-M, Melki J. The gene encoding p44, a subunit of the transcription factor TFIIH, is involved in large-scale deletions associated with Werdnig-Hoffmann disease. Am J Hum Genet. 1997;60:72–79. [PMC free article] [PubMed] [Google Scholar]
- Chui G, Linn S. Further characterization of HeLa DNA polymerase ε. J Biol Chem. 1995;270:7799–7808. doi: 10.1074/jbc.270.14.7799. [DOI] [PubMed] [Google Scholar]
- Coin F, Marinoni JC, Rodolfo C, Fribourg S, Pedrini AM, Egly JM. Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat Genet. 1998;20:184–188. doi: 10.1038/2491. [DOI] [PubMed] [Google Scholar]
- Coin F, Bergmann E, Tremeau-Bravard A, Egly JM. Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J. 1999;18:1357–1366. doi: 10.1093/emboj/18.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinou A, Gunz D, Evans E, Lalle P, Bates PA, Wood RD, Clarkson SG. Conserved residues of human XPG protein important for nuclease activity and function in nucleotide excision repair. J Biol Chem. 1999;274:5637–5648. doi: 10.1074/jbc.274.9.5637. [DOI] [PubMed] [Google Scholar]
- de Laat WL, Jaspers NGJ, Hoeijmakers JHJ. Molecular mechanism of nucleotide excision repair. Genes & Dev. 1999;13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- Drapkin R, Le Roy G, Cho H, Akoulitchev S, Reinberg D. Human cyclin-dependent kinase-activating kinase exists in three distinct complexes. Proc Natl Acad Sci. 1996;93:6488–6493. doi: 10.1073/pnas.93.13.6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois MF, Nguyen VT, Bellier S, Bensaude O. Inhibitors of transcription such as 5,6-dichloro-1-beta-D- ribofuranosylbenzimidazole and isoquinoline sulfonamide derivatives (H-8 and H-7) promote dephosphorylation of the carboxyl-terminal domain of RNA polymerase II largest subunit. J Biol Chem. 1994;269:13331–13336. [PubMed] [Google Scholar]
- Evans E, Fellows J, Coffer A, Wood RD. Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO J. 1997a;16:625–638. doi: 10.1093/emboj/16.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E, Moggs JG, Hwang JR, Egly J-M, Wood RD. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997b;16:6559–6573. doi: 10.1093/emboj/16.21.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feaver WJ, Svejstrup JQ, Henry NL, Kornberg RD. Relationship of CDK-activating kinase and RNA polymerase II CTD kinase TFIIH/TFIIK. Cell. 1994;79:1103–1109. doi: 10.1016/0092-8674(94)90040-x. [DOI] [PubMed] [Google Scholar]
- Gerard M, Fischer L, Moncollin V, Chipoulet J-M, Chambon P, Egly J-M. Purification and interaction properties of the human RNA polymerase B(II) general transcription factor BTF2. J Biol Chem. 1991;266:20940–20945. [PubMed] [Google Scholar]
- Guzder SN, Habraken Y, Sung P, Prakash L, Prakash S. Reconstitution of yeast nucleotide excision-repair with purified Rad proteins, replication protein-A, and transcription factor TFIIH. J Biol Chem. 1995;270:12973–12976. doi: 10.1074/jbc.270.22.12973. [DOI] [PubMed] [Google Scholar]
- Guzder SN, Sung P, Prakash L, Prakash S. Yeast Rad7-Rad16 complex, specific for the nucleotide excision-repair of the nontranscribed DNA strand, is an ATP-dependent DNA-damage sensor. J Biol Chem. 1997;272:21665–21668. doi: 10.1074/jbc.272.35.21665. [DOI] [PubMed] [Google Scholar]
- Guzder SN, Sung P, Prakash L, Prakash S. The DNA-dependent ATPase activity of yeast nucleotide excision-repair factor-4 and its role in DNA-damage recognition. J Biol Chem. 1998;273:6292–6296. doi: 10.1074/jbc.273.11.6292. [DOI] [PubMed] [Google Scholar]
- He ZG, Wong JMS, Maniar HS, Brill SJ, Ingles CJ. Assessing the requirements for nucleotide excision repair proteins of Saccharomyces cerevisiae in an in vitro system. J Biol Chem. 1996;271:28243–28249. doi: 10.1074/jbc.271.45.28243. [DOI] [PubMed] [Google Scholar]
- Henricksen L, Umbricht C, Wold M. Recombinant replication protein-A—Expression, complex-formation, and functional-characterization. J Biol Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- Hübscher U, Mossi R, Ferrari E, Stucki M, Jónsson ZO. Functional analysis of DNA replication accessory proteins. In: Cotterill S, editor. Eukaryotic DNA replication. Oxford, UK: Oxford University Press; 1999. pp. 119–137. [Google Scholar]
- Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci. 1999;96:424–428. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JR, Moncollin V, Vermeulen W, Seroz T, van Vuuren H, Hoeijmakers JHJ, Egly JM. A 3′-5′ XPB helicase defect in repair/transcription factor TFIIH of xeroderma pigmentosum group-B affects both DNA-repair and transcription. J Biol Chem. 1996;271:15898–15904. doi: 10.1074/jbc.271.27.15898. [DOI] [PubMed] [Google Scholar]
- Johnson RT, Squires S, Elliott GC, Koch GL, Rainbow AJ. Xeroderma pigmentosum D-HeLa hybrids with low and high ultraviolet sensitivity associated with normal and diminished DNA repair ability, respectively. J Cell Sci. 1985;76:115–133. doi: 10.1242/jcs.76.1.115. [DOI] [PubMed] [Google Scholar]
- Jones CJ, Wood RD. Preferential binding of the xeroderma pigmentosum group A complementing protein to damaged DNA. Biochemistry. 1993;32:12096–12104. doi: 10.1021/bi00096a021. [DOI] [PubMed] [Google Scholar]
- Lauder S, Bankmann M, Guzder SN, Sung P, Prakash L, Prakash S. Dual requirement for the yeast mms19 gene in DNA repair and RNA polymerase II transcription. Mol Cell Biol. 1996;16:6783–6793. doi: 10.1128/mcb.16.12.6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombaerts M, Tijsterman M, Verhage RA, Brouwer J. Saccharomyces cerevisiae mms19 mutants are deficient in transcription-coupled and global nucleotide excision repair. Nucleic Acids Res. 1997;25:3974–3979. doi: 10.1093/nar/25.20.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenney VJ, Barnes DJ, Lindahl T. Specific function of DNA ligase I in simian virus 40 DNA replication by human cell-free extracts is mediated by the amino-terminal non-catalytic domain. J Biol Chem. 1997;272:11550–11556. doi: 10.1074/jbc.272.17.11550. [DOI] [PubMed] [Google Scholar]
- Marinoni JC, Rossignol ME, Egly JM. Purification of the transcription/repair factor TFIIH and evaluation of its associated activities in vitro. Methods. 1997;12:235–253. doi: 10.1006/meth.1997.0476. [DOI] [PubMed] [Google Scholar]
- Masutani C, Araki M, Sugasawa K, van der Spek PJ, Yamada A, Uchida A, Maekawa T, Bootsma D, Hoeijmakers JHJ, Hanaoka F. Identification and characterization of XPC-binding domain of hHR23B. Mol Cell Biol. 1997;17:6915–6923. doi: 10.1128/mcb.17.12.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moggs JG, Yarema KJ, Essigmann JM, Wood RD. Analysis of incision sites produced by human cell extracts and purified proteins during nucleotide excision repair of a 1,3-intrastrand d(GpTpG)-cisplatin adduct. J Biol Chem. 1996;271:7177–7186. doi: 10.1074/jbc.271.12.7177. [DOI] [PubMed] [Google Scholar]
- Mu D, Park CH, Matsunaga T, Hsu DS, Reardon JT, Sancar A. Reconstitution of human DNA-repair excision nuclease in a highly defined system. J Biol Chem. 1995;270:2415–2418. doi: 10.1074/jbc.270.6.2415. [DOI] [PubMed] [Google Scholar]
- Mu D, Hsu DS, Sancar A. Reaction-mechanism of human DNA-repair excision nuclease. J Biol Chem. 1996;271:8285–8294. doi: 10.1074/jbc.271.14.8285. [DOI] [PubMed] [Google Scholar]
- Mu D, Wakasugi M, Hsu DS, Sancar A. Characterization of reaction intermediates of human excision-repair nuclease. J Biol Chem. 1997;272:28971–28979. doi: 10.1074/jbc.272.46.28971. [DOI] [PubMed] [Google Scholar]
- Podust VN, Hübscher U. Lagging strand DNA synthesis by calf thymus DNA polymerases α, β, δ, and ε in the presence of auxiliary proteins. Nucleic Acids Res. 1993;21:841–846. doi: 10.1093/nar/21.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Ge H, Gibbs E, Sancar A, Hurwitz J, Pan ZQ. Isolation and characterization of 2 human transcription factor IIH (TFIIH)-related complexes—ERCC2/CAK and TFIIH. Proc Natl Acad Sci. 1996;93:6482–6487. doi: 10.1073/pnas.93.13.6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SH, You ZY, Friedberg EC. The yeast RAD7 and RAD16 genes are required for postincision events during nucleotide excision-repair—In-vitro and in-vivo studies with rad7 and rad16 mutants and purification of a Rad7/Rad16-containing protein complex. J Biol Chem. 1998;273:29481–29488. doi: 10.1074/jbc.273.45.29481. [DOI] [PubMed] [Google Scholar]
- Robins P, Pappin DJC, Wood RD, Lindahl T. Structural and functional homology between mammalian DNase IV and the 5′ nuclease domain of Escherichia coli DNA polymerase I. J Biol Chem. 1994;269:28535–28538. [PubMed] [Google Scholar]
- Rossignol M, Kolb-Cheynel I, Egly JM. Substrate specificity of the cdk-activating kinase (CAK) is altered upon association with TFIIH. EMBO J. 1997;16:1628–1637. doi: 10.1093/emboj/16.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Adamczewski JP, Seroz T, Vermeulen W, Tassan JP, Schaeffer L, Nigg EA, Hoeijmakers JHJ, Egly JM. The MO15 cell-cycle kinase is associated with the TFIIH transcription DNA-repair factor. Cell. 1994;79:1093–1101. doi: 10.1016/0092-8674(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Shivji MKK, Podust VN, Hübscher U, Wood RD. Nucleotide excision repair DNA synthesis by DNA polymerase ε in the presence of PCNA, RFC, and RPA. Biochemistry. 1995;34:5011–5017. doi: 10.1021/bi00015a012. [DOI] [PubMed] [Google Scholar]
- Shivji MKK, Ferrari E, Ball K, Hübscher U, Wood RD. Resistance of human nucleotide excision repair synthesis in vitro to p21Cdn1. Oncogene. 1998;17:2827–2838. doi: 10.1038/sj.onc.1202352. [DOI] [PubMed] [Google Scholar]
- Shivji MKK, Moggs JG, Kuraoka I, Wood RD. Dual incision assays for nucleotide excision repair using DNA with a lesion at a specific site. In: Henderson DS, editor. DNA repair protocols: Eukaryotic systems. Totowa, NJ: Humana Press; 1999. pp. 373–392. [DOI] [PubMed] [Google Scholar]
- Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei Y-F, Moggs JG, Carter KC, Shell BK, Evans E, de Jong MC, et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell. 1996;86:811–822. doi: 10.1016/s0092-8674(00)80155-5. [DOI] [PubMed] [Google Scholar]
- Stefanini M, Vermeulen W, Weeda G, Giliani S, Nardo T, Mezzina M, Sarasin A, Harper JI, Arlett CF, Hoeijmakers JHJ, Lehmann AR. A new nucleotide-excision-repair gene associated with the disorder trichothiodystrophy. Am J Hum Genet. 1993;53:817–821. [PMC free article] [PubMed] [Google Scholar]
- Svejstrup JQ, Wang Z, Feaver WJ, Wu X, Bushnell DA, Donahue TF, Friedberg EC, Kornberg RD. Different forms of TFIIH for transcription and DNA repair: Holo-TFIIH and a nucleotide excision repairosome. Cell. 1995;80:21–28. doi: 10.1016/0092-8674(95)90447-6. [DOI] [PubMed] [Google Scholar]
- Svejstrup J, Vichi P, Egly J-M. The multiple roles of transcription/repair factor TFIIH. Trends Biochem Sci. 1996;21:346–350. [PubMed] [Google Scholar]
- Syväoja J, Linn S. Characterization of a large form of DNA polymerase δ from HeLa cells that is insensitive to proliferating cell nuclear antigen. J Biol Chem. 1989;264:2489–2497. [PubMed] [Google Scholar]
- Takayama K, Salazar EP, Lehmann A, Stefanini M, Thompson LH, Weber CA. Defects in the DNA repair and transcription gene ERCC2 in the cancer-prone disorder xeroderma pigmentosum group D. Cancer Res. 1995;55:5656–5663. [PubMed] [Google Scholar]
- Taylor EM, Broughton BC, Botta E, Stefanini M, Sarasin A, Jaspers NGJ, Fawcett H, Harcourt SA, Arlett CF, Lehmann AR. Xeroderma pigmentosum and trichothiodystrophy are associated with different mutations in the XPD (ERCC2) repair/transcription gene. Proc Natl Acad Sci. 1997;94:8658–8663. doi: 10.1073/pnas.94.16.8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirode F, Busso D, Coin F, Egly JM. Reconstitution of the transcription factor TFIIH: Assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell. 1999;3:87–95. doi: 10.1016/s1097-2765(00)80177-x. [DOI] [PubMed] [Google Scholar]
- Verhage R, Zeeman A, de Groot N, Gleig F, Bang D, van de Putte P, Brouwer J. The RAD7 and RAD16 genes, which are essential for pyrimidine dimer removal from the silent mating-type loci, are also required for repair of the nontranscribed strand of an active gene in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:6135–6142. doi: 10.1128/mcb.14.9.6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen W, van Vuuren AJ, Chipoulet M, Schaeffer L, Appeldoorn E, Weeda G, Jaspers NGJ, Priestly A, Arlett CF, Lehmann AR, et al. Three unusual repair deficiencies associated with transcription factor BTF2(TFIIH): Evidence for the existence of a transcription syndrome. Cold Spring Harbor Symp Quant Biol. 1994;59:317–329. doi: 10.1101/sqb.1994.059.01.036. [DOI] [PubMed] [Google Scholar]
- Wang ZG, Wei SG, Reed SH, Wu XH, Svejstrup JQ, Feaver WJ, Kornberg RD, Friedberg EC. The RAD7, RAD16, and RAD23 genes of Saccharomyces cerevisiae—Requirement for transcription-independent nucleotide excision repair in vitro and interactions between the gene products. Mol Cell Biol. 1997;17:635–643. doi: 10.1128/mcb.17.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda G, van Ham RCA, Vermeulen W, Bootsma D, van der Eb AJ, Hoeijmakers JHJ. A presumed DNA helicase encoded by the excision repair gene ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne's Syndrome. Cell. 1990;62:777–791. doi: 10.1016/0092-8674(90)90122-u. [DOI] [PubMed] [Google Scholar]
- Weiser T, Gassmann M, Thömmes P, Ferrari E, Hafkemeyer P, Hübscher U. Biochemical and functional comparison of DNA polymerases α, δ, and ε from calf thymus. J Biol Chem. 1991;266:10420–10428. [PubMed] [Google Scholar]
- Winkler, G.S., S.J. Araújo, U. Fiedler, W. Vermeulen, F. Coin, J.-M. Egly, J.H.J. Hoeijmakers, R.D. Wood, H.T.M. Timmers, and G. Weeda. 2000. TFIIH with inactive XPD helicase functions in transcription initiation but is defective in DNA repair. J. Biol. Chem. 275: (in press). [DOI] [PubMed]
- Wood RD, Robins P, Lindahl T. Complementation of the xeroderma pigmentosum DNA repair defect in cell-free extracts. Cell. 1988;53:97–106. doi: 10.1016/0092-8674(88)90491-6. [DOI] [PubMed] [Google Scholar]