

Abstract
Notch signal transduction appears to involve the ligand-induced intracellular processing of Notch, and the formation of a processed Notch-Suppressor of Hairless complex that binds DNA and activates the transcription of Notch target genes. This suggests that loss of either Notch or Su(H) activities should lead to similar cell fate changes. However, previous data indicate that, in the Drosophila blastoderm embryo, mesectoderm specification requires Notch but not Su(H) activity. The determination of the mesectodermal fate is specified by Single-minded (Sim), a transcription factor expressed in a single row of cells abutting the mesoderm. The molecular mechanisms by which the dorsoventral gradient of nuclear Dorsal establishes the single-cell wide territory of sim expression are not fully understood. We have found that Notch activity is required for sim expression in cellularizing embryos. In contrast, at this stage, Su(H) has a dual function. Su(H) activity was required to up-regulate sim expression in the mesectoderm, and to prevent the ectopic expression of sim dorsally in the neuroectoderm. We have shown that repression of sim transcription by Su(H) is direct and independent of Notch activity. Conversely, activation of sim transcription by Notch requires the Su(H)-binding sites. Thus, Notch signalling appears to relieve the repression exerted by Su(H) and to up-regulate sim transcription in the mesectoderm. We propose a model in which repression by Su(H) and derepression by Notch are essential to allow for the definition of a single row of mesectodermal cells in the blastoderm embryo.
Keywords: Notch, transcription, sim, Su(H), dorsoventral axis
The development of a multicellular organism from a single egg cell requires the specification of a wide range of cell types in a spatially regulated manner. Within a field of cells, generation of cell diversity can result from signals produced by a localized source. In the Drosophila embryo, the fundamental mechanisms by which positional information is translated into discrete spatial domains of gene expression are now well understood (St. Johnston and Nüsslein-Volhard 1992). Four localized maternal signals establish positional information along the anteroposterior and dorsoventral (DV) axes. Pattern formation along the DV axis begins with the establishment of asymmetries in the egg chamber of the ovary, leading to the formation of a DV gradient of nuclear localization of a transcriptional regulator, Dorsal, in the preblastoderm embryo. This gradient of nuclear Dorsal establishes distinct territories of gene activity that determine cell fate (for review, see Rusch and Levine 1996). High levels of Dorsal in ventral nuclei result in the transcriptional activation of the twist and snail genes that specify the mesoderm, whereas low levels of Dorsal in lateral nuclei activate neuroectoderm-determining genes, and the absence of Dorsal in dorsal nuclei allows the expression of genes specifying dorsal fates.
In contrast to these broad domains of gene expression, a single row of cells located between the mesoderm and the neuroectoderm express the single-minded (sim) gene, that specifies the mesectoderm (Crews et al. 1988; Thomas et al. 1988). The molecular mechanisms by which positional information selects, in a precise and reproducible way, a single cell on the basis of the graded distribution of Dorsal are not yet clear. However, some features of the transcriptional regulation of sim are understood. The sim gene contains two promoters. The early (E) promoter is activated in the mesectoderm during cellularization and remains active until stage 9. Genetic and molecular evidence indicate that the E promoter is activated by Dorsal and Twist, and repressed by Snail (Kosman et al. 1991; Leptin 1991; Kasai et al. 1992, 1998; Fig. 1A,B). The synergistic interaction between Dorsal and Twist contributes to define the sharp border of snail expression, which coincides with the boundary between the presumptive mesoderm and neuroectoderm (Kosman et al. 1991; Ip et al. 1992). Thus, repression by Snail appears to define the ventral border of sim expression. The late (L) promoter is activated in midline cells only after mesoderm invagination (Nambu et al. 1990, 1991; Muralidhar et al. 1993). From stage 8 onward, maintenance of sim transcription is regulated by a positive feedback loop (Wharton et al. 1994). Sim is a transcription factor of the bHLH–PAS family that heterodimerizes with Tango, another bHLH–PAS family member (Crews 1998). Sim/Tango heterodimers bind to the sim regulatory sequences and promote transcription from the E and L promoters in midline cells (Wharton et al. 1994; Crews 1998; Fig. 1C,D).
Figure 1.
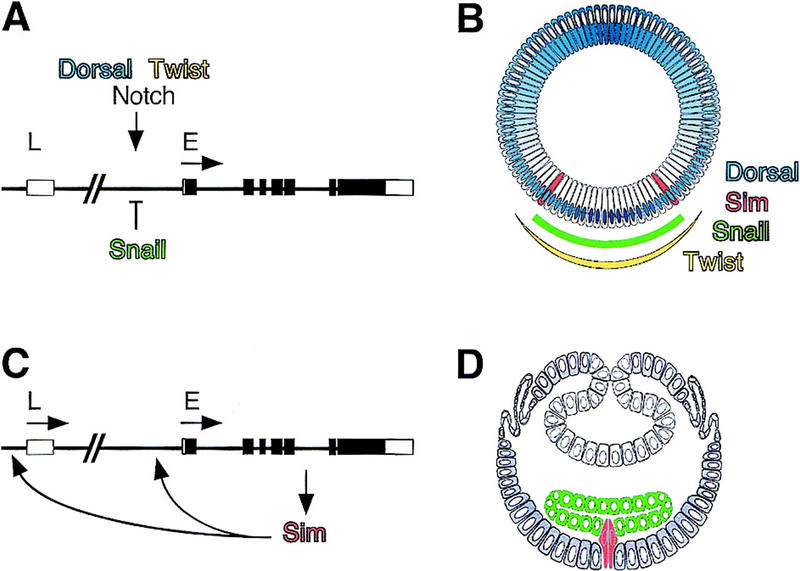
Regulation of sim transcription along the DV axis. (A) Genomic map of the sim gene showing positions of the early (E) and late (L) promoters and intron–exon structure. Open boxes correspond to untranslated regions, and closed boxes to coding sequence. At stage 5, transcriptional activation of the E promoter is positively regulated by Dorsal, Twist, and Notch signaling, and inhibited by Snail. (B) Schematic cross section of the Drosophila blastoderm embryo after cellularization (stage 5) (adapted from Campos-Ortega and Hartenstein 1997). The DV gradient of nuclear localization of Dorsal is shown in blue. Dorsal is mostly nuclear in ventral cells, and predominantly cytoplasmic in dorsal cells. In ventral nuclei, peak levels of Dorsal activate transcription of the mesoderm-determining genes twist and snail, and repress transcription of dorsal fate-determining genes. The domains of snail and twist expression are shown in green and yellow, respectively. In lateral nuclei, lower levels of Dorsal activate the transcription of neuroectoderm-determining genes, such as short gastrulation and rhomboid, which are repressed ventrally by Snail. Separating the ventral neuroectoderm from the mesoderm is the mesectoderm, a single row of cells on either side of the embryo. Mesectodermal cells, which express sim, are in red. In dorsal nuclei, levels of Dorsal are too low to repress the expression of genes determining dorsal fates. (C) Sim autoregulates its own transcription from both early (E) and Late (L) promoters from stage 8 onward. (D) Schematic cross section of a gastrulating embryo (stage 8). Adapted from Campos-Ortega and Hartenstein (1997). Following invagination of the mesoderm (in green), mesectodermal cells (in red) merge at the midline, and, after two rounds of cell division, differentiate into 22–26 midline neurons and glia per segment (Klambt et al. 1991).
Results from cell transplantation experiments indicate that cell–cell signaling between mesodermal and nonmesodermal cells is also required for the early expression of sim (Leptin and Roth 1994). A role for cell signaling in the regulation of the early expression of sim is further suggested by the finding that mesectoderm specification and expression of sim at gastrulation require Notch activity (Menne and Klambt 1994; Martin-Bermudo et al. 1995). Whether Notch signaling is required for the initial activation of the E promoter, or whether it participates in the Sim autoregulatory loop is unknown.
Cell–cell signaling mediated by Notch regulates the specification of a wide variety of cell types in Drosophila (Artavanis-Tsakonas et al. 1999). Activation of Notch appears to lead to its intracellular processing. The Notch intracellular domain (NICD) fragment that results from this proteolytic cleavage translocates into the nucleus and interacts with Suppressor of Hairless [Su(H)], a DNA-binding protein with no defined DNA-binding and activation/repression domains. DNA-bound complexes containing both NICD and Su(H) activate the transcription of Notch responsive genes (Jarriault et al. 1995; Kidd et al. 1998; Lecourtois and Schweisguth 1998; Schroeter et al. 1998; Struhl and Adachi 1998). In view of this observation, it is surprising that, in contrast with Notch, the activity of Su(H) was found to be largely dispensable for the formation of midline cells; in Su(H) mutant embryos, only a few midline cells failed to express sim at stage 10 (Lecourtois and Schweisguth 1995). This observation led to the hypothesis that Notch might signal in a Su(H)-independent manner to regulate sim expression.
Although Su(H) acts as a transcriptional activator in Notch signaling, its mammalian homolog, CBF1, was first identified as a transcriptional repressor (Dou et al. 1994; Henkel et al. 1994). Repression by CBF1 appears to be mediated by the binding of corepressors that facilitate the formation of repression complexes including histone deacetylase activity (Kao et al. 1998; Hsieh et al. 1999). It has been proposed that the binding of NICD to CBF1 disrupts this repression complex and facilitates the formation of an activation complex (Hsieh et al. 1996; Kao et al. 1998). However, the developmental significance of this transcriptional switch is unknown.
In this work we have examined the role that Notch and Su(H) play in regulating the early expression of sim in the mesectoderm. Using a newly isolated null allele of Su(H), we have found that Su(H) activity is required to up-regulate sim expression in the mesectoderm, and also to prevent the ectopic expression of sim dorsally in the neuroectoderm. Analysis of the sim promoter revealed that Su(H) directly represses, in a Notch-independent manner, the expression of sim in the mesectoderm, as well as in one to three rows of cells located dorsal to the mesectoderm. Notch was shown to antagonize the repression exerted by Su(H) and to up-regulate sim transcription in the mesectoderm. Regulation of sim expression by Notch appears to be mediated by Su(H). We propose a model in which uniform repression by Su(H) and local derepression by Notch contribute to define a single row of mesectodermal cells in the blastoderm embryo.
Results
Notch activity is required for sim expression in the mesectoderm
Notch activity is required for the expression of sim in gastrulating embryos (Martin-Bermudo et al. 1995). To test whether Notch signaling is required for the initial activation of the E promoter, the early expression of the sim gene was studied by in situ hybridization. In wild-type embryos, sim transcripts were first detected in a single row of mesectodermal cells at mid-cellularization (Fig. 2A). Expression of sim in these cells persisted during gastrulation, as they formed the midline (Fig. 2B,C). The role of Notch in activating sim transcription was analyzed in Notch mutant embryos derived from germ-line clones (GLCs), referred to as Notch mutant embryos hereafter. At stages 5–6, low levels of sim transcripts were detected in very few cells in the presumptive mesectoderm (Fig. 2D,E). By stage 8, sim was expressed in a few midline cells (Fig. 2F). These cells accumulated high levels of sim transcripts, possibly because sim autoregulation does not require Notch activity (see below). These data show that Notch signaling is required to activate sim expression in the mesectoderm at stage 5. High levels of sim transcripts were seen at the posterior pole, indicating that Notch activity is specifically required for sim expression in the mesectoderm.
Figure 2.
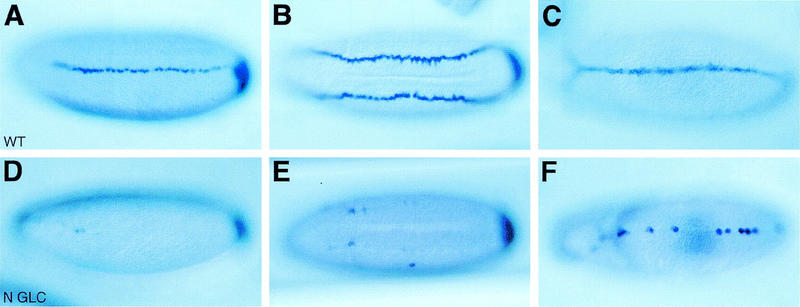
Regulation of sim expression by Notch signaling. In situ hybridization of wild-type (A–C) and Notch mutant (D–F) embryos showing the distribution of sim transcripts at stages 5 (A,D), 6 (B,E), and 8 (C,F). (A,B) Expression of sim was detected in a single row of mesectodermal cells in wild-type embryos [ventrolateral (A) and ventral (B) views]. Accumulation of sim transcripts at the posterior pole is out of focus. (C) Expression of sim in midline cells in a wild-type stage 8 embryo (ventral view). (D,E) In Notch mutant embryos, the expression of sim was restricted to a few cells in the mesectoderm. High levels of sim transcripts were seen at the posterior pole [ventrolateral (D) and ventral (E) views]. (F) The expression of sim was detected in very few midline cells in Notch mutant embryos at stage 8 (ventral view). Two null mutant alleles of Notch, N55e11 (D–F) and N5419 (not shown), were used in this study and gave similar results. In A–F anterior is to the left.
Expression of activated forms of Notch and Su(H) led to the ectopic expression of sim in the ventral neuroectoderm
We next studied the effect of ectopic activation of Notch signaling on sim expression. Ubiquitous expression of a constitutively activated form of Notch, Nintra, in a maternal-Gal4 [Matα4–GAL–VP16 (Hacker and Perrimon 1998)]/UAS–Nintra embryo led to the ectopic accumulation of sim transcripts in two to three rows of cells in the ventral neuroectoderm at stage 5 (Fig. 3A; see also F and G). Ectopic expression of sim was not observed in more dorsal neuroectodermal cells. Uniform expression of Nintra did not result in sim expression in the mesoderm. We conclude that the competence of the sim promoter to respond to Notch activation is patterned along the DV axis. For instance, a minimal concentration of nuclear Dorsal might be required for Nintra to ectopically activate sim expression, whereas repression by Snail in the mesoderm would prevent activation by Nintra. Ectopic expression of sim in the neuroectoderm persisted during gastrulation until stage 10 (Fig. 3B). At this stage, expression of sim could either be due to the direct effect of Nintra or to sim autoregulation. To distinguish between these two hypotheses, accumulation of Nintra was induced at different stages of development with a hs-Nintra transgene. Conditional expression of Nintra at stage 5 resulted in the ectopic expression of sim in the ventral neuroectoderm (Fig. 3C). As described above for UAS–Nintra, ectopic expression was not detected in the mesoderm, and, in the neuroectoderm, was found to gradually decrease dorsally. In contrast, heat-induced expression of Nintra at stages 6–10 did not alter sim expression (Fig. 3D). This result shows that the competence of the sim promoter to respond to Notch activation is temporally restricted to the early phase of sim activation at stage 5. Thus, the ectopic expression seen in Matα4–GAL–VP16/UAS–Nintra embryos at stages 8–10 (Fig. 3B) is likely to result from sim autoregulation.
Figure 3.
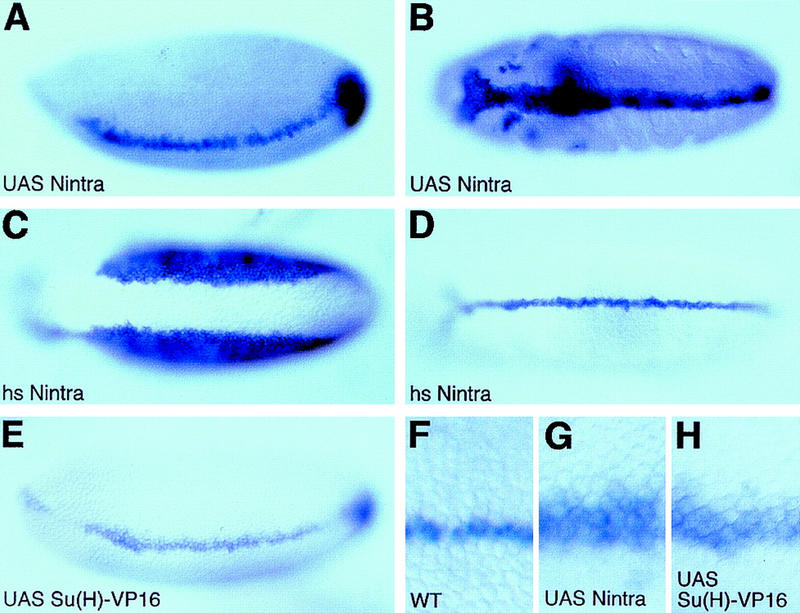
Ectopic expression of sim in the ventral neuroectoderm. In situ hybridization of Matα4–GAL–VP16/UAS–Nintra (A,B,G), hs-Nintra (C,D), Matα4–GAL–VP16/UAS–Su(H)–VP16 (E,H), and wild-type (F) embryos showing the distribution of sim transcripts. (A) Lateral view showing the ectopic expression of sim in the neuroectoderm of a stage 5 Matα4–GAL–VP16/UAS–Nintra embryo. Expression of sim was not detected in the mesoderm. Mesodermal cells were identified here as the cells that invaginate into the ventral furrow; in Matα4–GAL–VP16/UAS–Nintra embryos, all of the cells located between the two bands of sim-expressing cells invaginate to form the mesoderm. (B) Ventral view of a stage 10 embryo showing that this ectopic expression of sim was seen persisting after mesoderm invagination. (C) Transient overexpression of hs-Nintra at stage 5 induced the ectopic expression of sim in the neuroectoderm (ventral view). No sim expression was detected in the mesoderm. (D) Ventral view of a stage 8 hs-Nintra embryo showing that heat-induced expression of Nintra did not affect sim expression after mesoderm invagination. (E) Lateral view of a stage 5 Matα4–GAL–VP16/UAS–Su(H)–VP16 embryo. Nintra and Su(H)–VP16 similarly induced the ectopic expression of sim in the neuroectoderm. (F–H) Higher magnification views of the embryos shown in Figs. 2A (F; wild-type control), 3A (G; UAS–Nintra), and 3E [H; UAS–Su(H)–VP16].
Transcriptional activation by Nintra is thought to be mediated by Su(H). To investigate whether Su(H) can stimulate sim transcription, an activated form of Su(H), Su(H)–VP16, was expressed maternally. At stage 5, ectopic expression of sim was seen in the ventral neuroectoderm in embryos expressing Su(H)–VP16 in a uniform manner (Fig. 3E,H). In contrast, overexpression of wild-type Su(H) did not result in ectopic expression of sim in the neuroectoderm (data not shown), indicating that Su(H) does not, on its own, activate sim transcription. Thus, activated forms of Notch and Su(H) share the ability to deregulate the expression of the sim gene in a similarly restricted manner. These results indicate that the Notch–Su(H) pathway may be involved in the transcriptional activation of sim in the mesectoderm.
Molecular and genetical analysis of Su(H)del47, a null allele of Su(H)
The finding that Su(H)–VP16 activates sim transcription contrasts with our earlier hypothesis that Notch signaling regulates sim expression in a Su(H)-independent manner (Lecourtois and Schweisguth 1995). This hypothesis was based on our observation that most midline cells were correctly specified in Su(H)SF8 mutant embryos derived from GLC. However, because the molecular lesion in the SF8 allele of Su(H) is not known, it is possible that some residual Su(H) activity present in Su(H)SF8 embryos might be responsible for activating sim transcription.
To analyze the phenotype associated with a complete loss of Su(H) function, we first isolated deletion alleles of Su(H). A P element inserted in the 5′ UTR region of Su(H) was mobilized to recover small deletions at the Su(H) locus. One of the mutant alleles recovered in this screen, Su(H)del47, corresponds to a 1.9-kb deletion that removes the Su(H)–l(2)35Bg intergenic region, as well as the transcriptional start site and the ATG of both genes (Fig. 4A,B; see legend to Fig. 4 and Materials and Methods for details). Thus, del47 represents a null allele of Su(H).
Figure 4.

Molecular and phenotypical analysis of Su(H)del47. (A) Schematic representation of the l(2)35Bg–Su(H) genomic region. The four Su(H) exons appear as boxes. The ORF of Su(H) is shown in black. The positions of the Su(H) and l(2)35Bg transcriptional starts are indicated by arrows. Numbering refers to the transcriptional start of Su(H) (+1). The genomic structure of the l(2)35Bg gene has not been determined. The 4.2-kb EcoRI–EcoRI DNA fragment used as a probe for Southern blot analysis (B) is shown as a black line (EcoRI). The MR1 allele of Su(H) results from the insertion at position +65 of a defective P element. Its imprecise excision generated a 1.9-kb deletion called del47. Sequence analysis of a PCR product encompassing the deletion breakpoint showed that it actually corresponds to the substitution of a 1881-bp DNA fragment by 9 unrelated nucleotides. Su(H)del47 did not complement Su(H) and l(2)35Bg lethal alleles. In addition, a P element containing a 6.8-kb genomic DNA fragment encoding the transcription unit called B in Schweisguth and Posakony (1992), P[l(2)35Bg+], rescues the embryonic lethality associated with Su(H)del47 and l(2)35Bg mutant alleles (data not shown; the 6.8-kb DNA fragment used for genomic rescue is shown as a hatched bar). These results demonstrate that l(2)35Bg corresponds to the B transcription unit located 5′ to Su(H) and deleted in Su(H)del47. (B) Genomic Southern blot hybridization analysis of the del47 allele. EcoRI-digested genomic DNA was analyzed with the 4.2-kb EcoRI fragment shown in A as a hybridization probe. The 5.4-, 4.2-, and 2.3-kb bands correspond to the MR1, wild-type, and del47 alleles of Su(H), respectively. (C,D) Cuticular preparations of Su(H)SF8 (C) and Su(H)del47 P[l(2)35Bg+] (D) mutant embryos. The phenotype of Su(H)del47 appears to be slightly stronger than the one associated with Su(H)SF8, but is significantly milder than the one resulting from a loss of Notch activity (Zecchini et al. 1999).
The neurogenic cuticular phenotype associated with Su(H)del47 was analyzed in mutant embryos derived from maternal GLC and carrying a genomic copy of l(2)35Bg [see Materials and Methods for details; these embryos will be referred to as Su(H)del47 mutant embryos hereafter]. The dorsal cuticle produced by Su(H)del47 mutant embryos was found to be smaller than that of Su(H)SF8 mutant embryos (Fig. 4C,D), indicating that SF8 is probably not a null allele. Nevertheless, the cuticular phenotype associated with Su(H)del47 is significantly milder than the one associated with a complete loss of Notch activity (Zecchini et al. 1999).
Ectopic and reduced expression of sim in Su(H) mutant embryos
Next, we analyzed the expression of sim in Su(H)del47 null mutant embryos. At stage 5, lower levels of sim expression were observed in the mesectoderm of Su(H)del47 mutant embryos than were seen in wild-type embryos, and a few gaps were seen in the row of sim-expressing cells (cf. Figs. 5A,A′ and 2A). In addition, sim expression was no longer strictly restricted to a single row of cells, as low levels of sim transcripts were detected in one or two rows of cells in the dorsal neuroectoderm. Expression at the posterior pole, however, did not appear to be modified. At stages 6–8, two distinct phenotypes were observed. First, cells that did not express sim were occasionally found at the midline, creating gaps in the mesectoderm (Fig. 5B). These gaps might correlate with the partial loss of sim expression observed at stage 5. Secondly, cells expressing high levels of sim transcripts were observed in one to two rows of cells away from the midline at stage 8, forming small clusters (Fig. 5B,B′). This phenotype might result from the ectopic expression of sim in the neuroectoderm at stage 5. These results indicate that Su(H) activity is required both to restrict the expression of sim to a single row of cells, and to achieve a high level of sim expression in the mesectoderm at stage 5. However, unlike Notch, Su(H) activity is not strictly required for sim activation in the mesectoderm. Because null alleles were used, this reflects functional differences between Notch and Su(H) activities.
Figure 5.
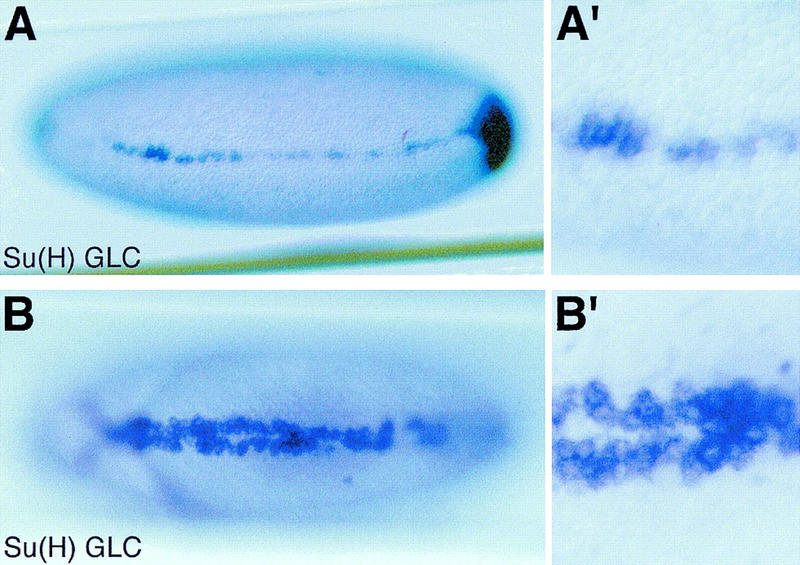
Reduced and ectopic expression of sim in Su(H) mutant embryos. In situ hybridization of Su(H)del47 P[l(2)35Bg+] mutant embryos showing the distribution of sim transcripts at stages 5 (A,A′) and 8 (B,B′). (A,A′) Low levels of sim expression were detected in two to three cell rows at stage 5 (lateral view; A′ is an enlarged view of the same embryo). Accumulation of sim transcripts at the posterior pole is out of focus. (B,B′) Occasionally, a few cells located at the midline failed to express sim, creating small gaps. In other regions, cells expressing high levels of sim transcripts were observed in one to two rows of cells away from the midline at stage 8, forming small clusters (ventral views). No discernable pattern of gaps and clusters was recognized.
Identification of 10 Su(H)-binding sites in the sim regulatory sequence
To gain insight into the molecular mechanisms by which Su(H) and Notch regulate sim expression, we first examined whether Su(H) regulates sim expression in a direct manner. Previous work established that the regulatory elements necessary for mesectodermal expression of sim are contained within a 2.8-kb genomic DNA region (Wharton et al. 1994; Kasai et al. 1998). Sequence analysis identified 10 putative Su(H)-binding sites, with 6 of these exactly matching the GTGRGAA consensus binding (sites Su4, Su5, Su7, Su8, Su9, and Su10 in Fig. 6A; Tun et al. 1994; Bailey and Posakony 1995; Lecourtois and Schweisguth 1995; Nellesen et al. 1999). In gel shift experiments, Su(H) was found to bind strongly to oligonucleotides corresponding to each of these sites (Fig. 6, C, lane 2, and D, lane 3; data not shown). Two additional sites, Su2 and Su6, matched the consensus RTGRGAR that accomodates nearly all sites that have been shown to bind Su(H) in vitro (Nellesen et al. 1999). These two sites were found to bind weakly to Su(H), both in direct binding assays (Fig. 6C, lanes 5,6,9,10) and in competition experiments (Fig. 6D, lanes 16–18,22–24). We also examined the ability of two noncanonical sites, Su1 and Su3, to bind Su(H) in vitro. Similar noncanonical sites have been shown previously to bind the mouse homolog of Su(H) in an in vitro selection experiment (Tun et al. 1994). We found that both Su1 and Su3 bound weakly to Su(H) (Fig. 6C, lanes 7,8,11,12, and D, lanes 19–21,25–27). Other sequences that differ from the RTGRGAR at a single position are not known to bind Su(H) in vitro (see site c in Fig. 6, A,C, lanes 13 and 14, and D, lanes 28–30). In these assays, binding specificity was demonstrated by point mutations in two nucleotides shown previously to be essential for target site recognition by Su(H) (Fig. 6, C, lane 4, and D, lanes 7–9; Tun et al. 1994; Bailey and Posakony 1995). Thus, these results indicate that the sim regulatory sequences contain at least 10 binding sites for Su(H). Eight of these sites are clustered in a 500-bp region that was shown previously to contain functional binding sites for Dorsal, Twist, and Snail (Kasai et al. 1992, 1998; Wharton et al. 1994). Moreover, the organization of this regulatory region has been conserved throughout evolution between D. melanogaster and D. virilis (Kasai et al. 1998; Fig. 6B). Together, these data strongly suggest that Su(H) regulates sim transcription directly.
Figure 6.
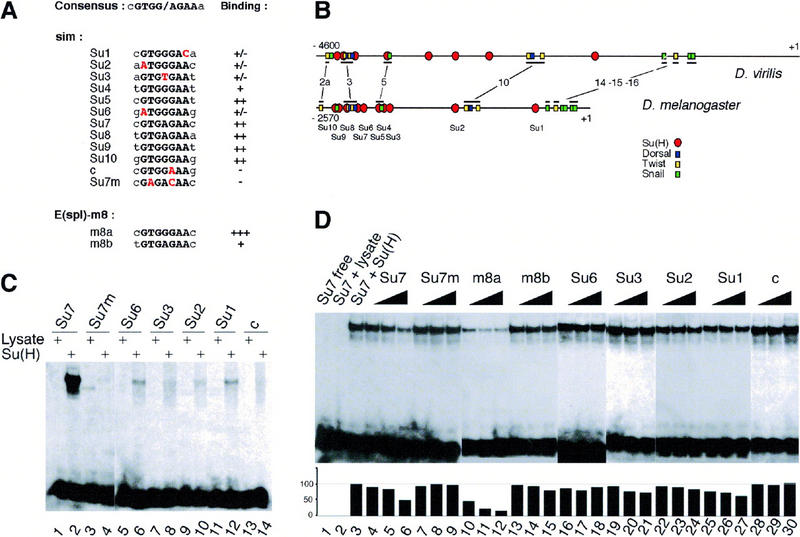
Identification of Su(H)-binding sites in the sim regulatory region. (A) Alignment of the predicted Su(H)-binding sites (Su1 to Su10) contained within a 2.8-kb upstream regulatory region of sim to the consensus Su(H)-binding site. The core consensus is shown in black uppercase letters; the two residues flanking the core consensus are less conserved. Sites Su4, Su5, Su7, Su8, Su9, and Su10 perfectly match the core consensus. Sites Su1, Su2, Su3, and Su6 differ at one conserved position (as indicated in red). Putative site c differs at a position shown previously to be essential for CBF-1/RBP-Jk binding (Tun et al. 1994). The sim regulatory sequence contains no other sites differing by less than two conserved nucleotides. Two binding sites from the Enhancer of split m8 gene [E(spl)-m8] were used as positive controls (Bailey and Posakony 1995; Lecourtois and Schweisguth 1995). For each putative site, the relative binding affinity, as estimated from gel shift assays, is indicated on the right. (+++, ++, +, +/−) Very high, high, medium, and weak binding affinity, respectively; (−) no detectable binding. Site c, which differs from the consensus at a strictly conserved position, did not bind Su(H). (B) Schematic diagrams of the upstream regions of the sim genes from D. virilis and D. melanogaster (Kasai et al. 1998). The position of the predicted Su(H)-binding sites is shown relative to the predicted Snail-, Twist-, and Dorsal-binding sites. The conserved regions that include known binding sites are underlined. These correspond to regions 2a, 3, 5, 10, 14, 15, and 16 described in Kasai et al. (1998). Four binding sites predicted to bind strongly Su(H) (Su9, Su8, Su7, and Su5) appeared to be clustered with predicted Snail-, Twist-, and Dorsal-binding sites in both D. virilis and D. melanogaster. Nucleotide numbering refers to the translation initiation codon. (C) Gel retardation analysis of Su(H) binding to putative sites from the sim regulatory region. Radiolabeled 17-mer oligonucleotides centered around putative Su(H) binding sites were tested for their ability to form retarded complex with Su(H) in an EMSA. One site perfectly matching the core consensus, Su7, as well as all the sites differing by one nucleotide to the core consensus (Su6, Su3, Su2, Su1, and c) were analyzed. For each probe, free lysate was used as a negative control (lanes 1,3,5,7,9,11,13). In vitro translated Su(H) proteins bound strongly to Su7 (lane 2). Weak binding was also observed with Su6, Su3, Su2, and Su1 (lanes 6,8,10,12, respectively). No detectable binding was observed with putative site c (lane 14). Binding specificity was demonstrated with an oligonucleotide containing two mutations in the Su7 site, Su7m (lane 4). These results are consistent with the binding specificity displayed by the mouse homolog of Su(H) (Tun et al. 1994), as the Su7m and c sites are the only ones that contain nucleotides differing from the consensus at strictly conserved position. (D) Determination of relative binding affinities by competition EMSA. Increasing amounts (5×, 10×, and 20×) of nonlabeled oligonucleotides were tested for their ability to compete with the formation of radiolabeled Su7–Su(H) complex (lanes 1–3). The m8a (lanes 10–12) and Su7 (lanes 4–6) oligonucleotides efficiently competed the binding of Su(H) to Su7. The m8b (lanes 13–15), Su6 (lanes 16–18), Su3 (lanes 19–21), Su2 (lanes 22–24), and Su1 (lanes 25–27) oligonucleotides competed only weakly. The Su7m (lanes 7–9) and c (lanes 28–30) oligonucleotides did not show significant competition activity. The plot underneath the EMSA gel shows the quantitation of the radioactivity contained within retarded complexes as measured by PhosphorImager analysis. The radioactivity measured in the absence of specific competitor was chosen as the 100% reference (lane 3).
Su(H) directly represses sim expression in the neuroectoderm
The role of these Su(H)-binding sites was examined in transgenic embryos. As shown previously (Wharton et al. 1994), the −2608/−127 sim promoter region directed the expression of a lacZ reporter gene in a single row of mesectodermal cells at stages 5 and 6 (Fig. 7A,A′). After mesoderm invagination, these cells were found to be the midline cells (not shown). The expression of sim–lacZ was then analyzed in Su(H)del47 and Notch mutant embryos. Ectopic expression of sim–lacZ was observed in the ventral neuroectoderm in Su(H)del47 mutant embryos (Fig. 8, cf. C,C′ with A,A′). In contrast, sim–lacZ was not expressed in Notch mutant embryos (Fig. 9A,A′). In addition, both Nintra and Su(H)–VP16 induced high levels of sim–lacZ expression in the ventral neuroectoderm (Figs. 8E,E′ and 9C,C′). Thus, sim–lacZ appeared to be regulated by Notch and Su(H) in a manner similar to the endogenous sim gene.
Figure 7.
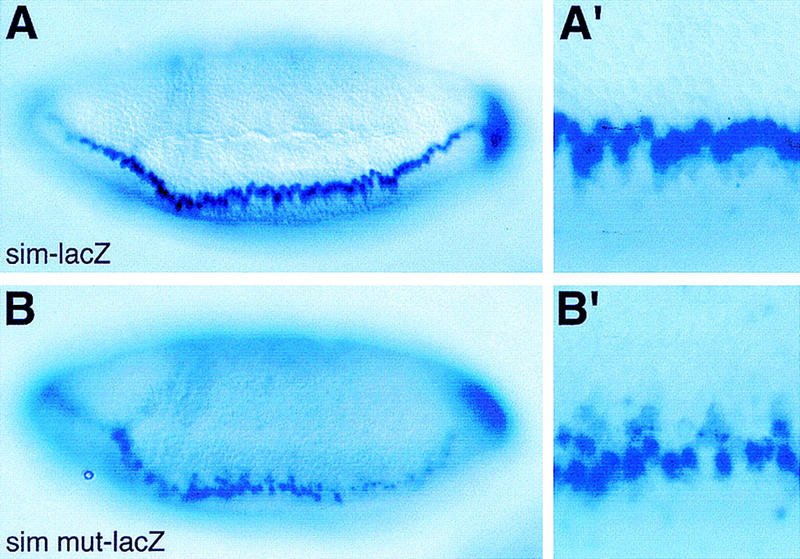
The Su(H)-binding sites are required to repress sim transcription in the neuroectoderm. Lateral views of wild-type sim–lacZ (A,A′) or simmut–lacZ (B,B′) embryos showing the distribution of lacZ transcripts at stage 6. lacZ transcripts accumulated in a single row of mesectodermal cells in sim–lacZ embryos (A,A′). In contrast, they were detected in several rows of cells extending into the ventral neuroectoderm in simmut–lacZ embryos (B,B′; cf. A′ and B′). (A′,B′) Enlarged views of the embryos shown in A and B. These embryos are homozygous and express two copies of the transgene (A,A′,B,B′).
Figure 8.
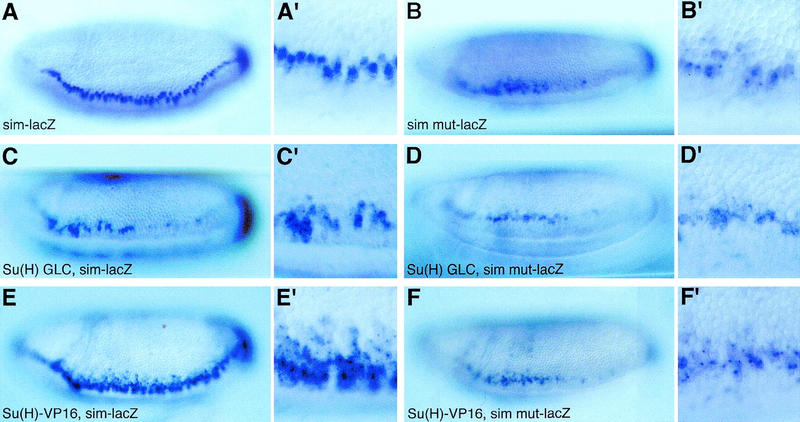
Su(H) acts via the Su(H)-binding sites to repress sim transcription. Lateral views of wild-type embryos (A,A′,B,B′) and ventrolateral views of Su(H)del47 P[l(2)35Bg+] mutant embryos (C,C′,D,D′) and Matα4–GAL–VP16/UAS–Su(H)–VP16 embryos (E,E′,F,F′) showing the expression pattern of sim–lacZ (A,A′,C,C′,E,E′) and simmut–lacZ (B,B′,D,D′,F,F′) transgenes at stage 6. All embryos have only one copy of the same transgene. A reduced level of staining was observed in embryos carrying one copy of the sim–lacZ or simmut–lacZ transgenes (A–B′) compared with embryos homozygous for the same transgenes (Fig. 7A–B′). For both sim–lacZ (C,C′) and simmut–lacZ (D,D′), low levels of lacZ expression were detected in two to three cell rows in Su(H) mutant embryos. Expression of Su(H)–VP16 resulted in the ectopic expression of sim–lacZ in the neuroectoderm (E,E′). In contrast, expression of simmut–lacZ did not appear to be significantly up-regulated by Su(H)–VP16 (cf. F,F′ with B,B′).
Figure 9.
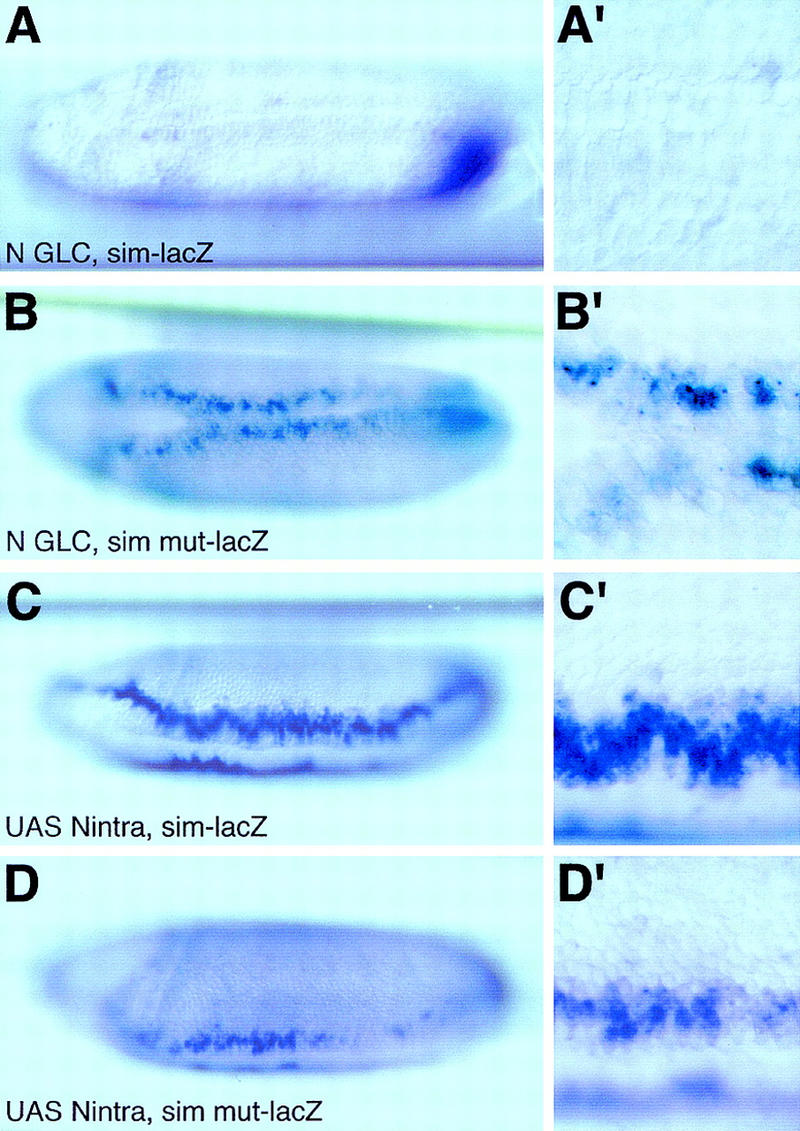
The repression mediated by the Su(H)-binding sites does not require Notch activity. In situ hybridization of N55e11 mutant embryos derived from GLC (A,A′,B,B′; ventral views) and Matα4–GAL–VP16/UAS–Nintra embryos (C,C′,D,D′; ventrolateral views) showing the expression pattern of sim–lacZ (A,A′,C,C′) and simmut–lacZ (B,B′,D,D′) transgenes at stage 6. Loss of Notch activity abolished sim–lacZ transcription (A,A′), but did not affect the expression of simmut–lacZ (cf. B,B′ with Fig. 7B,B′). Expression of activated Notch resulted in the ectopic expression of sim–lacZ in the neuroectoderm (C,C′). In contrast, expression of simmut–lacZ was not significantly modified by the expression of Nintra (cf. D,D′ with Fig. 7B,B′).
To test whether the binding of Su(H) to this promoter fragment is required to regulate sim expression, each Su(H)-binding site was mutated at two nucleotides, changing TG(G/A/T)GA into AG(G/A/T)CA, to produce simmut. These point mutations abolished the in vitro binding of Su(H) (Fig. 6C, lane 4; Bailey and Posakony 1995). Mutating all 10 Su(H)-binding sites resulted in lower levels of lacZ expression in the mesectoderm of simmut–lacZ embryos at stages 5 and 6 (Fig. 7B,B′). In addition, expression of simmut–lacZ was not restricted to the mesectoderm, but clearly extended into the ventral neuroectoderm. These results show that the Su(H)-binding sites are required to up-regulate sim expression in the mesectoderm, and to repress sim activation in the ventral neuroectoderm. A complete loss of Su(H) activity did not significantly modify the expression of simmut–lacZ (Fig. 8, cf. D,D′ with B,B′). Likewise, expression of simmut–lacZ was not changed following the ubiquitous expression of Su(H)–VP16 (Fig. 8F,F′). This indicates that Su(H)–VP16 acts via the Su(H)-binding sites identified above. We conclude that the ability of Su(H) to act on sim is greatly reduced when these binding sites are mutated, and that all the major Su(H) binding sites have been identified and mutated in simmut–lacZ. Together, these results show that Su(H) acts directly at the sim promoter both to promote its expression in mesectodermal cells and to repress its expression in the neuroectoderm.
Repression by Su(H) does not require Notch activity
We have established that the activity of Notch is required for the transcriptional activation of the sim gene in the mesectoderm, and that Su(H) directly regulates sim expression. However, both the sim gene and the simmut–lacZ construct that does not respond to activated Su(H) are expressed in mesectodermal cells in the complete absence of Su(H) activity (Figs. 5A,A′ and 8D,D′). These results might suggest that Notch signals, at least in part, in a Su(H)-independent manner to activate sim expression in the mesectoderm. Alternatively, our observation that Su(H) acts to repress sim expression raises the possibility that Notch might be required to antagonize repression by Su(H). To distinguish between these two possibilities, we have examined the expression of simmut–lacZ in Notch mutant embryos. We found that simmut–lacZ was expressed at a low level both in the mesectoderm and ectopically in the dorsal neuroectoderm (Fig. 9B,B′). This pattern is very similar to that observed for simmut–lacZ in wild-type embryos, and dramatically differs from the complete loss of sim–lacZ expression seen in Notch mutant embryos. This shows that the Su(H)-binding sites are required to repress sim transcription in the mesectoderm as well as in the neuroectoderm in the absence of Notch signaling. Furthermore, this demonstrates that repression of sim expression by Su(H), both in ventral neuroectodermal and mesectodermal cells, does not require Notch activity. We conclude that Su(H) acts as a Notch-independent repressor. Thus, we found no evidence for a Su(H)-independent function of Notch in the regulation of sim expression.
Finally, the expression of simmut–lacZ was not significantly up-regulated by Nintra in Matα4–GAL–VP16/UAS–Nintra embryos (Fig. 9D,D′), suggesting that activated Notch acts via the Su(H)-binding sites to regulate sim expression. This indicates that the up-regulation of sim transcription in the mesectoderm is mediated by Notch via Su(H).
Because Su(H) acts as a Notch-independent repressor, and because transcriptional activation by Notch requires the Su(H) binding sites, we conclude that activation of the Notch receptor in mesectodermal cells relieves the repression otherwise exerted by Su(H).
Discussion
This study demonstrates that Su(H) acts as a direct transcriptional repressor of the sim gene in blastoderm embryos, and that Notch signaling relieves this repression in cells directly juxtaposed to the mesoderm. This molecular switch is essential for precisely translating the DV gradient of nuclear localization of Dorsal into a single row of sim-expressing cells.
The sim gene is a direct transcriptional target of Notch signaling
Previous studies have established two phases in the transcriptional regulation of the sim gene in the mesectoderm. In the early activation phase, sim transcription is positively regulated by the transcription factors Dorsal and Twist, and repressed by Snail in the mesoderm. In the later phase, Sim regulates its own transcription in a positive feedback loop (Kasai et al. 1992; Wharton et al. 1994). Our results show that Notch acts as a positive regulator of sim transcription during the initial activation phase: The activity of Notch is required for the transcriptional activation of sim; whereas expression of Nintra results in the ectopic expression of sim. Using a heat-inducible promoter, however, we have shown that Nintra is able to activate sim expression only during the initial phase of sim regulation. This effect of Nintra is restricted to cells devoid of Snail but with a minimal amount of Dorsal and/or Twist, that is, cells of the ventral neuroectoderm. Thus, the initial activation of sim transcription appears to be under the combinatorial control of a signaling input from the Notch receptor and of selector proteins Dorsal, Twist, and Snail. This regulatory mechanism might ensure that the sim gene is responsive to Notch signaling in only a few cells and at a defined developmental stage.
Regulation of sim expression by Notch signaling is likely to be directly mediated by Su(H): (1) Ten Su(H)-binding sites were identified in a 2.8-kb DNA fragment containing the upstream regulatory sequences of the sim gene; (2) these binding sites are required for a high level of sim expression in mesectodermal cells; (3) these sites mediate the regulatory effects of Nintra; (4) finally, loss of Notch activity has no effect on a sim promoter carrying mutated Su(H)-binding sites. We propose that Su(H) and NICD form a DNA-bound complex that activates the transcription of sim in the mesectoderm. How activation of Notch signaling may be patterned itself along the DV axis is discussed below.
Su(H) as a transcriptional repressor in Drosophila
We have presented evidence that Su(H) not only mediates the Notch-dependent activation of sim transcription, but also acts as a transcriptional repressor. This latter conclusion is supported by the two following findings. First, a complete loss of Su(H) activity led to weak ectopic expression of sim in the neuroectoderm. Second, the deletion of all of the Su(H)-binding sites from the sim regulatory region also resulted in ectopic activation of the sim promoter in the ventral neuroectoderm. In Notch mutant embryos, repression by Su(H) was observed not only in the neuroectoderm, but also in the mesectoderm. Because Su(H) is expressed maternally (Schweisguth and Posakony 1992), we speculate that uniformly localized Su(H) might repress the activation of sim transcription in all of the cells in which Notch is not activated.
Ectopic expression of the simmut–lacZ reporter gene in the neuroectoderm was observed in wild-type embryos as well as in embryos that completely lacked Notch activity. This indicates that repression of sim expression by Su(H) in the neuroectoderm does not require Notch activity, and that a mechanism independent of both Notch and Su(H) directs the transcriptional activation of sim specifically in mesectodermal and ventral neuroectodermal cells. This mechanism probably involves activation by Dorsal and/or Twist. Thus, repression might only be revealed experimentally in cells that do not express the Snail repressor and in which low levels of sim transcription can be induced by low nuclear concentrations of Dorsal and/or Twist. Our results therefore suggest that Su(H) represses the transcription of the sim gene in a Notch-independent manner, and that Notch activates the expression of sim in the mesectoderm in a Su(H)-dependent manner.
Su(H) mediates a transcriptional switch in Notch signaling
This study provides the first evidence that Su(H) can act as a transcriptional repressor in Drosophila, and that its repression activity is inhibited by the activation of the Notch receptor. In mammals it has been suggested that the binding of processed Notch to CBF1 competes with the binding of corepressors to CBF1 to promote the formation of an activation complex (Hsieh et al. 1996; Kao et al. 1998). Our results suggest that Su(H) might mediate such a transcriptional switch at the sim promoter in mesectodermal cells.
This regulatory mechanism, in which transcriptional repression is inhibited by a signaling input, may be a general feature of Notch-mediated gene regulation. Consistent with this view, repression by Su(H) might contribute to the difference seen between Notch and Su(H) mutant cuticular phenotypes. Similarly, the cuticular phenotype associated with a deletion removing all of the bHLH–Enhancer of split genes, but not groucho, also appears to be more severe than the one associated with a complete loss of Su(H) function (V. Morel, unpubl.). Because the bHLH–Enhancer of split genes are direct transcriptional targets of Su(H) during neurogenesis (Bailey and Posakony 1995; Lecourtois and Schweisguth 1995), it is suggested that Su(H) might also act as a transcriptional repressor of the Enhancer of split genes.
Finally, our finding that Su(H) can repress a Notch target gene indicate that phenotypic differences between Notch and Su(H) mutations do not necessarily imply that Notch signals in a Su(H)-independent manner.
Defining a single row of mesectodermal cells along the DV axis: a model
How is a single-cell wide territory of sim expression established on the basis of the nuclear gradient of Dorsal? Our data, together with previous studies (Kosman et al. 1991; Leptin 1991; Ip et al. 1992; Kasai et al. 1992, 1998), suggest the following model (Fig. 10). In the mesoderm, transcriptional activation of sim by Dorsal and Twist is inhibited by Snail. Whether Su(H) and/or Notch play any role in these cells is not known. In more dorsal cells that do not accumulate Snail, we propose that positive regulation of sim by low levels of Dorsal and Twist is antagonized by Su(H). However, in cells bordering the mesoderm, negative regulation by Su(H) would be relieved locally by Notch signaling. This would lead to the specific expression of sim in these cells, which will then form the mesectoderm.
Figure 10.
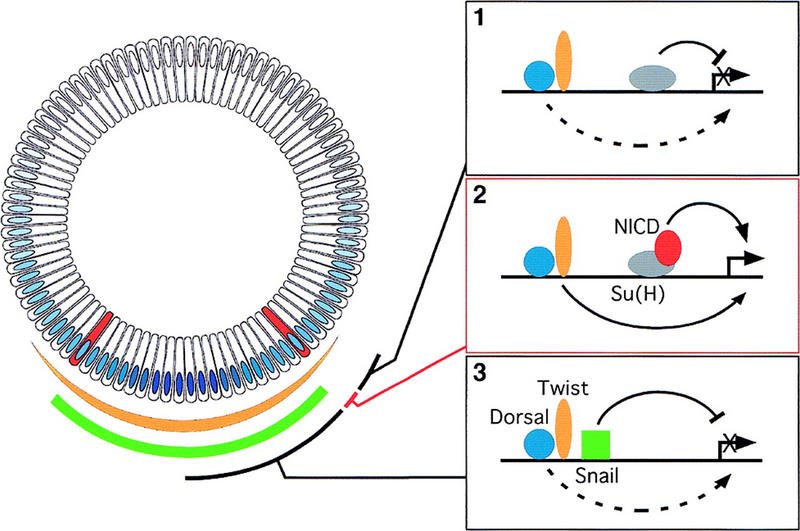
A model for the transcriptional activation of the sim gene in a single row of cells. Cross-section of a blastoderm embryo at stage 5. As in Fig. 1, the DV gradient of nuclear localization of Dorsal is shown in blue, the mesectoderm is in red. The sharp border of Snail accumulation (in green) coincides with the mesoderm–mesectoderm boundary. Accumulation of Twist (in yellow) gradually fades away in the neuroectoderm. In the neuroectoderm (1), transcriptional activation by low levels of Dorsal and Twist is inhibited by the Su(H)-mediated repression. In the mesectoderm (2), Notch activation relieves the repression mediated by Su(H) and, together with Dorsal and Twist, stimulates the expression of sim. In the mesoderm (3), Snail represses sim transcription, and overcomes the positive regulation mediated by Dorsal and Twist. Whether Su(H) and Notch participate in regulating sim in the mesoderm is unknown.
An important feature of this model is that Notch signaling overcomes repression by Su(H) only in the single row of cells abutting the mesoderm. One possible explanation for this is that Notch participates in the contact-dependent reception of a mesodermal signal. Results from nuclear transplantation experiments support the existence of a mesodermal signal. When transplanted into snail/twist double mutant embryos that do not express sim, wild-type nuclei can induce the expression of sim in neighboring mutant cells (Leptin and Roth 1994). This result suggests that, in wild-type embryos, mesodermal cells may produce an inductive signal that activates sim transcription in the mesectoderm. Although the molecular nature of this signal is not known, we speculate that this mesodermal signal might participate in the activation of Notch.
Consistent with the view that Notch is specifically activated in ventral cells, changes in the subcellular distribution of both Notch and Delta have been observed ventrally in stage 5 embryos. First, lower levels of Notch are found in ventral cells as the ventral furrow forms (Fehon et al. 1991). Second, in cellularized embryos, Delta is found at the cell membrane, except in ventral cells, in which it predominantly accumulates in vesicles (Kooh et al. 1993). Both down-regulation of Notch and vesicular accumulation of Delta are consistent with Delta activating Notch in ventral cells in stage 5 embryos (because Snail represses sim transcription, activation of Notch in the mesoderm may have no effect on sim transcription). It will thus be of interest to determine whether these changes in the subcellular distribution of Notch and Delta can be observed in both mesodermal and mesectodermal cells, but not in the more dorsal neuroectodermal cells.
In conclusion, repression by Su(H) can be viewed as a refining mechanism ensuring that Notch target genes are expressed only in cells reaching a high threshold of Notch activation. In the early embryo, repression of sim expression allows for the definition of a single row of mesectodermal cells. In these cells, a high level of Notch activity might be induced by a juxtacrine (contact-dependent) inductive signal produced by the mesoderm. In view of this hypothesis, the sharp mesodermal boundary defined by snail expression would be shifted dorsally by one cell, thereby defining a single row of mesectodermal cells.
Materials and methods
Flies
The del47 null allele of Su(H) was generated by the imprecise excision of an unmarked P element, MR1, inserted in the 5′ UTR of Su(H) (Schweisguth and Posakony 1992). This deletion allele was first selected as a strong dominant suppressor of the Hairless haplo-insufficient bristle phenotype. A rough eye phenotype was also observed in trans over a hypomorphic allele, Su(H)MR1, indicating that del47 is a strong loss of Su(H) function. From the 118 mutations that suppressed the Hairless bristle phenotype, 3 gave a rough eye phenotype over Su(H)MR1. These three alleles were analyzed at the molecular level. The del47 allele was the only mutation associated with a deletion detectable by Southern blot analysis.
Embryos homozygous for Su(H)del47 died as late embryos. This embryonic lethality resulted from the loss of l(2)35Bg activity, as it was rescued by a P[l(2)35Bg+] transgene. Because loss of l(2)35Bg activity blocked oogenesis, Su(H)del47 was recombined with a P[l(2)35Bg+] transgene. A Su(H)del47 FRT40A P[l(2)35Bg+] chromosome was used to produce Su(H)del47 mutant embryos derived from GLC as described previously (Lecourtois and Schweisguth 1995). Females producing GLC embryos were crossed with Su(H)del47/CyO ftz–lacZ, sim–lacZ Su(H)del47/CyO ftz–lacZ, or simmut–lacZ Su(H)del47/CyO ftz–lacZ males. Notch mutant embryos derived from GLC were produced by the FLP–DFS technique (Chou and Perrimon 1996) with N55e11 and N5419 [gifts of A. Martinez-Arias (Cambridge University, UK) and R. Nusse (Stanford University, CA), respectively]. Males carrying a PlacZ insertion at the polyhomeotic locus on the X chromosome were used to identify Notch hemizygous embryos. No paternal zygotic rescue of sim expression was detected.
Uniform accumulation of activated forms of Su(H) and Notch was obtained by crossing females carrying a maternal Gal4 driver, Matα4–GAL–VP16 (Hacker and Perrimon 1998), to males carrying a UAS–Su(H)–VP16 [gift of T. Lieber (Kidd et al. 1998)] or a UAS–Nintra (gift of M. Haenlin, CNRS, IGBMC, Strasbourg, France; Nintra includes amino acids 1789–2703). In some experiments, the Matα4–GAL–VP16 females were also homozygous for the sim–lacZ or simmut–lacZ reporter genes. Expression of Nintra was also induced by a 15-min heat shock at 37°C, followed by a 15-min recovery period at 25°C prior to fixation, in hs-Nintra embryos (Lieber et al. 1993).
Plasmids and germ-line transformation
P[l(2)35Bg+] corresponds to a 6.8-kb genomic DNA fragment that encodes the transcription unit called B in (Schweisguth and Posakony 1992). P[l(2)35Bg+] results from the insertion of BglII–XbaI and XbaI–PstI fragments purified from phage λ4–16 (Schweisguth and Posakony 1992) into CaSpeR opened by PstI and BamHI.
The −2608/−127 sim regulatory region was isolated from genomic DNA by PCR (numbering refers to the ATG of the sim ORF). All Su(H)-binding sites were mutated by the oligonucleotide-mediated mutagenesis method described by Kunkel (1985). At each site, two point mutations were introduced, as shown in boldface: (G/A)TG(G/A/T)GA(A/C) was changed into (G/A)AG(G/A/T)CA(A/C). Each mutation was verified by sequencing. The wild-type and mutated −2608/−127 sim regulatory regions were then subcloned into pCaSpeRβgal (Thummel et al. 1988) as EcoRI–KpnI fragments. P[w+] transposable elements were introduced into the germ line of w1118 recipient embryos by coinjection with a Δ2–3 helper plasmid. 8 P[sim–lacZ], 6 P[simmut–lacZ], and 3 P[l(2)35Bg+]-independent transformant lines were obtained.
Molecular biology
Southern blot analysis was performed as described previously (Schweisguth and Posakony 1992). The exact molecular structure of the del47 breakpoints was determined by sequencing a 0.8-kb PCR product generated with the following oligonucleotides: TGGGTGCTGTCCGACAAGATGCCGAC and TCCGTAATGTATGAAACCATCGCGCAC. The following internal primer was used for sequencing: CTTTGCGAATGACAACCTGGCTGAGG. The sequence of the breakpoint −1145/+737 is GAGACATACATACGACA. Underlined are the 9 nucleotides of unknown origin that replaced the 1881 nucleotides missing in del47.
Gel shift assays
The experimental conditions for the in vitro synthesis of the Su(H) protein and for gel retardation were as described previously (Brou et al. 1994). The ability of Su(H) to bind specific sites within the sim regulatory region was tested with 17-mer double-stranded oligonucleotides centered around each putative Su(H)-binding site. The sequences of all oligonucleotides used in this study are available on request. The amount of radioactivity in the retarded complexes were determined by PhosphorImager analysis (Fuji Bas 1000).
In situ hybridization
The synthesis of DIG-labeled RNA probes and in situ hybridization were as described previously (Lecourtois and Schweisguth 1995). Selected embryos were mounted in Spurr's embedding medium (Fullam Inc.) into glass capillaries (inner diam. 0.2 mm).
Acknowledgments
We thank S. Crews, M. Haenlin, F. Jimenez, T. Lieber, A. Martinez-Arias, F. Maschat, R. Nusse, and L. Seugnet for providing flies and DNA. We thank O. Massiani for technical assistance and all members of the laboratory for discussions and critical reading. This work was supported by grants from the CNRS (ATIPE Bio Dev), MENESR (ACC SV4), FRM, and ARC (ARC no. 9659).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL schweisg@wotan.ens.fr; FAX 33 1 44 32 38 87.
References
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: Cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Bailey AM, Posakony JW. Suppressor of Hairless directly activates transcription of Enhancer of split complex genes in response to Notch receptor activity. Genes & Dev. 1995;9:2609–2622. doi: 10.1101/gad.9.21.2609. [DOI] [PubMed] [Google Scholar]
- Brou C, Logeat F, Lecourtois M, Vandekerckhove J, Kourilsky P, Schweisguth F, Israel A. Inhibition of the DNA-binding activity of Drosophila Suppressor of Hairless and of its human homolog, KBF2/RBP-J κ, by direct protein–protein interaction with Drosophila hairless. Genes & Dev. 1994;8:2491–2503. doi: 10.1101/gad.8.20.2491. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega JA, Hartenstein V. The embryonic development of Drosophila melanogaster. Berlin, Germany: Springer; 1997. [Google Scholar]
- Chou T-B, Perrimon N. The autosomal FLP-DFS technique for generating germ-line mosaics in Drosophila melanogaster. Genetics. 1996;144:1673–1679. doi: 10.1093/genetics/144.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews ST. Control of cell lineage-specific development and transcription by bHLH-PAS proteins. Genes & Dev. 1998;12:607–620. doi: 10.1101/gad.12.5.607. [DOI] [PubMed] [Google Scholar]
- Crews ST, Thomas JB, Goodman CS. The Drosophila single-minded gene encodes a nuclear protein with sequence similarity to the per gene product. Cell. 1988;52:143–151. doi: 10.1016/0092-8674(88)90538-7. [DOI] [PubMed] [Google Scholar]
- Dou S, Zeng X, Cortes P, Erdjument-Bromage H, Tempst P, Honjo T, Vales LD. The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol Cell Biol. 1994;14:3310–3319. doi: 10.1128/mcb.14.5.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehon RG, Johansen K, Rebay I, Artavanis-Tsakonas S. Complex cellular and subcellular regulation of Notch expression during embryonic and imaginal development of Drosophila: Implications for Notch function. J Cell Biol. 1991;113:657–669. doi: 10.1083/jcb.113.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker U, Perrimon N. DRhoGEF2 encodes a member of the Dbl family of oncogenes and controls cell shape changes during Drosophila gastrulation. Genes & Dev. 1998;12:274–284. doi: 10.1101/gad.12.2.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel T, Ling PD, Hayward SD, Peterson MG. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science. 1994;265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- Hsieh JJ, Henkel T, Salmon P, Robey E, Peterson MG, Hayward SD. Truncated mammalian Notch1 activates CBF1/RBPJk-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol Cell Biol. 1996;16:952–959. doi: 10.1128/mcb.16.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh JJ, Zhou S, Chen L, Young DB, Hayward SD. CIR, a corepressor linking the DNA binding factor CBF1 to the histone deacetylase complex. Proc Natl Acad Sci. 1999;96:23–28. doi: 10.1073/pnas.96.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip YT, Park RE, Kosman D, Yazdanbakhsh K, Levine M. Dorsal-twist interactions establish snail expression in the presumptive mesoderm of the Drosophila embryo. Genes & Dev. 1992;6:1518–1530. doi: 10.1101/gad.6.8.1518. [DOI] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Kao HY, Ordentlich P, Koyano-Nakagawa N, Tang Z, Downes M, Kintner CR, Evans RM, Kadesch T. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes & Dev. 1998;12:2269–2277. doi: 10.1101/gad.12.15.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai Y, Nambu JR, Lieberman PM, Crews ST. Dorsal-ventral patterning in Drosophila: DNA binding of Snail protein to the single-minded gene. Proc Natl Acad Sci. 1992;89:3414–3418. doi: 10.1073/pnas.89.8.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai Y, Stahl S, Crews S. Specification of the Drosophila CNS midline cell lineage: Direct control of single-minded transcription by dorsal/ventral patterning genes. Gene Expr. 1998;7:171–189. [PMC free article] [PubMed] [Google Scholar]
- Kidd S, Lieber T, Young MW. Ligand-induced cleavage and regulation of nuclear entry of Notch in Drosophila melanogaster embryos. Genes & Dev. 1998;12:3728–3740. doi: 10.1101/gad.12.23.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klambt C, Jacobs JR, Goodman CS. The midline of the Drosophila central nervous system: A model for the genetic analysis of cell fate, cell migration, and growth cone guidance. Cell. 1991;64:801–815. doi: 10.1016/0092-8674(91)90509-w. [DOI] [PubMed] [Google Scholar]
- Kooh PJ, Fehon RG, Muskavitch MA. Implications of dynamic patterns of Delta and Notch expression for cellular interactions during Drosophila development. Development. 1993;117:493–507. doi: 10.1242/dev.117.2.493. [DOI] [PubMed] [Google Scholar]
- Kosman D, Ip YT, Levine M, Arora K. Establishment of the mesoderm-neuroectoderm boundary in the Drosophila embryo. Science. 1991;254:118–122. doi: 10.1126/science.1925551. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecourtois M, Schweisguth F. The neurogenic Suppressor of Hairless DNA-binding protein mediates the transcriptional activation of the Enhancer of split complex genes triggered by Notch signaling. Genes & Dev. 1995;9:2598–2608. doi: 10.1101/gad.9.21.2598. [DOI] [PubMed] [Google Scholar]
- ————— Indirect evidence for Delta-dependent intracellular processing of notch in Drosophila embryos. Curr Biol. 1998;8:771–774. doi: 10.1016/s0960-9822(98)70300-8. [DOI] [PubMed] [Google Scholar]
- Leptin M. Twist and Snail as positive and negative regulators during Drosophila mesoderm development. Genes & Dev. 1991;5:1568–1576. doi: 10.1101/gad.5.9.1568. [DOI] [PubMed] [Google Scholar]
- Leptin M, Roth S. Autonomy and non-autonomy in Drosophila mesoderm determination and morphogenesis. Development. 1994;120:853–859. doi: 10.1242/dev.120.4.853. [DOI] [PubMed] [Google Scholar]
- Lieber T, Kidd S, Alcamo E, Corbin V, Young MW. Antineurogenic phenotypes induced by truncated Notch proteins indicate a role in signal transduction and may point to a novel function for Notch in nuclei. Genes & Dev. 1993;7:1949–1965. doi: 10.1101/gad.7.10.1949. [DOI] [PubMed] [Google Scholar]
- Martin-Bermudo MD, Carmena A, Jimenez F. Neurogenic genes control gene expression at the transcriptional level in early neurogenesis and in mesectoderm specification. Development. 1995;121:219–224. doi: 10.1242/dev.121.1.219. [DOI] [PubMed] [Google Scholar]
- Menne TV, Klambt C. The formation of commissures in the Drosophila CNS depends on the midline cells and on the Notch gene. Development. 1994;120:123–133. doi: 10.1242/dev.120.1.123. [DOI] [PubMed] [Google Scholar]
- Muralidhar MG, Callahan CA, Thomas JB. Single-minded regulation of genes in the embryonic midline of the Drosophila central nervous system. Mech Dev. 1993;41:129–138. doi: 10.1016/0925-4773(93)90043-w. [DOI] [PubMed] [Google Scholar]
- Nambu JR, Franks RG, Hu S, Crews ST. The single-minded gene of Drosophila is required for the expression of genes important for the development of CNS midline cells. Cell. 1990;63:63–75. doi: 10.1016/0092-8674(90)90288-p. [DOI] [PubMed] [Google Scholar]
- Nambu JR, Lewis JO, Wharton KA, Jr, Crews ST. The Drosophila single-minded gene encodes a helix-loop-helix protein that acts as a master regulator of CNS midline development. Cell. 1991;67:1157–1167. doi: 10.1016/0092-8674(91)90292-7. [DOI] [PubMed] [Google Scholar]
- Nellesen DT, Lai EC, Posakony JW. Discrete enhnacer elements mediate selective responsiveness of Enhancer of split complex genes to common transcriptional activators. Dev Biol. 1999;213:33–53. doi: 10.1006/dbio.1999.9324. [DOI] [PubMed] [Google Scholar]
- Rusch J, Levine M. Threshold responses to the dorsal regulatory gradient and the subdivision of primary tissue territories in the Drosophila embryo. Curr Opin Genet Dev. 1996;6:416–423. doi: 10.1016/s0959-437x(96)80062-1. [DOI] [PubMed] [Google Scholar]
- Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- Schweisguth F, Posakony JW. Suppressor of Hairless, the Drosophila homolog of the mouse recombination signal-binding protein gene, controls sensory organ cell fates. Cell. 1992;69:1199–1212. doi: 10.1016/0092-8674(92)90641-o. [DOI] [PubMed] [Google Scholar]
- St. Johnston D, Nüsslein-Volhard C. The origin of pattern and polarity in the Drosophila embryo. Cell. 1992;68:201–219. doi: 10.1016/0092-8674(92)90466-p. [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A. Nuclear access and action of Notch in vivo. Cell. 1998;93:649–660. doi: 10.1016/s0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- Thomas JB, Crews ST, Goodman CS. Molecular genetics of the single-minded locus: A gene involved in the development of the Drosophila nervous system. Cell. 1988;52:133–141. doi: 10.1016/0092-8674(88)90537-5. [DOI] [PubMed] [Google Scholar]
- Thummel CS, Boulet AM, Lipshitz HD. Vectors for Drosophila P-element-mediated transformation and tissue culture transfection. Gene. 1988;74:445–456. doi: 10.1016/0378-1119(88)90177-1. [DOI] [PubMed] [Google Scholar]
- Tun T, Hamaguchi Y, Matsunami N, Furukawa T, Honjo T, Kawaichi M. Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res. 1994;22:965–971. doi: 10.1093/nar/22.6.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton KA, Jr, Franks RG, Kasai Y, Crews ST. Control of CNS midline transcription by asymmetric E-box-like elements: Similarity to xenobiotic responsive regulation. Development. 1994;120:3563–3569. doi: 10.1242/dev.120.12.3563. [DOI] [PubMed] [Google Scholar]
- Zecchini V, Brennan K, Martinez-Arias A. An activity of Notch regulates JNK signalling and affects dorsal closure in Drosophila. Curr Biol. 1999;9:460–469. doi: 10.1016/s0960-9822(99)80211-5. [DOI] [PubMed] [Google Scholar]